Abstract
The synthesis of sydnones heteroarylated at C-4 with an indolizine was achieved by Chichibabin (Tschitschibabin) indolizine synthesis starting from the corresponding sydnone-N-pyridinium bromides. The latter compounds were also transformed to sydnone-indolizines connected through a keto group at the C-4 position by refluxing them in 1,2-epoxybutane with an activated alkyne. The structures of the new compounds were assigned by FTIR, NMR spectroscopy and X-ray analysis.

Graphical Abstract
Introduction
In recent decades, interest in the syntheses of biheteroaryls has been focused on the creation of new hetaryl–hetaryl C(sp2)–C(sp2) bonds, in particular through cross-coupling reactions. These reactions are catalyzed by palladium or other metals and represent a versatile tool for obtaining hybrid heterocycles with properties that are useful for the development of new advanced materials and for medicinal purposes [1-4].
Recently, we obtained, by 1,3-dipolar cycloaddition reactions of sydnone-ylides as bis(1,3-dipoles) with activated acetylenes, the hybrid structures 1 containing a pyrroloazine (pyrroloisoquinoline or pyrrolophthalazine) connected to the sydnone ring through a keto group [5]. As a result, it was of interest to prepare analogous compounds containing a pyrroloazine directly attached to the C-4 position of a sydnone ring. From a survey of the literature data [6-15] and based on our previous findings [16-19] it was concluded that the heterocycle in the pyrroloazine series most suitable for this study would be indolizine (2).
Indolizine or pyrrolo[1,2-a]pyridine (2) and its derivatives exhibit remarkable biological activities and optical properties. Indolizine is a privileged structure for the design of new drugs and its framework is present in many natural compounds. As a consequence, the synthesis, chemical, physicochemical and biological properties of indolizines have been the subject of various reviews [6-15].
Sydnones 3 are mesoionic heterocyclic compounds with a 1,2,3-oxadiazole skeleton and their chemistry and biological properties are well represented and documented in the literature [20-26]. The most interesting chemical reactions of sydnones are their electrophilic substitution at C-4 and their participation in 1,3-dipolar cycloaddition reactions with olefinic and acetylenic dipolarophiles to form pyrazoles and functional transformation at C-4 [20-26].
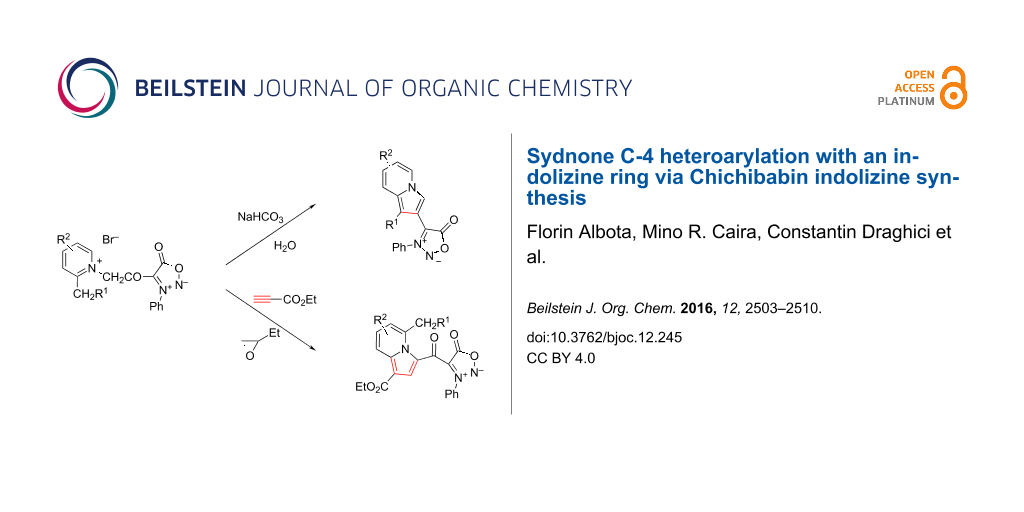
Herein we present the synthesis of indolizines 4 (Figure 1) with the indolizine directly attached to C-4 of a sydnone ring by the indolizine Chichibabin approach. In addition, the new indolizine structures 5, connected through a keto group to a sydnone ring, were prepared by 1,3-dipolar cycloaddition reaction starting from the same pyridinium salts used in the indolizine Chichibabin synthesis.
![[1860-5397-12-245-1]](/bjoc/content/figures/1860-5397-12-245-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Sydnone-pyrroloazines hybrids 1, indolizine (2), sydnone 3, indolizines attached directly to C-4 of a sydnone 4, indolizines attached via a keto group to C-4 of a sydnone 5.
Figure 1: Sydnone-pyrroloazines hybrids 1, indolizine (2), sydnone 3, indolizines attached directly to C-4 of...
Results and Discussion
In order to obtain the compounds 4 containing an indolizine ring attached directly to the C-4 carbon atom of a sydnone ring (i.e., indolizination of sydnones at C-4), the Chichibabin indolizine synthesis was employed. The Chichibabin reaction was discovered ninety years ago [27] and consists in the cyclization of pyridinium salts that require the presence of a CH2R group (e.g., R = H, Alk, Ar, CN, CO2Et, SO2Ar) at the position adjacent to the nitrogen atom. Despite this limitation, the method remains one of the most efficient syntheses of indolizines [28-31] owing to the simple working procedure and the large number of 2-substituted pyridines that are commercially available. An overview of Chichibabin indolizine syntheses has been included in the reviews dedicated to indolizine syntheses and their applications in medicine and advanced materials [6-15].
