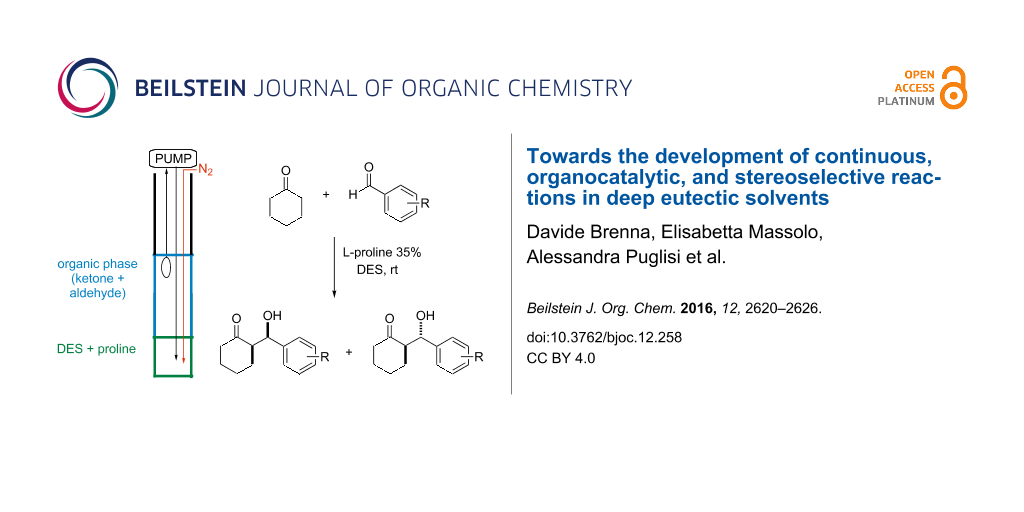
Abstract
Different deep eutectic solvent (DES) mixtures were studied as reaction media for the continuous synthesis of enantiomerically enriched products by testing different experimental set-ups. L-Proline-catalysed cross-aldol reactions were efficiently performed in continuo, with high yield (99%), anti-stereoselectivity, and enantioselectivity (up to 97% ee). Moreover, using two different DES mixtures, the diastereoselectivity of the process could be tuned, thereby leading to the formation, under different experimental conditions, to both the syn- and the anti-isomer with very high enantioselectivity. The excess of cyclohexanone was recovered and reused, and the reaction could be run and the product isolated without the use of any organic solvent by a proper choice of DES components. The dramatic influence of the reaction media on the reaction rate and stereoselectivity of the process suggests that the intimate architecture of DESs deeply influences the reactivity of different species involved in the catalytic cycle.

Graphical Abstract
Introduction
The aldol reaction is a powerful synthetic tool to create new C–C bonds [1]. It offers several possibilities to control the stereochemical outcome of the process and to afford stereochemically defined chiral products [2]. Among all the possible options, the L-proline-catalysed stereoselective cross-aldol reaction remains the greener choice. After the pioneering works by List and Barbas [3], a huge effort was made by the scientific community to improve both the yield and the stereoselectivity of the reaction. The most explored strategies involve the development of a new class of catalysts (mainly prolinamide derivatives) [4-6], the study of additives in combination with proline itself [7-13], and the use of unusual reaction media [14-19].
In this context, it was recently reported that L-proline-catalysed direct aldol reactions may be successfully carried out also in deep eutectic solvents (DESs) [20-22]. Recently, our group reported on the possibility of running organocatalyzed, stereoselective reactions in DESs, promoted by an enantiopure primary amine, with advantages in terms of reaction sustainability. In particular, the possibility to strongly reduce the amounts of organic solvent and the recyclability of the catalyst were demonstrated [23]. Moreover, in this approach, no structural modification of the precious chiral catalyst was necessary.
A well-explored strategy aimed at positively realizing the recovery and the reuse of the catalyst is represented by the immobilization of the catalytic species [24-27]. Synthetic modifications of the original catalyst, however, are required in order to attach the catalyst to the material of choice. The aim of the present study was to develop a catalytic system working in continuo, whereas DES acts at the same time as catalyst trap and as reaction medium, immiscible with the organic reactants. The main advantage of this approach is that the catalyst (i.e., L-proline) would be kept in an environmentally benign reaction medium, without the need of any synthetic modification. Of note, in the herein proposed system, readily assembled using standard glassware, the use of the organic solvent, both for the reaction and for the isolation process, would be strongly reduced or even, ideally, eliminated.
