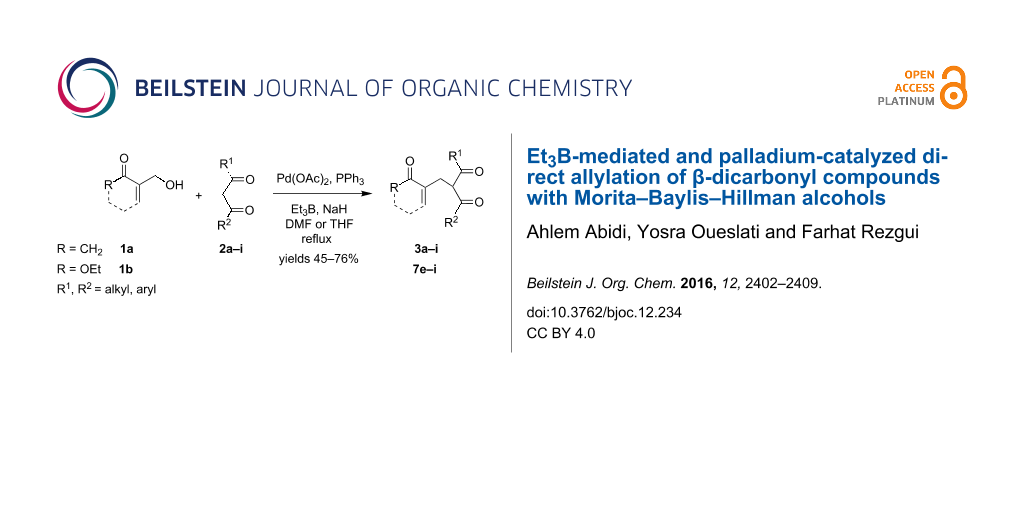
Abstract
A practical and efficient palladium-catalyzed direct allylation of β-dicarbonyl compounds with both cyclic and acyclic Morita–Baylis–Hillman (MBH) alcohols, using Et3B as a Lewis acid promoter, is described herein. A wide range of the corresponding functionalized allylated derivatives have been obtained in good yields and with high selectivity.

Graphical Abstract
Introduction
In nucleophilic allylic substitutions, π-allylpalladium complexes are useful intermediates for the construction of carbon–carbon and carbon–heteroatom bonds in organic synthesis [1]. Usually, palladium species are used as catalysts in the Tsuji–Trost reaction involving, as substrates, allyl carboxylates [2], carbonates [3], and phosphates [4]. Obviously, the direct nucleophilic allylic substitution of allyl alcohols is a more attractive process especially from an economical and environmental point of view [5], as water, generated by this reaction, is a non-toxic byproduct. However, the poor ability of the hydroxy moiety, as a leaving group, has limited the use of the allyl alcohols as substrates. Correlatively, some efforts have been made in this direction by the use of transition metals such as copper [6], nickel [7], ruthenium (I, II) [8], and palladium (0, II) [9,10] as the catalyst or by converting the allylic alcohols into esters of inorganic acids, e.g., As2O3 [11], B2O3 [12], CO2 [13,14]. More recently the Lewis acids, such as, Ti(OiPr )4 [15], BEt3 [16-19], BPh3 [20], SnCl2 [21], and FeCl3 [22], have also been reported to catalyze these reactions by coordination with the hydroxy moiety, thereby increasing its leaving group ability [23-28].
Recently, Tamaru and co-workers have intensively investigated the use of triethylborane as an additive with either Pd(PPh3)4 or Pd(OAc)2 as catalysts for the allylation of a variety of active methylene compounds [29], aldehydes [30], ketones [31], and imines [32] with only common allylic alcohols.
As part of an ongoing program studying the behavior of MBH derivatives [33] towards β-dicarbonyl compounds, our research group [34-37] has reported an interesting synthesis of bicyclic dienones in a one-pot process involving the reaction of 2-(acetoxymethyl)cyclohex-2-enone with 1,3-dicarbonyl compounds using K2CO3 as a weak base.
Later, Chamakh and Amri [38] have described a one-pot synthesis of (E)-4-alkylidene-2-cyclohexen-1-ones through a cross coupling of the MBH carboxylates with aliphatic 1,3-diketones in the presence of K2CO3. A drawback of these synthetic approaches is the need to first perform the acylation step of the corresponding allyl MBH alcohols. For this reason, we herein report an efficient direct method for the allylation of β-dicarbonyl compounds with MBH alcohols [39,40] 1a and 1b (Figure 1) considered as multi-functionalized starting materials bearing both allyl alcohol and Michael acceptor moieties.