Synthesis of pyridinium bromides 8
The pyridinium bromides 8 were the starting materials for the synthesis of indolizines attached either directly or through a keto group to the sydnone ring. These salts were obtained in good yields by heating pyridines 6a–d with 4-(bromoacetyl)-3-phenylsydnone (7, Scheme 1) in refluxing acetone [5]. The obtained pyridinium salts 8 were used in the next step without further purification.
![[1860-5397-12-245-i1]](/bjoc/content/inline/1860-5397-12-245-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of pyridinium bromides 8 and sydnone-indolizine hybrids 9 through the Chichibabin reaction.
Scheme 1: Synthesis of pyridinium bromides 8 and sydnone-indolizine hybrids 9 through the Chichibabin reactio...
The structures of the pyridinium bromides 8a–d were assigned by 1H NMR, 13C NMR and IR spectroscopy. The chemical shifts for the protons of the pyridine ring match the proposed structures 8. In the 1H NMR spectra of the pyridinium bromides recorded in DMSO-d6 the protons of the methylene groups appear as singlets with chemical shifts in the range of 5.96–6.06 ppm. The pyridine ring protons are strongly deshielded and were assigned based on their multiplicity and by comparison with literature data for compounds with similar structures. Thus, the chemical shifts of H-4 (8.44–8.60 ppm) and H-6 (8.72–8.93 ppm) are more deshielded due to their γ and α positions, respectively, to N-1 in comparison with H-3 (7.91–8.10 ppm) and H-5 (7.82–8.04), which are both in β position relative to N-1.
In the 13C NMR spectrum pyridinium bromides 8 representative chemical shifts are found for the methylene group (62.7–63.4 ppm), C-4 from the sydnone ring (106.2–106.3 ppm) and the two carbonyl groups. Here, the chemical shifts for the endocyclic C=O of the sydnone ring lie in the range of 165.7–165.8 ppm whereas for the exocyclic C=O the chemical shifts are at ~176.5 ppm. The chemical shifts for C-2, C-4 and C-6 (145.4–160.4 ppm) in the α and γ positions of the pyridine ring are easily identified as they are deshielded in comparison with C-3 and C-5 which are in the β position (125.4–129.7 ppm). An exception to the rule is the chemical shift for C-5 of compound 8d, which has a high value of 141.3 ppm due to the influence of the ethyl substituent attached to C-5. In the ATR-IR spectra of compounds 8 the functional group absorption frequencies are representative for the CO groups attached to C-4 of the sydnone ring (range 1678–1683 cm−1) and the absorption bands associated with CO groups from the endocyclic sydnone ring (range 1771–1779 cm−1).
Indolizination of sydnones at C-4 by the Chichibabin reaction
Generally, the formation of single bonds between two aromatic or heteroaromatic rings involves palladium-catalyzed cross-coupling reactions and cyclization of functional groups attached to an aromatic and heteroaromatic ring. Thus, arylation and heteroarylation at the C-4 position of sydnones has been performed by palladium-catalyzed cross-coupling reactions [32-36] starting from 4-unsubstituted sydnones, 4-halosydnones or by chemical transformation of the substituents attached to C-4 [32-36].
Herein, using the Chichibabin indolizine reaction one of the simplest and most efficient methods for indolizine synthesis, the “indolizination” at the C-4 position of sydnones was achieved using a procedure involving quaternization of pyridines 6 with sydnone derivative 7 to form pyridinium bromides 8, followed by their intramolecular cyclization in the presence of a base. In order to improve the yield, the transformation of pyridinium bromides 8 into the sydnone-indolizine biheteroaryls 9 (Scheme 1) was optimized by some variations in the Chichibabin indolizine synthesis for the representative salt 8a. Thus, the treatment of the quaternary salts 8 dissolved in hot water with sodium bicarbonate followed by solvent extraction gave the sydnone-indolizines 9 in yields of over 50%. If the dissolution of the quaternary salts was incomplete, a small quantity of ethanol could be added. Alternatively, replacement of sodium bicarbonate with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and the use of acetonitrile as solvent gave, in the case of the representative pyridinium bromide 8a, compound 9a in 53% yield. Similarly, 9a could be prepared in 54% yield by heating pyridine 6a under reflux with sydnone derivative 7 for four hours followed by the addition of DBU for cyclization. Although all three procedures for the cyclization of bromides 8 finally gave good results, the method involving water as reaction medium and sodium bicarbonate as cyclization reagent was preferred because it used non-toxic components. The cyclization of pyridinium bromides 8 to biheteroaryls 9 allows the isolation of compounds with substituents in both rings and has the advantage over cross-coupling reactions insofar as it overcomes side reactions and uses inexpensive materials.
The structures of the new sydnone-indolizine hybrids produced by the Chichibabin synthesis were confirmed by elemental analysis, NMR and IR spectroscopy, with further supporting X-ray structural elucidation of the representative compound 9d, which displays an interesting packing feature described below. The molecular structure and numbering scheme for 9d are shown in Figure 2a. Principal torsion angles that describe the molecular conformation include C5–C4–C13–C14, −18.2(2)° and C4–N3–C7–C8, 111.2(2)°, defining the orientations of the indolizine and phenyl rings relative to the sydnone ring. Bond distances in the latter ring (see caption of Figure 2) compare favorably with previously published values in the Cambridge Structural Database (CSD) [37]. The five-membered ring is planar, with a maximum deviation from the least-squares plane of 0.011(1) Å for C5, while atom O6 deviates from the plane by 0.022(1) Å. Full listings of geometrical parameters are available in the CIF file (Supporting Information File 2).