Results and Discussion
Among the plethora of possible DES mixtures [28-33], based on our previous experience [34-39] and preliminary studies on the physicochemical properties of DES combinations, we decided to focus our attention on the use of a few choline chloride (ChCl)-based eutectic mixtures as reaction media (Table 1) [40].
The behaviour of DES mixtures A–E in the proline-catalysed model aldol reaction between cyclohexanone and 4-nitrobenzaldehyde was preliminarily investigated under standard batch conditions (Scheme 1).
![[1860-5397-12-258-i1]](/bjoc/content/inline/1860-5397-12-258-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: L-Proline-promoted stereoselective aldol reaction in DES.
Scheme 1: L-Proline-promoted stereoselective aldol reaction in DES.
In our hands, the reaction proceeded completely in 20 hours and with high conversion (≥95%) in all tested DESs (A–E, Table 2, entries 1–5). While low diastereoselectivity was observed in DES A (Table 2, entry 1), anti-stereoselectivity (up to 85:15) and high enantiomeric excess in favour of the anti isomer (up to 92% ee) were instead detected running the reaction in DESs B–E (Table 2, entries 2–5).
Table 2: DES screening for the proline-catalyzed in batch aldol reaction.
| Entry | DES | Conv. (%)a | dr (anti:syn)a | ee % (anti/syn)b |
|---|---|---|---|---|
| 1 | A | 99 | 57:43 | 81/80 |
| 2 | B | 98 | 82:18 | 89/69 |
| 3 | C | 96 | 85:15 | 92/54 |
| 4 | D | 95 | 75:25 | 84/67 |
| 5 | E | 96 | 70:30 | 82/67 |
aConversion and dr were evaluated by NMR technique on the crude reaction mixture; bee was evaluated by using an HPLC with a chiral stationary phase.
Based on these results, we turned our attention to design and realize a home-made system, to be easily assembled with common glassware, for the continuous synthesis of the aldol product, using a DES mixture as reaction media able to hold back the proline.
In these very explorative studies, different experimental set-ups were investigated, focusing especially on some points, such as (a) the phase contact between the organic phase, composed by cyclohexanone and the aldehyde, and the DES phase, (b) the ratio between DES and L-proline, and, finally, (c) the possible interaction between the aldol product and the DES network (Figure 1). Due to its favourable physical and mechanical properties, DES A was selected for the initial screening of the different experimental conditions in continuo.
![[1860-5397-12-258-1]](/bjoc/content/figures/1860-5397-12-258-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Experimental set-up I: test tube (d = 0.5 cm); flow 1 mL/min; DES (1.5 mL); L-proline/DES = 130 mg/mL. Experimental set-up II: test tube (d = 2.5 cm); flow 1 mL/min; DES (1.5 mL); L-proline/DES = 130 mg/mL. Experimental set-up III: test tube (d = 2.5 cm); flow 1 mL/min; DES (1.5 mL); L-proline/DES = 130 mg/mL.
Figure 1: Experimental set-up I: test tube (d = 0.5 cm); flow 1 mL/min; DES (1.5 mL); L-proline/DES = 130 mg/...
The first experimental set-up that was studied (Figure 1, I) was built using a test tube of reduced diameter (green color in the picture) containing the DES and L-proline, surrounded by an external, larger cylinder filled with a solution of cyclohexanone and 4-nitrobenzaldehyde. The organic solution, fluxed by a HPLC pump onto the bottom of the internal smaller tube, went back through DES due to the difference in the viscosity of the two phases, thereby generating a upper organic phase (blue in the picture) which finally ended into the organic phase of the larger tube, that was continuously pumped into the DES phase to realize a closed cycle.
In set-up II, the mixture of DES and L-proline was covered with the solution of ketone and aldehyde in a 10 mL graduated cylinder. The organic phase was continuously pumped on the bottom of the DES phase and recirculated (Figure 1, II). In order to improve the contact surface between the two phases and favour the phases interaction, nitrogen was used as a diffusor, thus realizing in set-up III a better mixing of the two phases (Figure 1, III).
By monitoring the transformations performed with the above-described different set-ups, it was observed that both the diastereoselection and the enantioselectivity were constant during the reaction time (Table 3). With set up I (Table 3, entries 1–5), after 20 h, a 39% conversion was reached, while full conversion was obtained after 48 h of reaction. Remarkably, high ee values for the syn adduct were observed (up to 94% ee), unfortunately, with a low diastereoisomeric ratio (dr). Using set-up II (Table 3, entries 6 and 7), after 24 h, the conversion was still very low (35%) and the ee for the syn aldol was up to 90%, the complete conversion was achieved after 48 h. Interestingly, the analysis of the mass of the crude mixture showed that a part of the product was trapped into the DES phase. In order to quantitatively collect the aldol adduct, the DES was diluted with 1 mL of water and extracted five times with 2 mL of ethyl acetate. Using this procedure, all the aldol adduct was completely recovered.