![[1860-5397-12-234-1]](/bjoc/content/figures/1860-5397-12-234-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cyclic and acyclic MBH alcohols.
Figure 1: Cyclic and acyclic MBH alcohols.
The interest of this reaction is the fact that it would be possible to directly convert, both cyclic 2-(hydroxymethyl)cyclohex-2-enone (1a) and acyclic ethyl 2-(hydroxymethyl)acrylate (1b), under the action of 1,3-dicarbonyl compounds 2, in the presence of an appropriate palladium catalyst and Et3B as a Lewis acid promoter, into the allylation compounds 3–8 with the formation of only water as a byproduct. These derivatives can be further used as synthetic intermediates in numerous synthetic routes to heterocyclic compounds and molecules of biological interest [41,42].
Results and Discussion
We first investigated the allylation of diethyl malonate (2a) with the allylic alcohol 1a in DMF in the presence of Pd(OAc)2 (10 mol %), PPh3 (20 mol %). Under these conditions, no reaction took place at 80 °C for 3 days (Table 1, entry 1).
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-234-i3.svg?max-width=637&scale=1.0)
On the other hand, the allylation slowly proceeded in the presence of NaH (0.5 equiv) but only a trace of the expected product 3a was obtained (Table 1, entry 2). Interestingly, the allylation reaction, carried out in DMF at 80 °C, gave a better result (30% yield in 12 h), using, in addition to NaH (0.5 equiv), 1 equiv of Et3B (Table 1, entry 3). Moreover, a remarkable improvement in yield (60% in 6 h) was also observed for the allylation of diethyl malonate (2a) with the MBH alcohol 1a using an excess of Et3B (3 equiv) and 1 equiv of NaH (Table 1, entry 4).
Mechanistic considerations
Scheme 1 illustrates the most probable catalytic cycle for the allylation of diethyl malonate (2a) with allyl alcohol 1a. We assume that there is first an activation of 1a through its conversion into I1 using Et3B [29], which further gives, in the presence of Pd(0), the π-allylpalladium complex I2. This intermediate reacts then with the diethyl malonate carbanion I3, in situ formed, to generate the promoter Et3B of this nucleophilic allylic substitution and the desired allylated product 3a.
![[1860-5397-12-234-i1]](/bjoc/content/inline/1860-5397-12-234-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed catalytic cycle involving palladium catalysis for Et3B-promoted allylation of diethyl malonate with MBH alcohol 1a.
Scheme 1: Proposed catalytic cycle involving palladium catalysis for Et3B-promoted allylation of diethyl malo...
Next, under the previously optimized conditions (Table 1, entry 4), we examined the scope of the catalytic system, Et3B/ Pd(OAc)2/PPh3, for a wide range of 1,3-dicarbonyl compounds and related derivatives (pKa = 9–14) [29,31] using two typical MBH alcohols 1a and 1b. The results of this study are summarized in Table 2.
Table 2: Palladium-catalyzed allylation of 1,3-dicarbonyl compounds with MBH alcohol 1aa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-234-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Pronucleophile 2 | Time (h) | Yield 3 (%)a | Yield 4 (%)b |
|---|---|---|---|---|
| 1 | CH2(CO2Et)2, 2a | 6 | 3a (60) [48] | – |
| 2 | CH2(CO2Me)2, 2b | 6 | 3b (65) [48] | – |
| 3 | NCCH2CO2Et, 2c | 3 | 3c (45) [48] | 4c (23) |
| 4 | MeCOCH2CO2Me, 2d | 4 | 3d (62) [34,44] | – |
| 5 | MeCOCH2CO2Et, 2e | 6 | 3e (72) [34,44] | – |
| 6 | PhCOCH2CO2Et, 2f | 3 | 3f (76) [34,44] | – |
| 7 | MeCOCH2COMe, 2g | 3 | 3g (57) [34,44]c | |
| 8 | PhCOCH2COPh, 2h | 3 | 3h (62) [34,44] | |
| 9 | PhCOCH2COMe, 2i | 3 | 3i (60) [34,44] | |
aReaction conditions: dicarbonyl compounds 2 (1.1 mmol), allylic alcohol 1a (1.0 mmol), Et3B (3 mmol), Pd(OAc)2 (10 mol %), PPh3 (20 mol %), NaH (1 mmol) in DMF (5 mL) under N2. bisolated yield; ccontaining the enolic form 5g.