![[1860-5397-12-245-2]](/bjoc/content/figures/1860-5397-12-245-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The molecular structure of 9d with thermal ellipsoids drawn at the 50% probability level. (a) Inversion-related molecules of 9d showing the antiparallel alignment of the dipolar sydnone rings and short contacts C5···C5i (i = −x, 1−y, −z) and O6···ring centroid, represented by dashed lines. (b) Bond distances (Å) in the sydnone moiety are: O1–N2 1.382(1), N2–N3 1.322(2), N3–C4 1.349(2), C4–C5 1.414(2), C5–O1 1.402(2), and C5–O6 1.222(2).
Figure 2: The molecular structure of 9d with thermal ellipsoids drawn at the 50% probability level. (a) Inver...
A search of the CSD revealed 104 crystal structures that contain the sydnone moiety. Of these, eleven were found to display the same anti-parallel sydnone–sydnone motif as is occurring in 9d (Figure 2b), reflecting the dipolar alignment of these moieties consistent with the positive charge centered on the ring and the negative charge located on the exocyclic oxygen atom. This robust supramolecular motif is characterized by an abnormally short C5···C5′ distance for the entries listed in the CSD, namely 3.088–3.338 Å. In the crystal of 9d, this distance is only 3.054(2) Å, i.e. 0.35 Å less than the sum of the van der Waals radii (the O-6···ring centroid distance is nominally 3.052 Å, i.e., indistinguishable from the C5···C5′ distance).
The molecules of 9d associate into centrosymmetric dimers via two identical pairs of intermolecular bifurcated C–H···O hydrogen bonds (Figure 3), namely C14–H14···O6ii and C16–H16···O6ii (ii = −x, −y, −z) with C···O distances of 3.271(2) and 3.216(2) Å, respectively.
![[1860-5397-12-245-3]](/bjoc/content/figures/1860-5397-12-245-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Centrosymmetric C–H···O hydrogen bonded dimeric motif in the crystal of 9d.
Figure 3: Centrosymmetric C–H···O hydrogen bonded dimeric motif in the crystal of 9d.
In the 1H NMR spectra the chemical shifts for the indolizine protons were assigned on the basis of HH and HC correlations and by comparison with NMR data for the parent indolizine. The chemical shifts of hydrogen atoms of compound 9a substituted at position 2 with a sydnone have different values for H-1 (Δ = 0.6 ppm) and H-3 (Δ = 0.6 ppm) when compared to unsubstituted indolizine whereas the variations in chemical shift data for the other protons are less than 0.2 ppm. It should be mentioned that in the case of compounds 9a,c,d the signals for protons H-3 and H-5 overlap with those of the phenyl group protons. The 13C NMR spectra of sydnone-indolizines show a good agreement with the parent indolizine 2 and its derivatives. Also noteworthy are the values of the chemical shifts for C-4 (δ = 106.9–107.9 ppm) and C-5 (CO group, δ = 166.9–167.9 ppm) from the sydnone ring. The IR spectra of biheteroaryls 9 show the characteristic absorbance bands of the endocyclic CO group of the sydnone ring in the range 1724–1765 cm−1.
Synthesis of sydnone-indolizines by 1,3-dipolar cycloaddition reaction
As indolizines exhibit more than 30 different types of biological activity and they are highly fluorescent compounds the synthesis of new compounds with an indolizine framework is of great interest. The Chichibabin method and 1,3-dipolar cycloaddition reactions of pyridinium N-ylides with acetylenic dipolarophiles are two of the most versatile syntheses for the preparation of indolizines. Both methods use as starting materials pyridinium salts whose preparation is simple and in many cases commercially available pyridines and halogenated compounds are employed.
Therefore, the pyridinium bromides 8 used in the synthesis of bihetaryls 9 can also serve as starting materials for the generation of sydnone-pyridinium N-ylides 10 (Scheme 2) in the presence of acetylenic dipolarophiles, to form sydnone-indolizine hybrids 12. In the new compounds 12 (Scheme 2) the indolizine moiety is connected by a keto group to C-4 of the sydnone. The reaction was achieved in a one-pot procedure by refluxing in 1,2-epoxybutane the bromides 8 with ethyl propiolate as acetylenic dipolarophile. The formation of cycloadducts 12 implies in the first step the generation of pyridinium N-ylides under the action of 1,2-epoxybutane. The mechanism for the transformation of N-heteroaromatic salts into the corresponding N-ylides with epoxides was reported recently [5,17,18]. The next step represents a 1,3-dipolar cycloaddition of the bis(1,3-dipoles) 10 with formation of dihydro-indolizines 11 which, under the reaction conditions, are dehydrogenated to the final products 12a–c with yields in the range of 41–52%.
![[1860-5397-12-245-i2]](/bjoc/content/inline/1860-5397-12-245-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis and mechanism of formation of sydnone-indolizines 12.
Scheme 2: The synthesis and mechanism of formation of sydnone-indolizines 12.
Bearing in mind that the formation of indolizines through Chichibabin synthesis and 1,3-dipolar cycloaddition reaction requires in both cases the formation of an intermediate pyridinium N-ylide, it is expected that in these cycloadditions a mixture of sydnone-indolizine 9 and cycloadduct 12 might result. In order to verify this hypothesis the crude products resulting from the cycloaddition reactions were carefully investigated by NMR spectroscopy. No trace of product 9 that might have resulted from Chichibabin cyclization was observed. This result indicates that the reaction rate of pyridinium N-ylides 10 with the acetylenic dipolarophile is fast in comparison with an intramolecular cyclization to form compounds 9.
The structures of the sydnone-indolizines 12 were assigned by NMR spectroscopy, IR and elemental analysis. As representative compound, 12c was selected for X-ray structural elucidation. Figure 4 shows the molecular structure, whose conformation is partially stabilized by two significant intramolecular hydrogen bonds. These are found between C24–H24A···O14 and C25–H25B···O28, with remarkably short C···O distances of 2.787(2) and 2.912(2) Å, respectively. Torsion angles that define the relative orientations of the indolizine moiety, the sydnone ring and the linking keto group include C23–C15–C13–C4 −38.7(2)°, C23–C15–C13–O14 138.0(1)° and C15–C13–C4–C5 −37.3(2)°. The relative orientations of the phenyl and sydnone rings are defined by the torsion angle C4–N3–C7–C12 −55.7(2)°.