In the set-up III (Table 3, entries 8–11) the presence of a more efficient phase mixing led to a faster conversion. After only 5 h (Table 3, entry 8), 26% conversion was observed, with interesting diastereoselection and high enantioselection (up to 92% for the syn adduct). After 48 h, the aldehyde was almost quantitatively converted into the desired aldol product, with high enantioselectivity for both the syn (up to 92%) and the anti (up to 90%) isomers.
Table 3: Three different set-ups for the aldol reaction in continuo.
| Entry | Set-up | Time (h) | Conv. (%)a | anti:syna | ee% (anti/syn)b |
|---|---|---|---|---|---|
| 1 | I | 20 | 39 | 59:41 | 70/94 |
| 2 | I | 24 | 47 | 58:42 | 68/92 |
| 3 | I | 40 | 87 | 55:45 | 79/92 |
| 4 | I | 48 | 99 | 53:47 | 76/88 |
| 5 | I | washc | 99 | 52:48 | 70/84 |
| 6 | II | 24 | 35 | 49:51 | 78/90 |
| 7 | II | 48 | 96 | 64:36 | 84/83 |
| 8 | III | 5 | 26 | 62:38 | 86/92 |
| 9 | III | 24 | 48 | 63:37 | 90/91 |
| 10 | III | 48 | 90 | 64:36 | 84/85 |
| 11 | III | washc | 91 | 67:33 | 84/85 |
aConversion and dr were evaluated after removing cyclohexanone from samples taken at indicated reaction times; bee was evaluated by HPLC on chiral stationary phase. cin order to wash the pump 2 mL of cyclohexanone were used.
Having identified the system III as the best experimental set-up, the general scope was briefly investigated by running the reaction with a few different aldehydes and comparing the activities of DES mixtures A and B in the reactions performed in continuo (Scheme 2).
![[1860-5397-12-258-i2]](/bjoc/content/inline/1860-5397-12-258-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Aldol reaction under continuous flow conditions in DESs.
Scheme 2: Aldol reaction under continuous flow conditions in DESs.
In the case of 4-nitrobenzaldeyde, the use of DES B (a ternary mixture of ChCl, urea and water, 1:2:1.5 ratio) led to impressive results, both in reaction rate and stereoselectivity, compared to the reaction run in DES A (Table 4, entries 1–4). The reaction proceeded completely in only 15 h, and afforded a clean product (aldol 1, Scheme 2) that was easily isolated by evaporation of excess cyclohexanone, with high anti-diastereoselectivity (up to 90:10), and enantioselectivity (up to 92%) for the major anti isomer.
Table 4: In continuo aldol reactions of different aldehydes in DES A and DES B.
| Entry | DES | Aldol | R | Time (h) | Conv. (%)a | anti:syna | ee % (anti/syn)b |
|---|---|---|---|---|---|---|---|
| 1 | A | 1 | 4-NO2 | 5 | 26 | 62:38 | 86/92 |
| 2 | A | 1 | 4-NO2 | 20 | 48 | 63:37 | 90/91 |
| 3 | B | 1 | 4-NO2 | 5 | 73 | 85:15 | 92/70 |
| 4 | B | 1 | 4-NO2 | 15 | 99 | 90:10 | 90/70 |
| 5 | A | 2 | 4-Cl | 3 | 13 | 71:29 | 73/78 |
| 6c | A | 2 | 4-Cl | 24 | 99 | 57:43 | 73/78 |
| 7 | B | 2 | 4-Cl | 3 | 50 | 88:12 | 88/73 |
| 8 | B | 2 | 4-Cl | 24 | 91 | 80:20 | 88/77 |
| 9 | A | 3 | 4-Br | 24 | 67 | 65:35 | 81/70 |
| 10 | A | 3 | 4-Br | 42 | 99 | 65:35 | 80/70 |
| 11 | B | 3 | 4-Br | 3 | 10 | 70:30 | 91/85 |
| 12 | B | 3 | 4-Br | 24 | 75 | 70:30 | 93/86 |
| 13 | B | 4 | H | 42 | 20 | 90:10 | 87/64 |
| 14 | B | 5 | 2-NO2 | 3 | 9 | 90:10 | 95/50 |
| 15 | B | 5 | 2-NO2 | 24 | 51 | 93:7 | 97/52 |
aConversion and dr were evaluated after removing cyclohexanone from samples taken at indicated reaction times; bee was evaluated using an HPLC with a chiral stationary phase; cin this case, it was necessary to use 10 mL of EtOAc to quantitatively recover the aldol adduct (Supporting Information File 1).