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-234-i5.svg?max-width=637&scale=1.18182)
Like diethyl malonate (2a), the malonate derivative 2b (pKa = 13) [29] similarly reacted with the allylic alcohol 1a in the presence of the catalytic system Pd(0)/Et3B to exclusively give the mono-allylated product 3a in 65% yield, whereas the same reaction with ethyl 3-cyano-3-oxopropanoate (2c, pKa = 10.7) [43] whose acidity is relatively higher, led to a mixture of the mono- and bis-alkylation products 3c and 4c in 45 and 23% yields, respectively (Table 2, entries 1–3).
Under the same conditions, the allylation of a variety of β-keto esters and β-diketones (Table 2, entries 4–9), in DMF at 80 °C, selectively gave the monoallylation products 3d–i in moderate to good yields [44].
The analysis of 1H NMR spectra of the β-dicarbonyl derivatives 3a–i in CDCl3 revealed that a keto–enol tautomerism exists only for the acetylacetone derivative 3g and its enolic form 5g in a 54:46 ratio, respectively (Table 2, entry 7), whereas the other compounds 3a–f, 3h and 3i are exclusively in the β-dicarbonyl form [45-47].
Moreover, the allylation of ethyl cyclopentanone-2-carboxylate (2j) [49] as a cyclic β-keto ester, with alcohol 1a, catalyzed by the same system, smoothly proceeded at 80 °C in DMF, providing, after 2 h, the mono-allylated product 3j in 12% yield, along with the tricyclic compound 6j, in good yield, which is resulting from the intramolecular conjugate addition of 3j carbanion on the enone moiety (Table 3, entry 2). Under the same conditions, the keto ester 2j reacted with alcohol 1a, in DMF at 80 °C for 6 h longer reaction time, to selectively afford the compound 6j in 76% yield (Table 3, entry 3).
Table 3: Palladium-catalyzed allylation of β-keto ester 2j with the MBH alcohol 1aa.
| Entry | Nucleophile | T (°C)/Time (h) | Yield (%) |
|---|---|---|---|
| 1 | 2j | 0 to rt/24 | N.R. |
| 2 | 80/2 | 3j: 12% 6j: 75% | |
| 3 | 80/6 | 6j: 76% | |
aReaction conditions: dicarbonyl compounds 2j (1.1 mmol), allylic MBH alcohol 1a (1.0 mmol), Et3B (3 mmol), Pd(OAc)2 (10 mol %), PPh3 (20 mol %), NaH (1 mmol) in DMF (5 mL) under N2.
A plausible reaction mechanism for the formation of the tricyclic compound 6j from the MBH alcohol 1a is presented in Scheme 2. We believe that the treatment of the MBH alcohol 1a with Et3B in the presence of Pd(OAc)2 may generate a π-allylpalladium intermediate I that further undergoes a nucleophilic substitution reaction with the β-keto ester carbanion derived from 2j, affording the monoallylated compound 3j. The conversion of the keto ester 3j into the tricyclic product 6j was further performed through an intramolecular conjugate addition of the β-keto ester carbanion onto the enone moiety (Scheme 2) [50,51].
![[1860-5397-12-234-i2]](/bjoc/content/inline/1860-5397-12-234-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanistic pathway leading to the tricyclic compound 6j.
Scheme 2: Mechanistic pathway leading to the tricyclic compound 6j.
The structure of the tricyclic compound 6j was elucidated on the basis of X-ray diffraction analysis (Figure 2).
![[1860-5397-12-234-2]](/bjoc/content/figures/1860-5397-12-234-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of tricyclic compound 6j.
Figure 2: X-ray crystal structure of tricyclic compound 6j.
In a previous report, Alexakis and co-workers have demonstrated that copper-catalyzed conjugate addition of Grignard reagents onto α-methyl cyclic enones, affording mainly the trans-2,3-disubstituted cyclohexanones as being the thermodynamic products [52]. Similarly, we believe that the intramolecular conjugate addition of carbanion 3j onto α-substituted enone moiety is under thermodynamic control, leading to the tricyclic compound 6j, in which the two hydrogens H2 and H3, are on a trans-ring junction whereas the second ring junction is cis (Figure 2).
Encouraged by these successful results on the allylation of β-dicarbonyl compounds with the alcohol 1a in the presence of Pd/Et3B, we attempted to extend this methodology to the acyclic MBH alcohol 1b (Table 4). We initially reacted this substrate with ethyl acetoacetate (2e, 1.1 equiv) in the presence of the catalyst Pd(OAc)2 without any additive. After stirring the reaction mixture for 24 h, either at room temperature or in refluxing THF, the starting alcohol 1b was recovered (Table 4, entry 1). Next, the use of NaH as a strong base with ethyl acetoacetate (2e) allowed the carbanion formation that then reacted with alcohol 1b, affording a trace of the allylation product 7e with only a 60% conversion of the starting material (Table 4, entry 2).