![[1860-5397-12-245-4]](/bjoc/content/figures/1860-5397-12-245-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure of 12c with atoms represented as thermal ellipsoids at the 50% probability level. Dotted lines indicate intramolecular hydrogen bonds. Bond distances (Å) in the sydnone moiety are: O1–N2 1.370(2), N2–N3 1.307(2),N3–C4 1.361(2), C4–C5 1.427(2), C5–O1 1.424(2), and C5–O6 1.202(2).
Figure 4: Molecular structure of 12c with atoms represented as thermal ellipsoids at the 50% probability leve...
There is evidence of significant steric hindrance in the ‘bay region’ of the indolizine moiety of 12c, with very short non-bonded C···C and C···O distances (Figure 5). In particular, steric repulsion between the methyl carbon C24 and the keto oxygen O14 causes the bond C17–C24 to be rotated by 6.5° below the indolizine plane and the bond C15–C13 to be rotated above the indolizine plane by as much as 20.8°. The net effect is that the chain of atoms C24–C17–N16–C15–C13–O14 resembles a spiral ramp.
![[1860-5397-12-245-5]](/bjoc/content/figures/1860-5397-12-245-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Intramolecular distortion in 12c.
Figure 5: Intramolecular distortion in 12c.
The introduction of the linking keto group in 12c also results in small but statistically significant changes in the interatomic distances in the sydnone ring relative to compound 9d. The most pronounced effect is observed for the distance C5–O1, which is 0.022 Å longer in 12c, i.e., a difference of ~8σ (σ = combined standard deviation). Furthermore, the sydnone-ring environment in the crystal of 12c differs significantly from that in 9d. The dipolar alignment of the sydnone moieties observed in 9d does not occur in 12c. The exocyclic atom O6 engages as an acceptor in an intermolecular C–H···O hydrogen bond. Further stabilizing interactions in the crystal of 12c include two additional intermolecular C–H···O bonds and an offset π-stacking interaction between the indolizine ring and its inversion-related counterpart, with the shortest centroid···centroid distance 3.718(1) Å.
The 1H NMR spectra of compounds 12 confirmed the indolizine structure of the cycloadducts. The representative signals for the protons H-2 appear at 8.16–8.18 ppm as strongly deshielded singlets. The protons from the pyridine moiety were assigned unequivocally and could be distinguished from the protons of the sydnone-attached phenyl ring. The 13C NMR data are in agreement with the proposed structures for the hybrid heterocycles 12. The signals for the endocyclic and exocyclic CO groups of the sydnone moiety are strongly deshielded and the chemical shifts are very close, appearing in the range of 165.5–166.1 ppm, whereas the carbonyls of the ester groups are shielded by 2–3 ppm. Both, C-1 from the indolizine and C-4 from the sydnone rings are shielded and resonate in the range of 106.8–106.9 ppm. In the two series of indolizines 9 and 12 the chemical shifts for the carbon atoms of the indolizine moiety are strongly dependent on the nature of the substituents. Thus, the differences between the chemical shifts for the corresponding carbon atoms of the two indolizine series are generally higher than 5 ppm (except for C-3 and C-8). In the ATR-IR spectra the main characteristic absorption frequencies are those for the three carbonyl groups.
Conclusion
The indolizination of sydnones at C-4 was achieved by the simple Chichibabin indolizine synthesis involving the formation of pyridinium salts from commercially available pyridines and 4-bromoacetyl-3-phenylsydnone, followed by cyclization of the pyridinium bromides in the presence of a base.
Depending on the accessibility of the compounds of type Het-COCH2Hal the indolizination of aromatic heterocycles by the Chichibabin synthesis could be extended to link two heteroarenes. This method overcomes the difficulties encountered in obtaining biheteroaryls through palladium-catalyzed cross-coupling reactions.
In addition, the pyridinium salts used in the synthesis of sydnone-indolizine hybrids could be transformed into sydnone-indolizines connected via a keto group. The transformation was achieved by a one-pot procedure implying the in situ generation of sydnone-pyridinium N-ylides followed by 1,3-dipolar cycloaddition reaction with an acetylenic dipolarophile.
The structures of the new hybrid heterocyclic sydnone-indolizines were assigned by elemental analysis, NMR and IR spectroscopy and X-ray analyses of representative compounds. The latter technique revealed the dipolar alignment of the sydnone moieties in one case and a significant intramolecular distortion in the other.
Supporting Information
Experimental details of synthetic procedures and experimental details of X-ray structure analyses. Crystallographic information files (CIF) for compound 9d and 12c, listing crystal data, experimental and refinement details, and molecular parameters (bond lengths, bond angles and torsion angles) are provided.