By performing the reaction with 4-chlorobenzaldehyde in DES A (entries 5 and 6, Table 4), the desired aldol product 2 was obtained in 99% yield after only 24 h, with up to 73% enantioselectivity for the anti isomer. Notably, using DES B (Table 4, entries 7 and 8) a high anti diastereoselectivity (up to 88:12) jointly with a very high ee for the major isomer (up to 88% ee) was detected. It is worth mentioning that when working in DES A, the aldol adduct 2 was partially retained in the DES phase and an extraction with ethyl acetate was necessary to quantitatively recover the product. However, as for the reaction in DES B, the whole aldol product was recovered simply by evaporating the organic phase (distilling off the excess of cyclohexanone; for experimental details see Supporting Information File 1).
Analogous results were obtained in the reaction with 4-bromobenzaldehyde. In DES B, the aldol product 3 was isolated in higher yield and stereoselectivity than in DES A (Table 4, entries 9–12; 93% ee for the major anti isomer). While the reaction with benzaldehyde led to poor results, the conversion of 2-nitrobenzaldehyde in the expected aldol adduct 5 proceeded in moderate yield (51% after 24 h), but with a remarkable anti-diastereoselectivity (93:7) and enantioselectivity (up to 97%).
The different stereoselectivities of the reaction observed in different DES phases could be related to the creation of different tridimensional networks between DES and L-proline, and thus of different chiral reaction environments possibly affecting the stereochemistry of the intermediate species involved in the catalytic cycle [41]. The equilibrating nature of the aldol reaction and the influence of such reversibility on its stereochemical outcome has recently been studied [42]. It has also been reported that the use of additives may have a dramatic influence on the diastereoselectivity and the enantioselectivity in proline-catalyzed aldol transformations [43].
Typically, reactions run in DES mixtures lead to a very clean crude mixture. The recovery of the final aldol adduct can be, indeed, achieved using a reduced quantity of cyclohexanone (12 mL for 1.3 grams of crude aldol), that could be recovered by distillation and reused in new reactions (for experimental details on the product recovery, mass balance and 1H NMR spectra of the crude mixture see Supporting Information File 1).
Finally, we also performed preliminary recycling experiments using two different DESs and set-up III. DES mixtures A or B (1.5 mL), containing L-proline (0.35 equiv, 195 mg), previously used for 48 h in the aldol reaction of cyclohexanone with 4-nitrobenzaldehyde, were recycled in the same transformation. At the end of the reaction, the pump was washed with 3 mL of cyclohexanone, in order to recover the product present in the pump system, then the supernatant (cyclohexanone and aldol product) was separated from the DES phase, containing the catalyst, and analyzed. To the DES phase, new reagents (cyclohexanone and aldehyde) were added and the reaction was started again. While the catalytic system in DES A showed a lower activity, thus affording the product in a significant lower yield, the L-proline/DES B system afforded results comparable to the first run, both in terms of chemical yield and stereoselectivity (93% yield, 92% ee for the major anti isomer; see Table S2 in Supporting Information File 1).
Conclusion
In conclusion, the possibility of a continuous, organocatalyzed, stereoselective process in DES was, for the first time, studied and successfully developed. Using different experimental set-ups, it was possible to realize efficient proline-catalysed cross-aldol reactions in continuo with high yield (99%), anti-stereoselectivity, and enantioselectivity (up to 97% ee). Moreover, using two different DES mixtures, the diastereoselection of the process could be tuned, to obtain both the syn- and the anti-isomer with very high ee values working under different experimental conditions.
DESs were successfully employed as reaction media for continuous production of enantioenriched aldol products, and the excess of cyclohexanone could be recovered and reused. It is worth noting that the reaction can be run and the product isolated without the use of any organic solvent by a proper choice of DES components. The dramatic influence of the reaction media, both on the reaction rate and the stereoselectivity of the process, is consistent with an unprecedented influence of 3D DES architecture on the reactivity of the different species involved in the catalytic cycle, even when using an apparently simple organocatalyst such as L-proline. These observations have important implications in the future design of chiral catalysts, thereby opening the floodgates to new intriguing opportunities for organocatalysis in unconventional reaction media.