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-234-i6.svg?max-width=637&scale=1.0)
The addition of a Lewis acid (1 equiv of Et3B) started the reaction by activating the hydroxy group, affording the allylation product 7e in 30% isolated yield (Table 4, entry 3). To further improve the performance of this allylation reaction, we employed 2 equiv of Et3B (Table 4, entry 4). The reaction afforded within 20 h, at room temperature, the compound 7e in 60% yield. Interestingly, in refluxing THF, this reaction was achieved in shorter reaction time (2 h), leading to the same compound in the same yield (Table 4, entry 5).
Encouraged by these results and those of cyclic MBH alcohol 1a, we selected the acyclic alcohol 1b to react with stabilized carbanions derived from the β-keto esters 2e–f and β-diketones 2g–i in anhydrous THF using 1 equiv of NaH and 2 equiv of Et3B. Under these conditions, all these reactions worked well in refluxing THF, affording in 2 h the corresponding monoallylation products 7e–i [41,44] in 60–70% yields (Table 5, entries 1–4).
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-234-i7.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-234-i8.svg?max-width=637&scale=1.18182)
Conclusion
In summary, we have developed a mild and direct process for the C–C bond formation from the reaction of the MBH alcohols 1a and 1b with β-dicarbonyl compounds 2 in the presence of a palladium catalyst and Et3B (promoter) with the formation of only water as a side product. This method provides a straightforward and practical route to a range of allylated compounds. Further work is in progress in our laboratory to investigate the Pd catalysis of the reaction of the MBH adducts with various pronucleophiles including nitroalkanes, amines and thiols.
Experimental
General
IR spectra were recorded on a Bruker IFS 66v/S spectrometer. 1H NMR and 13C NMR spectra were recorded either on a Bruker AC-300 spectrometer (300 MHz for 1H and 75 MHz for 13C) in CDCl3, using TMS as an internal standard (chemical shifts in δ values, J in Hz). Mass spectra (EI) were recorded on an Hewlett-Packard (70 eV) apparatus. Analytical thin-layer chromatography (TLC) was performed using Fluka Kieselgel 60 F254 precoated silica gel plates. Visualization was achieved by UV light (254 nm). Flash chromatography was performed using Merck silica gel 60 and a gradient solvent system (petroleum ether/ether as eluents).
General procedure for the allylation of β-dicarbonyl compounds with the MBH alcohols 1a and 1b
Into a nitrogen-purged two-necked flask, equipped with a reflux condenser, containing 5 mL of DMF or THF, Pd(OAc)2 (10 mol %), and PPh3 (20 mol %) were added successively the 1,3-dicarbonyl compound 2 (1.1 mmol), NaH (1 mmol) and an Et3B solution 1.0 M (2 to 3 mmol) in THF. The mixture was stirred for 5 to 10 min at room temperature, then the MBH allylic alcohol 1a or 1b (1 mmol) was added. The mixture was stirred and heated at 80 °C for 3 to 6 h during which the mixture was colored black. When the reaction, followed by TLC, was finished, the mixture was diluted with CH2Cl2 (20 mL) and washed with 2 M HCl, sat. NaHCO3 and then brine. The organic extracts were dried over MgSO4 and concentrated in vacuo and the residual oil was subjected to column chromatography over silica gel (gradient: petroleum ether/ether = 4:1) to give the pure allylated products 3–8 in moderate to good yields.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization and spectral data for synthesized compounds and X-ray data for compound 6j. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Trost, B. M.; Crawley, M. Chem. Rev. 2003, 103, 2921–2944. doi:10.1021/cr020027w
Return to citation in text: [1] -
Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395–422. doi:10.1021/cr9409804
Return to citation in text: [1] -
Monopoli, A.; Cotugno, P.; Zambonin, C. G.; Ciminale, F.; Nacci, A. Beilstein J. Org. Chem. 2015, 11, 994–999. doi:10.3762/bjoc.11.111
Return to citation in text: [1] -
Tsuji, J. Transition Metal Reagents and Catalysts; Wiley: New York, 2000; Chapter 4.