| Supporting Information File 1: Experimental. | ||
| Format: PDF | Size: 223.8 KB | Download |
| Supporting Information File 2: Crystallographic information file (CIF) for compound 9d. | ||
| Format: CIF | Size: 258.8 KB | Download |
| Supporting Information File 3: Crystallographic information file (CIF) for compound 12c. | ||
| Format: CIF | Size: 582.0 KB | Download |
References
-
Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174–238. doi:10.1021/cr0509760
Return to citation in text: [1] -
Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Adv. Synth. Catal. 2014, 356, 17–117. doi:10.1002/adsc.201300922
Return to citation in text: [1] -
Stephens, D. E.; Larionov, O. V. Tetrahedron 2015, 71, 8683–8716. doi:10.1016/j.tet.2015.08.034
Return to citation in text: [1] -
Hao, W.; Liu, Y. Beilstein J. Org. Chem. 2015, 11, 2132–2144. doi:10.3762/bjoc.11.230
Return to citation in text: [1] -
Albota, F.; Drăghici, C.; Caira, M. R.; Dumitrascu, F. Tetrahedron 2015, 71, 9095–9100. doi:10.1016/j.tet.2015.10.021
Return to citation in text: [1] [2] [3] -
Borrows, E. T.; Holland, D. O. Chem. Rev. 1948, 42, 611–643. doi:10.1021/cr60133a005
Return to citation in text: [1] [2] [3] -
Flitsch, W. Pyrroles with fused six-membered Heterocyclic Rings: (i) a-Fused. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Katritzky, A.; Rees, C. W., Eds.; Comprehensive Heterocyclic Chemistry, Vol. 4; Pergamon: Oxford, 1984; pp 443–495. doi:10.1016/B978-008096519-2.00058-8
Return to citation in text: [1] [2] [3] -
Mosby, W. L. Fused 5/5 Ring Systems. Heterocyclic Systems with Bridgehead Nitrogen Atoms, Part I; The Chemistry of Heterocyclic Compounds, Vol. 15; Interscience: New York, 1961. doi:10.1002/9780470186732.ch4
Return to citation in text: [1] [2] [3] -
Prostakov, N. S.; Baktibaev, O. B. Usp. Khim. 1975, 9, 1649–1687.
Return to citation in text: [1] [2] [3] -
Sharma, V.; Kumar, V. Med. Chem. Res. 2014, 23, 3593–3606. doi:10.1007/s00044-014-0940-1
Return to citation in text: [1] [2] [3] -
Shipman, M. Product Class 16: Indolizines. In Fused Five-Membered Hetarenes with One Heteroatom; Thomas, E. J., Ed.; Science of Synthesis, Vol. 10; Thieme: Stuttgart, 2010; pp 745–787.
Return to citation in text: [1] [2] [3] -
Singh, G. S.; Mmatli, E. E. Eur. J. Med. Chem. 2011, 46, 5237–5257. doi:10.1016/j.ejmech.2011.08.042
Return to citation in text: [1] [2] [3] -
Swinborne, P.-J.; Hunt, J. H.; Klinkert, G. Adv. Heterocycl. Chem. 1979, 23, 103–170. doi:10.1016/S0065-2725(08)60842-9
Return to citation in text: [1] [2] [3] -
Uchida, T.; Matsumoto, K. Synthesis 1976, 209–236. doi:10.1055/s-1976-23994
Return to citation in text: [1] [2] [3] -
Kheder, N. A.; Darwish, E. S.; Dawood, K. M. Heterocycles 2009, 78, 177–188. doi:10.3987/COM-08-11527
Return to citation in text: [1] [2] [3] -
Caira, M. R.; Popa, M. M.; Drăghici, C.; Barbu, L.; Dumitrescu, D.; Dumitrascu, F. Tetrahedron Lett. 2014, 55, 5635–5638. doi:10.1016/j.tetlet.2014.08.054
Return to citation in text: [1] -
Dumitrascu, F.; Căproiu, M. T.; Georgescu, F.; Drăghici, B.; Popa, M. M.; Georgescu, E. Synlett 2010, 2407–2410. doi:10.1055/s-0030-1258035
Return to citation in text: [1] [2] -
Dumitrascu, F.; Georgescu, E.; Georgescu, F.; Popa, M. M.; Dumitrescu, D. Molecules 2013, 18, 2635–2645. doi:10.3390/molecules18032635
Return to citation in text: [1] [2] -
Popa, M. M.; Georgescu, E.; Caira, M. R.; Georgescu, F.; Draghici, C.; Stan, R.; Deleanu, C.; Dumitrascu, F. Beilstein J. Org. Chem. 2015, 11, 1079–1088. doi:10.3762/bjoc.11.121
Return to citation in text: [1] -
Asundaria, S. T.; Pannecouque, C.; De Clercq, E.; Supuran, C. T.; Patel, K. C. Med. Chem. Res. 2013, 22, 5752–5763. doi:10.1007/s00044-013-0567-7
Return to citation in text: [1] [2] -
Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 533–568. doi:10.1016/j.tet.2009.10.085
Return to citation in text: [1] [2] -
Dumitraşcu, F.; Drăghici, C.; Dumitreşcu, D.; Tarko, L.; Râileanu, D. Liebigs Ann./Recl. 1997, 2613–2616. doi:10.1002/jlac.199719971229
Return to citation in text: [1] [2] -