Supporting Information
| Supporting Information File 1: Experimental set-up and general procedures for the continuous reactions and in batch reactions; product characterization. | ||
| Format: PDF | Size: 698.2 KB | Download |
Acknowledgements
We thank Mr. Raffaele Cocquio for valuable assistance in the experimental work. E.M. and D.B. thank the Università degli Studi di Milano for a Ph.D. fellowship. A.P thanks the Università degli Studi di Milano for the grant “Piano di Sostegno alla Ricerca 2015-17 - LINEA 2 Azione A (Giovani Ricercatori)”. V.C. thanks the Interuniversities Consortium C.I.N.M.P.I.S. for financial support.
References
-
Mahrwald, R. Modern Aldol Reactions; Wiley-VCH: Weinheim, Germany, 2004; Vol. 1 and 2. doi:10.1002/9783527619566
Return to citation in text: [1] -
Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527604677
Return to citation in text: [1] -
List, B.; Lerner, R. A.; Barbas, C. F., III. J. Am. Chem. Soc. 2000, 122, 2395–2396. doi:10.1021/ja994280y
Return to citation in text: [1] -
Liu, X.; Lin, L.; Feng, X. Chem. Commun. 2009, 6145–6158. doi:10.1039/b913411e
Return to citation in text: [1] -
Orlandi, M.; Benaglia, M.; Raimondi, L.; Celentano, G. Eur. J. Org. Chem. 2013, 2346–2354. doi:10.1002/ejoc.201201643
Return to citation in text: [1] -
Guizzetti, S.; Benaglia, M.; Pignataro, L.; Puglisi, A. Tetrahedron: Asymmetry 2006, 17, 2754–2760. doi:10.1016/j.tetasy.2006.10.018
Return to citation in text: [1] -
Cho, E.; Kim, T. H. Tetrahedron Lett. 2014, 55, 6470–6473. doi:10.1016/j.tetlet.2014.10.009
Return to citation in text: [1] -
Karmakar, A.; Maji, T.; Wittmann, S.; Reiser, O. Chem. – Eur. J. 2011, 17, 11024–11029. doi:10.1002/chem.201101299
Return to citation in text: [1] -
Opalka, S. M.; Steinbacher, J. L.; Lambiris, B. A.; McQuade, D. T. J. Org. Chem. 2011, 76, 6503–6517. doi:10.1021/jo200838v
Return to citation in text: [1] -
El-Hamdouni, N.; Companyó, X.; Rios, R.; Moyano, A. Chem. – Eur. J. 2010, 16, 1142–1148. doi:10.1002/chem.200902678
Return to citation in text: [1] -
Reis, O.; Eymur, S.; Reis, B.; Demir, A. S. Chem. Commun. 2009, 1088–1090. doi:10.1039/b817474a
Return to citation in text: [1] -
Mandal, T.; Zhao, C.-G. Angew. Chem., Int. Ed. 2008, 47, 7714–7717. doi:10.1002/anie.200803236
Return to citation in text: [1] -
Clarke, M. L.; Fuentes, J. A. Angew. Chem., Int. Ed. 2007, 46, 930–933. doi:10.1002/anie.200602912
Return to citation in text: [1] -
Rodriguez, B.; Bruckmann, A.; Bolm, C. Chem. – Eur. J. 2007, 13, 4710–4722. doi:10.1002/chem.200700188
See for ball mill approach.
Return to citation in text: [1] -
Clegg, W.; Harrington, R. W.; North, M.; Pizzato, F.; Villuendas, P. Tetrahedron: Asymmetry 2010, 21, 1262–1271. doi:10.1016/j.tetasy.2010.03.051
See for the use of carbamates.
Return to citation in text: [1] -
Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C. F., III. J. Am. Chem. Soc. 2006, 128, 734–735. doi:10.1021/ja0573312
See for aldol reaction on water.
Return to citation in text: [1] -
Hayashi, Y.; Sumiya, T.; Takahashi, J.; Gotoh, H.; Urushima, T.; Shoji, M. Angew. Chem., Int. Ed. 2006, 45, 958–961. doi:10.1002/anie.200502488
Return to citation in text: [1] -
Guizzetti, S.; Benaglia, M.; Raimondi, L.; Celentano, G. Org. Lett. 2007, 9, 1247–1250. doi:10.1021/ol070002p
Return to citation in text: [1] -
Mlynarski, J.; Bas, S. Chem. Soc. Rev. 2014, 43, 577–587. doi:10.1039/C3CS60202H
See for a recent review on organocatalyzed reactions in/on water.
Return to citation in text: [1] -
Ilgen, F.; König, B. Green Chem. 2009, 11, 848–854. doi:10.1039/B816551C
See for pioneer studies of L-proline in DESs as catalysts of Diels-Alder reactions.