Return to citation in text: [1] -
Ozawa, F.; Okamoto, H.; Kawagishi, S.; Yamamoto, S.; Minami, T.; Yoshifuji, M. J. Am. Chem. Soc. 2002, 124, 10968–10969. doi:10.1021/ja0274406
Return to citation in text: [1] -
Mazerolles, P.; Laurent, C.; Faucher, A. J. Organomet. Chem. 1989, 366, 57–60. doi:10.1016/0022-328X(89)87315-2
Return to citation in text: [1] -
Blieck, R.; Azizi, M. S.; Mifleur, A.; Roger, M.; Persyn, C.; Sauthier, M.; Bonin, H. Eur. J. Org. Chem. 2016, 1194–1198. doi:10.1002/ejoc.201501556
Return to citation in text: [1] -
He, J.; Kim, J. W.; Yamaguchi, K.; Mizuno, N. Angew. Chem. 2009, 121, 10072–10075. doi:10.1002/ange.200905385
Return to citation in text: [1] -
Kinoshita, H.; Shinokubo, H.; Oshima, K. Org. Lett. 2004, 6, 4085–4088. doi:10.1021/ol048207a
Return to citation in text: [1] -
Huo, X.; Yang, G.; Liu, D.; Liu, Y.; Gridnev, I. D.; Zhang, W. Angew. Chem., Int. Ed. 2014, 53, 6776–6780. doi:10.1002/anie.201403410
Return to citation in text: [1] -
Lu, X.; Lu, L.; Sun, J. J. Mol. Catal. 1987, 41, 245–251. doi:10.1016/0304-5102(87)80032-9
Return to citation in text: [1] -
Lu, X.; Jiang, X.; Tao, X. J. Organomet. Chem. 1988, 344, 109–118. doi:10.1016/0022-328X(88)80217-1
Return to citation in text: [1] -
Sakamoto, M.; Shimizu, I.; Yamamoto, A. Bull. Chem. Soc. Jpn. 1996, 69, 1065–1078. doi:10.1246/bcsj.69.1065
Return to citation in text: [1] -
Lang, S. B.; Locascio, T. M.; Tunge, J. A. Org. Lett. 2014, 16, 4308–4311. doi:10.1021/ol502023d
Return to citation in text: [1] -
Shue, Y.-J.; Yang, S.-C.; Lai, H.-C. Tetrahedron Lett. 2003, 44, 1481–1485. doi:10.1016/S0040-4039(02)02861-7
Return to citation in text: [1] -
Kimura, M.; Horino, Y.; Mukai, R.; Tanaka, S.; Tamaru, Y. J. Am. Chem. Soc. 2001, 123, 10401–10402. doi:10.1021/ja011656a
Return to citation in text: [1] -
Kimura, M.; Futamata, M.; Shibata, K.; Tamaru, Y. Chem. Commun. 2003, 234–235. doi:10.1039/b210920d
Return to citation in text: [1] -
Tamaru, Y. Eur. J. Org. Chem. 2005, 2647–2656. doi:10.1002/ejoc.200500076
Return to citation in text: [1] -
Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501–1507. doi:10.1039/B823217B
Return to citation in text: [1] -
Starý, I.; Stara, I. G.; Kočovský, P. Tetrahedron Lett. 1993, 34, 179–182. doi:10.1016/S0040-4039(00)60088-6
Return to citation in text: [1] -
Masuyama, Y.; Kagawa, M.; Kurusu, Y. Chem. Lett. 1995, 24, 1121–1122. doi:10.1246/cl.1995.1121
Return to citation in text: [1] -
Zhang, X.; Rao, W.; Sally; Chan, P. W. Org. Biomol. Chem. 2009, 7, 4186–4193. doi:10.1039/b908447a
Return to citation in text: [1] -
Tamaru, Y.; Horino, Y.; Araki, M.; Tanaka, S.; Kimura, M. Tetrahedron Lett. 2000, 41, 5705–5709. doi:10.1016/S0040-4039(00)00934-5
Return to citation in text: [1] -
Manabe, K.; Kobayashi, S. Org. Lett. 2003, 5, 3241–3244. doi:10.1021/ol035126q
Return to citation in text: [1] -
Basavaiah, D.; Rao, K. V.; Reddy, R. J. Chem. Soc. Rev. 2007, 36, 1581–1588. doi:10.1039/B613741P
Return to citation in text: [1] -
Krishna, P. R.; Sachwani, R.; Reddy, P. S. Synlett 2008, 2897–2912. doi:10.1055/s-0028-1087338
Return to citation in text: [1] -
Butt, N. A.; Zhang, W. Chem. Soc. Rev. 2015, 44, 7929–7967. doi:10.1039/C5CS00144G
Return to citation in text: [1] -
Dryzhakov, M.; Richmond, E.; Moran, J. Synthesis 2016, 48, 935–959. doi:10.1055/s-0035-1560396
Return to citation in text: [1] -
Kimura, M.; Mukai, R.; Tanigawa, N.; Tanaka, S.; Tamaru, Y. Tetrahedron 2003, 59, 7767–7777. doi:10.1016/S0040-4020(03)01234-1
Return to citation in text: [1] [2] [3] [4] -
Kimura, M.; Tomizawa, T.; Horino, Y.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 2000, 41, 3627–3629. doi:10.1016/S0040-4039(00)00429-9
Return to citation in text: [1] -
Horino, Y.; Naito, M.; Kimura, M.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 2001, 42, 3113–3116. doi:10.1016/S0040-4039(01)00381-1
Return to citation in text: [1] [2] -
Shimizu, M.; Kimura, M.; Watanabe, T.; Tamaru, Y. Org. Lett. 2005, 7, 637–640. doi:10.1021/ol047609f
Return to citation in text: [1] -
Basavaiah, D.; Veeraraghavaiah, G. Chem. Soc. Rev. 2012, 41, 68–78. doi:10.1039/c1cs15174f
And references therein.