Wentrup, K.; Kvaskoff, D.; Veedu, R. N. J. Photochem. Photobiol., A 2015, 298, 62–63. doi:10.1016/j.jphotochem.2014.10.010
Return to citation in text: [1] [2] -
Weisner, A. J.; Turnbull, K. ARKIVOC 2014, No. iv, 164–169. doi:10.3998/ark.5550190.p008.474
Return to citation in text: [1] [2] -
Dürüst, Y.; Sağırlı, A.; Kariuki, B. M.; Knight, D. W. Tetrahedron 2014, 70, 6012–6019. doi:10.1016/j.tet.2014.04.083
Return to citation in text: [1] [2] -
Dumitrascu, F.; Mitan, C. I.; Dumitrescu, D.; Draghici, C.; Caproiu, M. T. ARKIVOC 2002, No. ii, 80–86. doi:10.3998/ark.5550190.0003.209
Return to citation in text: [1] [2] -
Tschitschibabin, A. E. Ber. Dtsch. Chem. Ges. 1927, 60, 1607–1617. doi:10.1002/cber.19270600724
Return to citation in text: [1] -
Chai, W.; Kwok, A.; Wong, V.; Carruthers, N. I.; Wu, J. Synlett 2003, 2086–2088. doi:10.1055/s-2003-42042
Return to citation in text: [1] -
Dinculescu, A.; Balaban, T.-S.; Balaban, A. T. Tetrahedron Lett. 1987, 28, 3145–3146. doi:10.1016/S0040-4039(00)96307-X
Return to citation in text: [1] -
Kulikova, Yu. A.; Surikova, O. V.; Mikhailovskii, A. G.; Vakhrin, M. I. Chem. Heterocycl. Compd. 2011, 47, 290–293. doi:10.1007/s10593-011-0755-7
Return to citation in text: [1] -
Adachi, S.; Liew, S. K.; Lee, C. F.; Lough, A.; He, Z.; St Denis, J. D.; Poda, G.; Yudin, A. K. Org. Lett. 2015, 17, 5594–5597. doi:10.1021/acs.orglett.5b02741
Return to citation in text: [1] -
Brown, A. W.; Harrity, J. P. A. J. Org. Chem. 2015, 80, 2467–2472. doi:10.1021/acs.joc.5b00143
Return to citation in text: [1] [2] -
Rodriguez, A.; Moran, W. J. Synthesis 2009, 650–654. doi:10.1055/s-0028-1083344
Return to citation in text: [1] [2] -
Shih, M.-H.; Tsai, C.-H.; Wang, Y.-C.; Shieh, M.-Y.; Lin, G.-L.; Wei, C.-Y. Tetrahedron 2007, 63, 2990–2999. doi:10.1016/j.tet.2007.01.066
Return to citation in text: [1] [2] -
Yang, Y.; Lu, C.; Zhong, W. ARKIVOC 2015, No. vii, 122–130. doi:10.3998/ark.5550190.p009.180
Return to citation in text: [1] [2] -
Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2009, 74, 396–400. doi:10.1021/jo802240e
Return to citation in text: [1] [2] -
Cambridge Structural Database and Cambridge Structural Database system, Version 5.37; Cambridge Crystallographic Data Centre: University Chemical Laboratory; Cambridge, England, 2015.
Return to citation in text: [1]
| 1. | Alberico, D.; Scott, M. E.; Lautens, M. Chem. Rev. 2007, 107, 174–238. doi:10.1021/cr0509760 |
| 2. | Rossi, R.; Bellina, F.; Lessi, M.; Manzini, C. Adv. Synth. Catal. 2014, 356, 17–117. doi:10.1002/adsc.201300922 |
| 3. | Stephens, D. E.; Larionov, O. V. Tetrahedron 2015, 71, 8683–8716. doi:10.1016/j.tet.2015.08.034 |
| 4. | Hao, W.; Liu, Y. Beilstein J. Org. Chem. 2015, 11, 2132–2144. doi:10.3762/bjoc.11.230 |
| 6. | Borrows, E. T.; Holland, D. O. Chem. Rev. 1948, 42, 611–643. doi:10.1021/cr60133a005 |
| 7. | Flitsch, W. Pyrroles with fused six-membered Heterocyclic Rings: (i) a-Fused. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Katritzky, A.; Rees, C. W., Eds.; Comprehensive Heterocyclic Chemistry, Vol. 4; Pergamon: Oxford, 1984; pp 443–495. doi:10.1016/B978-008096519-2.00058-8 |
| 8. | Mosby, W. L. Fused 5/5 Ring Systems. Heterocyclic Systems with Bridgehead Nitrogen Atoms, Part I; The Chemistry of Heterocyclic Compounds, Vol. 15; Interscience: New York, 1961. doi:10.1002/9780470186732.ch4 |
| 9. | Prostakov, N. S.; Baktibaev, O. B. Usp. Khim. 1975, 9, 1649–1687. |
| 10. | Sharma, V.; Kumar, V. Med. Chem. Res. 2014, 23, 3593–3606. doi:10.1007/s00044-014-0940-1 |
| 11. | Shipman, M. Product Class 16: Indolizines. In Fused Five-Membered Hetarenes with One Heteroatom; Thomas, E. J., Ed.; Science of Synthesis, Vol. 10; Thieme: Stuttgart, 2010; pp 745–787. |
| 12. | Singh, G. S.; Mmatli, E. E. Eur. J. Med. Chem. 2011, 46, 5237–5257. doi:10.1016/j.ejmech.2011.08.042 |
| 13. | Swinborne, P.-J.; Hunt, J. H.; Klinkert, G. Adv. Heterocycl. Chem. 1979, 23, 103–170. doi:10.1016/S0065-2725(08)60842-9 |
| 14. | Uchida, T.; Matsumoto, K. Synthesis 1976, 209–236. doi:10.1055/s-1976-23994 |