Return to citation in text: [1] -
Müller, C. R.; Meiners, I.; Domínguez de María, P. RSC Adv. 2014, 4, 46097–46101. doi:10.1039/C4RA09307K
See for proline-promoted aldol reaction.
Return to citation in text: [1] -
Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D. J. Green Chem. 2016, 18, 1724–1730. doi:10.1039/C5GC02526E
Return to citation in text: [1] -
Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F. M. Green Chem. 2016, 18, 792–797. doi:10.1039/C5GC01855B
See for primary amine-catalysed transformations in DESs.
Return to citation in text: [1] -
Kondo, K.; Yamano, T.; Takemoto, K. Makromol. Chem. 1985, 186, 1781–1785. doi:10.1002/macp.1985.021860906
See for selected studies on proline immobilization, on polystyrene.
Return to citation in text: [1] -
Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C. F., III. J. Am. Chem. Soc. 2001, 123, 5260–5267. doi:10.1021/ja010037z
See for selected studies on proline immobilization, on silica.
Return to citation in text: [1] -
Benaglia, M.; Celentano, G.; Cozzi, F. Adv. Synth. Catal. 2001, 343, 171–173. doi:10.1002/1615-4169(20010226)343:2<171::AID-ADSC171>3.3.CO;2-V
See for PEG-supported proline.
Return to citation in text: [1] -
Calderón, F.; Fernández, R.; Sánchez, F.; Fernández-Mayoralas, A. Adv. Synth. Catal. 2005, 347, 1395–1403. doi:10.1002/adsc.200505058
See for mesoporous silica.
Return to citation in text: [1] -
Smith, E. L.; Abbott, A. P.; Ryder, K. S. Chem. Rev. 2014, 114, 11060–11082. doi:10.1021/cr300162p
Return to citation in text: [1] -
Liu, P.; Hao, J.-W.; Mo, L.-P.; Zhang, Z.-H. RSC Adv. 2015, 5, 48675–48704. doi:10.1039/C5RA05746A
Return to citation in text: [1] -
Ruß, C.; König, B. Green Chem. 2012, 14, 2969–2982. doi:10.1039/c2gc36005e
Return to citation in text: [1] -
Gu, Y.; Jérôme, F. Chem. Soc. Rev. 2013, 42, 9550–9570. doi:10.1039/c3cs60241a
Return to citation in text: [1] -
Francisco, M.; van den Bruinhorst, A.; Kroon, M. C. Angew. Chem., Int. Ed. 2013, 52, 3074–3085. doi:10.1002/anie.201207548
Return to citation in text: [1] -
Alonso, D. A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Eur. J. Org. Chem. 2016, 612–632. doi:10.1002/ejoc.201501197
Return to citation in text: [1] -
Mallardo, V.; Rizzi, R.; Sassone, F. C.; Mansueto, R.; Perna, F. M.; Salomone, A.; Capriati, V. Chem. Commun. 2014, 50, 8655–8658. doi:10.1039/C4CC03149K
Return to citation in text: [1] -
Sassone, F. C.; Perna, F. M.; Salomone, M.; Florio, S.; Capriati, V. Chem. Commun. 2015, 51, 9459–9462. doi:10.1039/C5CC02884A
Return to citation in text: [1] -
García-Álvarez, J.; Hevia, E.; Capriati, V. Eur. J. Org. Chem. 2015, 6779–6799. doi:10.1002/ejoc.201500757
Return to citation in text: [1] -
Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F. M.; Salomone, M.; Florio, S.; Capriati, V. Chem. Sci. 2016, 7, 1192–1199. doi:10.1039/C5SC03436A
Return to citation in text: [1] -
Mancuso, R.; Maner, A.; Cicco, L.; Perna, F. M.; Capriati, V.; Gabriele, B. Tetrahedron 2016, 72, 4239–4244. doi:10.1016/j.tet.2016.05.062
Return to citation in text: [1] -
Capua, M.; Perrone, S.; Perna, F. M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. Molecules 2016, 21, 924. doi:10.3390/molecules21070924
Return to citation in text: [1] -
García-Álvarez, J. Deep Eutectic Solvents and Their Applications as New Green and Biorenewable Reaction Media. In Use, Health, and Environment, 2nd ed.; Wypych, G., Ed.; Handbook of Solvents, Vol. 2; ChemTec Publishing: Toronto, 2014.
Return to citation in text: [1] -
Hammond, O. S.; Bowron, D. T.; Edler, K. J. Green Chem. 2016, 18, 2736–2744. doi:10.1039/C5GC02914G
See for the first 3D liquid-phase structure of a ChCl-based DES.