Return to citation in text: [1] -
Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106
Return to citation in text: [1] -
Rezgui, F.; El Gaïed, M. M. Tetrahedron Lett. 1998, 39, 5965–5966. doi:10.1016/S0040-4039(98)01206-4
Return to citation in text: [1] -
Rezgui, F.; El Gaïed, M. M. J. Chem. Res., Synop. 1999, 510–511. doi:10.1039/A902012H
Return to citation in text: [1] -
Chamakh, A.; Amri, H. Tetrahedron Lett. 1998, 39, 375–378. doi:10.1016/S0040-4039(97)10593-7
Return to citation in text: [1] -
Villiéras, J.; Rambaud, M. Org. Synth. 1988, 66, 220. doi:10.15227/orgsyn.066.0220
Return to citation in text: [1] -
Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–892. doi:10.1021/cr010043d
Return to citation in text: [1] -
Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Basavaiah, D.; Reddy, B. S.; Lingam, H. Tetrahedron 2013, 69, 1994–2003. doi:10.1016/j.tet.2012.12.069
Return to citation in text: [1] -
Ono, Y. CATTECH 1997, 1, 31.
Return to citation in text: [1] -
Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] -
An experimental and theoretical study of the tautomeric equilibria between the β-dicarbonyl compounds 3a–i and the corresponding enol forms is currently in progress in our laboratory.
Return to citation in text: [1] -
Smith, K. T.; Young, S. C.; DeBlasio, J. W.; Hamann, C. S. J. Chem. Educ. 2016, 93, 790–794. doi:10.1021/acs.jchemed.5b00170
Return to citation in text: [1] -
Cook, A. G.; Feltman, P. M. J. Chem. Educ. 2007, 84, 1827–1829. doi:10.1021/ed084p1827
Return to citation in text: [1] -
Tamura, R.; Katayamaa, H.; Watabea, K.; Suzuki, H. Tetrahedron 1990, 46, 7557–7568. doi:10.1016/S0040-4020(01)89067-0
Return to citation in text: [1] [2] [3] -
Iglesias, E. New J. Chem. 2002, 26, 1352–1359. doi:10.1039/b202981m
Return to citation in text: [1] -
Abbiati, G.; Beccalli, E. M.; Broggini, G.; Zoni, C. J. Org. Chem. 2003, 68, 7625–7628. doi:10.1021/jo034636v
Return to citation in text: [1] -
Kim, S. Y.; Kim, K. H.; Moon, H. R.; Kim, J. N. Bull. Korean Chem. Soc. 2016, 37, 95–98. doi:10.1002/bkcs.10610
Return to citation in text: [1] -
Vuagnoux-d'Augustin, M.; Alexakis, A. Chem. – Eur. J. 2007, 13, 9647–9662. doi:10.1002/chem.200701001
Return to citation in text: [1]
| 29. | Kimura, M.; Mukai, R.; Tanigawa, N.; Tanaka, S.; Tamaru, Y. Tetrahedron 2003, 59, 7767–7777. doi:10.1016/S0040-4020(03)01234-1 |
| 29. | Kimura, M.; Mukai, R.; Tanigawa, N.; Tanaka, S.; Tamaru, Y. Tetrahedron 2003, 59, 7767–7777. doi:10.1016/S0040-4020(03)01234-1 |
| 31. | Horino, Y.; Naito, M.; Kimura, M.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 2001, 42, 3113–3116. doi:10.1016/S0040-4039(01)00381-1 |
| 48. | Tamura, R.; Katayamaa, H.; Watabea, K.; Suzuki, H. Tetrahedron 1990, 46, 7557–7568. doi:10.1016/S0040-4020(01)89067-0 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 48. | Tamura, R.; Katayamaa, H.; Watabea, K.; Suzuki, H. Tetrahedron 1990, 46, 7557–7568. doi:10.1016/S0040-4020(01)89067-0 |
| 48. | Tamura, R.; Katayamaa, H.; Watabea, K.; Suzuki, H. Tetrahedron 1990, 46, 7557–7568. doi:10.1016/S0040-4020(01)89067-0 |