| 15. | Kheder, N. A.; Darwish, E. S.; Dawood, K. M. Heterocycles 2009, 78, 177–188. doi:10.3987/COM-08-11527 |
| 5. | Albota, F.; Drăghici, C.; Caira, M. R.; Dumitrascu, F. Tetrahedron 2015, 71, 9095–9100. doi:10.1016/j.tet.2015.10.021 |
| 17. | Dumitrascu, F.; Căproiu, M. T.; Georgescu, F.; Drăghici, B.; Popa, M. M.; Georgescu, E. Synlett 2010, 2407–2410. doi:10.1055/s-0030-1258035 |
| 18. | Dumitrascu, F.; Georgescu, E.; Georgescu, F.; Popa, M. M.; Dumitrescu, D. Molecules 2013, 18, 2635–2645. doi:10.3390/molecules18032635 |
| 16. | Caira, M. R.; Popa, M. M.; Drăghici, C.; Barbu, L.; Dumitrescu, D.; Dumitrascu, F. Tetrahedron Lett. 2014, 55, 5635–5638. doi:10.1016/j.tetlet.2014.08.054 |
| 17. | Dumitrascu, F.; Căproiu, M. T.; Georgescu, F.; Drăghici, B.; Popa, M. M.; Georgescu, E. Synlett 2010, 2407–2410. doi:10.1055/s-0030-1258035 |
| 18. | Dumitrascu, F.; Georgescu, E.; Georgescu, F.; Popa, M. M.; Dumitrescu, D. Molecules 2013, 18, 2635–2645. doi:10.3390/molecules18032635 |
| 19. | Popa, M. M.; Georgescu, E.; Caira, M. R.; Georgescu, F.; Draghici, C.; Stan, R.; Deleanu, C.; Dumitrascu, F. Beilstein J. Org. Chem. 2015, 11, 1079–1088. doi:10.3762/bjoc.11.121 |
| 6. | Borrows, E. T.; Holland, D. O. Chem. Rev. 1948, 42, 611–643. doi:10.1021/cr60133a005 |
| 7. | Flitsch, W. Pyrroles with fused six-membered Heterocyclic Rings: (i) a-Fused. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Katritzky, A.; Rees, C. W., Eds.; Comprehensive Heterocyclic Chemistry, Vol. 4; Pergamon: Oxford, 1984; pp 443–495. doi:10.1016/B978-008096519-2.00058-8 |
| 8. | Mosby, W. L. Fused 5/5 Ring Systems. Heterocyclic Systems with Bridgehead Nitrogen Atoms, Part I; The Chemistry of Heterocyclic Compounds, Vol. 15; Interscience: New York, 1961. doi:10.1002/9780470186732.ch4 |
| 9. | Prostakov, N. S.; Baktibaev, O. B. Usp. Khim. 1975, 9, 1649–1687. |
| 10. | Sharma, V.; Kumar, V. Med. Chem. Res. 2014, 23, 3593–3606. doi:10.1007/s00044-014-0940-1 |
| 11. | Shipman, M. Product Class 16: Indolizines. In Fused Five-Membered Hetarenes with One Heteroatom; Thomas, E. J., Ed.; Science of Synthesis, Vol. 10; Thieme: Stuttgart, 2010; pp 745–787. |
| 12. | Singh, G. S.; Mmatli, E. E. Eur. J. Med. Chem. 2011, 46, 5237–5257. doi:10.1016/j.ejmech.2011.08.042 |
| 13. | Swinborne, P.-J.; Hunt, J. H.; Klinkert, G. Adv. Heterocycl. Chem. 1979, 23, 103–170. doi:10.1016/S0065-2725(08)60842-9 |
| 14. | Uchida, T.; Matsumoto, K. Synthesis 1976, 209–236. doi:10.1055/s-1976-23994 |
| 15. | Kheder, N. A.; Darwish, E. S.; Dawood, K. M. Heterocycles 2009, 78, 177–188. doi:10.3987/COM-08-11527 |
| 32. | Brown, A. W.; Harrity, J. P. A. J. Org. Chem. 2015, 80, 2467–2472. doi:10.1021/acs.joc.5b00143 |
| 33. | Rodriguez, A.; Moran, W. J. Synthesis 2009, 650–654. doi:10.1055/s-0028-1083344 |
| 34. | Shih, M.-H.; Tsai, C.-H.; Wang, Y.-C.; Shieh, M.-Y.; Lin, G.-L.; Wei, C.-Y. Tetrahedron 2007, 63, 2990–2999. doi:10.1016/j.tet.2007.01.066 |
| 35. | Yang, Y.; Lu, C.; Zhong, W. ARKIVOC 2015, No. vii, 122–130. doi:10.3998/ark.5550190.p009.180 |
| 36. | Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2009, 74, 396–400. doi:10.1021/jo802240e |
| 5. | Albota, F.; Drăghici, C.; Caira, M. R.; Dumitrascu, F. Tetrahedron 2015, 71, 9095–9100. doi:10.1016/j.tet.2015.10.021 |
| 37. | Cambridge Structural Database and Cambridge Structural Database system, Version 5.37; Cambridge Crystallographic Data Centre: University Chemical Laboratory; Cambridge, England, 2015. |
| 28. | Chai, W.; Kwok, A.; Wong, V.; Carruthers, N. I.; Wu, J. Synlett 2003, 2086–2088. doi:10.1055/s-2003-42042 |
| 29. | Dinculescu, A.; Balaban, T.-S.; Balaban, A. T. Tetrahedron Lett. 1987, 28, 3145–3146. doi:10.1016/S0040-4039(00)96307-X |
| 30. | Kulikova, Yu. A.; Surikova, O. V.; Mikhailovskii, A. G.; Vakhrin, M. I. Chem. Heterocycl. Compd. 2011, 47, 290–293. doi:10.1007/s10593-011-0755-7 |
| 31. | Adachi, S.; Liew, S. K.; Lee, C. F.; Lough, A.; He, Z.; St Denis, J. D.; Poda, G.; Yudin, A. K. Org. Lett. 2015, 17, 5594–5597. doi:10.1021/acs.orglett.5b02741 |