Return to citation in text: [1] -
Orlandi, M.; Ceotto, M.; Benaglia, M. Chem. Sci. 2016, 7, 5421–5427. doi:10.1039/C6SC01328G
Return to citation in text: [1] -
Martínez-Castañeda, A.; Rodríguez-Solla, H.; Concellón, C.; del Amo, V. J. Org. Chem. 2012, 77, 10375–10381. doi:10.1021/jo3020352
Return to citation in text: [1]
| 1. | Mahrwald, R. Modern Aldol Reactions; Wiley-VCH: Weinheim, Germany, 2004; Vol. 1 and 2. doi:10.1002/9783527619566 |
| 7. | Cho, E.; Kim, T. H. Tetrahedron Lett. 2014, 55, 6470–6473. doi:10.1016/j.tetlet.2014.10.009 |
| 8. | Karmakar, A.; Maji, T.; Wittmann, S.; Reiser, O. Chem. – Eur. J. 2011, 17, 11024–11029. doi:10.1002/chem.201101299 |
| 9. | Opalka, S. M.; Steinbacher, J. L.; Lambiris, B. A.; McQuade, D. T. J. Org. Chem. 2011, 76, 6503–6517. doi:10.1021/jo200838v |
| 10. | El-Hamdouni, N.; Companyó, X.; Rios, R.; Moyano, A. Chem. – Eur. J. 2010, 16, 1142–1148. doi:10.1002/chem.200902678 |
| 11. | Reis, O.; Eymur, S.; Reis, B.; Demir, A. S. Chem. Commun. 2009, 1088–1090. doi:10.1039/b817474a |
| 12. | Mandal, T.; Zhao, C.-G. Angew. Chem., Int. Ed. 2008, 47, 7714–7717. doi:10.1002/anie.200803236 |
| 13. | Clarke, M. L.; Fuentes, J. A. Angew. Chem., Int. Ed. 2007, 46, 930–933. doi:10.1002/anie.200602912 |
| 43. | Martínez-Castañeda, A.; Rodríguez-Solla, H.; Concellón, C.; del Amo, V. J. Org. Chem. 2012, 77, 10375–10381. doi:10.1021/jo3020352 |
| 4. | Liu, X.; Lin, L.; Feng, X. Chem. Commun. 2009, 6145–6158. doi:10.1039/b913411e |
| 5. | Orlandi, M.; Benaglia, M.; Raimondi, L.; Celentano, G. Eur. J. Org. Chem. 2013, 2346–2354. doi:10.1002/ejoc.201201643 |
| 6. | Guizzetti, S.; Benaglia, M.; Pignataro, L.; Puglisi, A. Tetrahedron: Asymmetry 2006, 17, 2754–2760. doi:10.1016/j.tetasy.2006.10.018 |
| 3. | List, B.; Lerner, R. A.; Barbas, C. F., III. J. Am. Chem. Soc. 2000, 122, 2395–2396. doi:10.1021/ja994280y |
| 41. |
Hammond, O. S.; Bowron, D. T.; Edler, K. J. Green Chem. 2016, 18, 2736–2744. doi:10.1039/C5GC02914G
See for the first 3D liquid-phase structure of a ChCl-based DES. |
| 2. | Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527604677 |
| 42. | Orlandi, M.; Ceotto, M.; Benaglia, M. Chem. Sci. 2016, 7, 5421–5427. doi:10.1039/C6SC01328G |
| 24. |
Kondo, K.; Yamano, T.; Takemoto, K. Makromol. Chem. 1985, 186, 1781–1785. doi:10.1002/macp.1985.021860906
See for selected studies on proline immobilization, on polystyrene. |
| 25. |
Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C. F., III. J. Am. Chem. Soc. 2001, 123, 5260–5267. doi:10.1021/ja010037z
See for selected studies on proline immobilization, on silica. |
| 26. |
Benaglia, M.; Celentano, G.; Cozzi, F. Adv. Synth. Catal. 2001, 343, 171–173. doi:10.1002/1615-4169(20010226)343:2<171::AID-ADSC171>3.3.CO;2-V
See for PEG-supported proline. |
| 27. |
Calderón, F.; Fernández, R.; Sánchez, F.; Fernández-Mayoralas, A. Adv. Synth. Catal. 2005, 347, 1395–1403. doi:10.1002/adsc.200505058
See for mesoporous silica. |
| 34. | Mallardo, V.; Rizzi, R.; Sassone, F. C.; Mansueto, R.; Perna, F. M.; Salomone, A.; Capriati, V. Chem. Commun. 2014, 50, 8655–8658. doi:10.1039/C4CC03149K |
| 35. | Sassone, F. C.; Perna, F. M.; Salomone, M.; Florio, S.; Capriati, V. Chem. Commun. 2015, 51, 9459–9462. doi:10.1039/C5CC02884A |