| 29. | Kimura, M.; Mukai, R.; Tanigawa, N.; Tanaka, S.; Tamaru, Y. Tetrahedron 2003, 59, 7767–7777. doi:10.1016/S0040-4020(03)01234-1 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 41. | Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 41. | Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 41. | Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 41. | Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 50. | Abbiati, G.; Beccalli, E. M.; Broggini, G.; Zoni, C. J. Org. Chem. 2003, 68, 7625–7628. doi:10.1021/jo034636v |
| 51. | Kim, S. Y.; Kim, K. H.; Moon, H. R.; Kim, J. N. Bull. Korean Chem. Soc. 2016, 37, 95–98. doi:10.1002/bkcs.10610 |
| 52. | Vuagnoux-d'Augustin, M.; Alexakis, A. Chem. – Eur. J. 2007, 13, 9647–9662. doi:10.1002/chem.200701001 |
| 45. | An experimental and theoretical study of the tautomeric equilibria between the β-dicarbonyl compounds 3a–i and the corresponding enol forms is currently in progress in our laboratory. |
| 46. | Smith, K. T.; Young, S. C.; DeBlasio, J. W.; Hamann, C. S. J. Chem. Educ. 2016, 93, 790–794. doi:10.1021/acs.jchemed.5b00170 |
| 47. | Cook, A. G.; Feltman, P. M. J. Chem. Educ. 2007, 84, 1827–1829. doi:10.1021/ed084p1827 |
| 41. | Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441 |
| 44. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 1. | Trost, B. M.; Crawley, M. Chem. Rev. 2003, 103, 2921–2944. doi:10.1021/cr020027w |
| 5. | Ozawa, F.; Okamoto, H.; Kawagishi, S.; Yamamoto, S.; Minami, T.; Yoshifuji, M. J. Am. Chem. Soc. 2002, 124, 10968–10969. doi:10.1021/ja0274406 |
| 20. | Starý, I.; Stara, I. G.; Kočovský, P. Tetrahedron Lett. 1993, 34, 179–182. doi:10.1016/S0040-4039(00)60088-6 |
| 4. | Tsuji, J. Transition Metal Reagents and Catalysts; Wiley: New York, 2000; Chapter 4. |
| 21. | Masuyama, Y.; Kagawa, M.; Kurusu, Y. Chem. Lett. 1995, 24, 1121–1122. doi:10.1246/cl.1995.1121 |
| 3. | Monopoli, A.; Cotugno, P.; Zambonin, C. G.; Ciminale, F.; Nacci, A. Beilstein J. Org. Chem. 2015, 11, 994–999. doi:10.3762/bjoc.11.111 |
| 15. | Shue, Y.-J.; Yang, S.-C.; Lai, H.-C. Tetrahedron Lett. 2003, 44, 1481–1485. doi:10.1016/S0040-4039(02)02861-7 |
| 2. | Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395–422. doi:10.1021/cr9409804 |
| 16. | Kimura, M.; Horino, Y.; Mukai, R.; Tanaka, S.; Tamaru, Y. J. Am. Chem. Soc. 2001, 123, 10401–10402. doi:10.1021/ja011656a |
| 17. | Kimura, M.; Futamata, M.; Shibata, K.; Tamaru, Y. Chem. Commun. 2003, 234–235. doi:10.1039/b210920d |
| 18. | Tamaru, Y. Eur. J. Org. Chem. 2005, 2647–2656. doi:10.1002/ejoc.200500076 |
| 19. | Bandini, M.; Tragni, M. Org. Biomol. Chem. 2009, 7, 1501–1507. doi:10.1039/B823217B |
| 9. | Kinoshita, H.; Shinokubo, H.; Oshima, K. Org. Lett. 2004, 6, 4085–4088. doi:10.1021/ol048207a |
| 10. | Huo, X.; Yang, G.; Liu, D.; Liu, Y.; Gridnev, I. D.; Zhang, W. Angew. Chem., Int. Ed. 2014, 53, 6776–6780. doi:10.1002/anie.201403410 |
| 12. | Lu, X.; Jiang, X.; Tao, X. J. Organomet. Chem. 1988, 344, 109–118. doi:10.1016/0022-328X(88)80217-1 |