| 5. | Albota, F.; Drăghici, C.; Caira, M. R.; Dumitrascu, F. Tetrahedron 2015, 71, 9095–9100. doi:10.1016/j.tet.2015.10.021 |
| 27. | Tschitschibabin, A. E. Ber. Dtsch. Chem. Ges. 1927, 60, 1607–1617. doi:10.1002/cber.19270600724 |
| 32. | Brown, A. W.; Harrity, J. P. A. J. Org. Chem. 2015, 80, 2467–2472. doi:10.1021/acs.joc.5b00143 |
| 33. | Rodriguez, A.; Moran, W. J. Synthesis 2009, 650–654. doi:10.1055/s-0028-1083344 |
| 34. | Shih, M.-H.; Tsai, C.-H.; Wang, Y.-C.; Shieh, M.-Y.; Lin, G.-L.; Wei, C.-Y. Tetrahedron 2007, 63, 2990–2999. doi:10.1016/j.tet.2007.01.066 |
| 35. | Yang, Y.; Lu, C.; Zhong, W. ARKIVOC 2015, No. vii, 122–130. doi:10.3998/ark.5550190.p009.180 |
| 36. | Browne, D. L.; Taylor, J. B.; Plant, A.; Harrity, J. P. A. J. Org. Chem. 2009, 74, 396–400. doi:10.1021/jo802240e |
| 20. | Asundaria, S. T.; Pannecouque, C.; De Clercq, E.; Supuran, C. T.; Patel, K. C. Med. Chem. Res. 2013, 22, 5752–5763. doi:10.1007/s00044-013-0567-7 |
| 21. | Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 533–568. doi:10.1016/j.tet.2009.10.085 |
| 22. | Dumitraşcu, F.; Drăghici, C.; Dumitreşcu, D.; Tarko, L.; Râileanu, D. Liebigs Ann./Recl. 1997, 2613–2616. doi:10.1002/jlac.199719971229 |
| 23. | Wentrup, K.; Kvaskoff, D.; Veedu, R. N. J. Photochem. Photobiol., A 2015, 298, 62–63. doi:10.1016/j.jphotochem.2014.10.010 |
| 24. | Weisner, A. J.; Turnbull, K. ARKIVOC 2014, No. iv, 164–169. doi:10.3998/ark.5550190.p008.474 |
| 25. | Dürüst, Y.; Sağırlı, A.; Kariuki, B. M.; Knight, D. W. Tetrahedron 2014, 70, 6012–6019. doi:10.1016/j.tet.2014.04.083 |
| 26. | Dumitrascu, F.; Mitan, C. I.; Dumitrescu, D.; Draghici, C.; Caproiu, M. T. ARKIVOC 2002, No. ii, 80–86. doi:10.3998/ark.5550190.0003.209 |
| 20. | Asundaria, S. T.; Pannecouque, C.; De Clercq, E.; Supuran, C. T.; Patel, K. C. Med. Chem. Res. 2013, 22, 5752–5763. doi:10.1007/s00044-013-0567-7 |
| 21. | Browne, D. L.; Harrity, J. P. A. Tetrahedron 2010, 66, 533–568. doi:10.1016/j.tet.2009.10.085 |
| 22. | Dumitraşcu, F.; Drăghici, C.; Dumitreşcu, D.; Tarko, L.; Râileanu, D. Liebigs Ann./Recl. 1997, 2613–2616. doi:10.1002/jlac.199719971229 |
| 23. | Wentrup, K.; Kvaskoff, D.; Veedu, R. N. J. Photochem. Photobiol., A 2015, 298, 62–63. doi:10.1016/j.jphotochem.2014.10.010 |
| 24. | Weisner, A. J.; Turnbull, K. ARKIVOC 2014, No. iv, 164–169. doi:10.3998/ark.5550190.p008.474 |
| 25. | Dürüst, Y.; Sağırlı, A.; Kariuki, B. M.; Knight, D. W. Tetrahedron 2014, 70, 6012–6019. doi:10.1016/j.tet.2014.04.083 |
| 26. | Dumitrascu, F.; Mitan, C. I.; Dumitrescu, D.; Draghici, C.; Caproiu, M. T. ARKIVOC 2002, No. ii, 80–86. doi:10.3998/ark.5550190.0003.209 |
| 6. | Borrows, E. T.; Holland, D. O. Chem. Rev. 1948, 42, 611–643. doi:10.1021/cr60133a005 |
| 7. | Flitsch, W. Pyrroles with fused six-membered Heterocyclic Rings: (i) a-Fused. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Katritzky, A.; Rees, C. W., Eds.; Comprehensive Heterocyclic Chemistry, Vol. 4; Pergamon: Oxford, 1984; pp 443–495. doi:10.1016/B978-008096519-2.00058-8 |
| 8. | Mosby, W. L. Fused 5/5 Ring Systems. Heterocyclic Systems with Bridgehead Nitrogen Atoms, Part I; The Chemistry of Heterocyclic Compounds, Vol. 15; Interscience: New York, 1961. doi:10.1002/9780470186732.ch4 |
| 9. | Prostakov, N. S.; Baktibaev, O. B. Usp. Khim. 1975, 9, 1649–1687. |
| 10. | Sharma, V.; Kumar, V. Med. Chem. Res. 2014, 23, 3593–3606. doi:10.1007/s00044-014-0940-1 |
| 11. | Shipman, M. Product Class 16: Indolizines. In Fused Five-Membered Hetarenes with One Heteroatom; Thomas, E. J., Ed.; Science of Synthesis, Vol. 10; Thieme: Stuttgart, 2010; pp 745–787. |
| 12. | Singh, G. S.; Mmatli, E. E. Eur. J. Med. Chem. 2011, 46, 5237–5257. doi:10.1016/j.ejmech.2011.08.042 |
| 13. | Swinborne, P.-J.; Hunt, J. H.; Klinkert, G. Adv. Heterocycl. Chem. 1979, 23, 103–170. doi:10.1016/S0065-2725(08)60842-9 |
| 14. | Uchida, T.; Matsumoto, K. Synthesis 1976, 209–236. doi:10.1055/s-1976-23994 |
| 15. | Kheder, N. A.; Darwish, E. S.; Dawood, K. M. Heterocycles 2009, 78, 177–188. doi:10.3987/COM-08-11527 |
© 2016 Albota et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)