| 36. | García-Álvarez, J.; Hevia, E.; Capriati, V. Eur. J. Org. Chem. 2015, 6779–6799. doi:10.1002/ejoc.201500757 |
| 37. | Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F. M.; Salomone, M.; Florio, S.; Capriati, V. Chem. Sci. 2016, 7, 1192–1199. doi:10.1039/C5SC03436A |
| 38. | Mancuso, R.; Maner, A.; Cicco, L.; Perna, F. M.; Capriati, V.; Gabriele, B. Tetrahedron 2016, 72, 4239–4244. doi:10.1016/j.tet.2016.05.062 |
| 39. | Capua, M.; Perrone, S.; Perna, F. M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. Molecules 2016, 21, 924. doi:10.3390/molecules21070924 |
| 23. |
Massolo, E.; Palmieri, S.; Benaglia, M.; Capriati, V.; Perna, F. M. Green Chem. 2016, 18, 792–797. doi:10.1039/C5GC01855B
See for primary amine-catalysed transformations in DESs. |
| 40. | García-Álvarez, J. Deep Eutectic Solvents and Their Applications as New Green and Biorenewable Reaction Media. In Use, Health, and Environment, 2nd ed.; Wypych, G., Ed.; Handbook of Solvents, Vol. 2; ChemTec Publishing: Toronto, 2014. |
| 20. |
Ilgen, F.; König, B. Green Chem. 2009, 11, 848–854. doi:10.1039/B816551C
See for pioneer studies of L-proline in DESs as catalysts of Diels-Alder reactions. |
| 21. |
Müller, C. R.; Meiners, I.; Domínguez de María, P. RSC Adv. 2014, 4, 46097–46101. doi:10.1039/C4RA09307K
See for proline-promoted aldol reaction. |
| 22. | Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D. J. Green Chem. 2016, 18, 1724–1730. doi:10.1039/C5GC02526E |
| 14. |
Rodriguez, B.; Bruckmann, A.; Bolm, C. Chem. – Eur. J. 2007, 13, 4710–4722. doi:10.1002/chem.200700188
See for ball mill approach. |
| 15. |
Clegg, W.; Harrington, R. W.; North, M.; Pizzato, F.; Villuendas, P. Tetrahedron: Asymmetry 2010, 21, 1262–1271. doi:10.1016/j.tetasy.2010.03.051
See for the use of carbamates. |
| 16. |
Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C. F., III. J. Am. Chem. Soc. 2006, 128, 734–735. doi:10.1021/ja0573312
See for aldol reaction on water. |
| 17. | Hayashi, Y.; Sumiya, T.; Takahashi, J.; Gotoh, H.; Urushima, T.; Shoji, M. Angew. Chem., Int. Ed. 2006, 45, 958–961. doi:10.1002/anie.200502488 |
| 18. | Guizzetti, S.; Benaglia, M.; Raimondi, L.; Celentano, G. Org. Lett. 2007, 9, 1247–1250. doi:10.1021/ol070002p |
| 19. |
Mlynarski, J.; Bas, S. Chem. Soc. Rev. 2014, 43, 577–587. doi:10.1039/C3CS60202H
See for a recent review on organocatalyzed reactions in/on water. |
| 28. | Smith, E. L.; Abbott, A. P.; Ryder, K. S. Chem. Rev. 2014, 114, 11060–11082. doi:10.1021/cr300162p |
| 29. | Liu, P.; Hao, J.-W.; Mo, L.-P.; Zhang, Z.-H. RSC Adv. 2015, 5, 48675–48704. doi:10.1039/C5RA05746A |
| 30. | Ruß, C.; König, B. Green Chem. 2012, 14, 2969–2982. doi:10.1039/c2gc36005e |
| 31. | Gu, Y.; Jérôme, F. Chem. Soc. Rev. 2013, 42, 9550–9570. doi:10.1039/c3cs60241a |
| 32. | Francisco, M.; van den Bruinhorst, A.; Kroon, M. C. Angew. Chem., Int. Ed. 2013, 52, 3074–3085. doi:10.1002/anie.201207548 |
| 33. | Alonso, D. A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Eur. J. Org. Chem. 2016, 612–632. doi:10.1002/ejoc.201501197 |
© 2016 Brenna et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)