| 8. | He, J.; Kim, J. W.; Yamaguchi, K.; Mizuno, N. Angew. Chem. 2009, 121, 10072–10075. doi:10.1002/ange.200905385 |
| 13. | Sakamoto, M.; Shimizu, I.; Yamamoto, A. Bull. Chem. Soc. Jpn. 1996, 69, 1065–1078. doi:10.1246/bcsj.69.1065 |
| 14. | Lang, S. B.; Locascio, T. M.; Tunge, J. A. Org. Lett. 2014, 16, 4308–4311. doi:10.1021/ol502023d |
| 7. | Blieck, R.; Azizi, M. S.; Mifleur, A.; Roger, M.; Persyn, C.; Sauthier, M.; Bonin, H. Eur. J. Org. Chem. 2016, 1194–1198. doi:10.1002/ejoc.201501556 |
| 6. | Mazerolles, P.; Laurent, C.; Faucher, A. J. Organomet. Chem. 1989, 366, 57–60. doi:10.1016/0022-328X(89)87315-2 |
| 11. | Lu, X.; Lu, L.; Sun, J. J. Mol. Catal. 1987, 41, 245–251. doi:10.1016/0304-5102(87)80032-9 |
| 29. | Kimura, M.; Mukai, R.; Tanigawa, N.; Tanaka, S.; Tamaru, Y. Tetrahedron 2003, 59, 7767–7777. doi:10.1016/S0040-4020(03)01234-1 |
| 22. | Zhang, X.; Rao, W.; Sally; Chan, P. W. Org. Biomol. Chem. 2009, 7, 4186–4193. doi:10.1039/b908447a |
| 23. | Tamaru, Y.; Horino, Y.; Araki, M.; Tanaka, S.; Kimura, M. Tetrahedron Lett. 2000, 41, 5705–5709. doi:10.1016/S0040-4039(00)00934-5 |
| 24. | Manabe, K.; Kobayashi, S. Org. Lett. 2003, 5, 3241–3244. doi:10.1021/ol035126q |
| 25. | Basavaiah, D.; Rao, K. V.; Reddy, R. J. Chem. Soc. Rev. 2007, 36, 1581–1588. doi:10.1039/B613741P |
| 26. | Krishna, P. R.; Sachwani, R.; Reddy, P. S. Synlett 2008, 2897–2912. doi:10.1055/s-0028-1087338 |
| 27. | Butt, N. A.; Zhang, W. Chem. Soc. Rev. 2015, 44, 7929–7967. doi:10.1039/C5CS00144G |
| 28. | Dryzhakov, M.; Richmond, E.; Moran, J. Synthesis 2016, 48, 935–959. doi:10.1055/s-0035-1560396 |
| 39. | Villiéras, J.; Rambaud, M. Org. Synth. 1988, 66, 220. doi:10.15227/orgsyn.066.0220 |
| 40. | Basavaiah, D.; Rao, A. J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811–892. doi:10.1021/cr010043d |
| 41. | Singh, V.; Batra, S. Synthesis 2006, 63–72. doi:10.1055/s-2005-918441 |
| 42. | Basavaiah, D.; Reddy, B. S.; Lingam, H. Tetrahedron 2013, 69, 1994–2003. doi:10.1016/j.tet.2012.12.069 |
| 34. | Rezgui, F.; El Gaïed, M. M. Tetrahedron 1997, 53, 15711–15716. doi:10.1016/S0040-4020(97)10023-0 |
| 35. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 36. | Rezgui, F.; El Gaïed, M. M. Tetrahedron Lett. 1998, 39, 5965–5966. doi:10.1016/S0040-4039(98)01206-4 |
| 37. | Rezgui, F.; El Gaïed, M. M. J. Chem. Res., Synop. 1999, 510–511. doi:10.1039/A902012H |
| 38. | Chamakh, A.; Amri, H. Tetrahedron Lett. 1998, 39, 375–378. doi:10.1016/S0040-4039(97)10593-7 |
| 32. | Shimizu, M.; Kimura, M.; Watanabe, T.; Tamaru, Y. Org. Lett. 2005, 7, 637–640. doi:10.1021/ol047609f |
| 33. |
Basavaiah, D.; Veeraraghavaiah, G. Chem. Soc. Rev. 2012, 41, 68–78. doi:10.1039/c1cs15174f
And references therein. |
| 30. | Kimura, M.; Tomizawa, T.; Horino, Y.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 2000, 41, 3627–3629. doi:10.1016/S0040-4039(00)00429-9 |
| 31. | Horino, Y.; Naito, M.; Kimura, M.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 2001, 42, 3113–3116. doi:10.1016/S0040-4039(01)00381-1 |
© 2016 Abidi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)