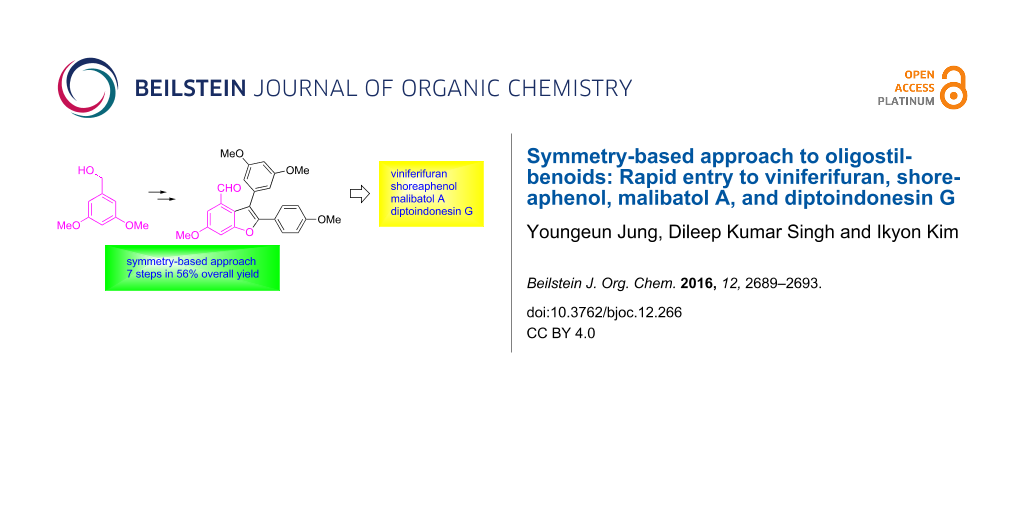
Abstract
The recognition of the local symmetric image within benzofuran-based natural oligostilbenoids guided us to design a modular synthetic approach to these molecules by utilizing a three-step sequence consisting of Sonogashira coupling, iodocyclization, and Suzuki coupling. During our synthesis, the relative reactivities of ester, aldehyde, and alkoxy groups on the same aryl ring toward the neighboring alkyne in the iodine-mediated cyclization reactions were explored. Starting from the symmetrical 3,5-dimethoxybenzyl alcohol, this route allowed rapid access to 2,3-diarylbenzofuran, a key intermediate to several oligostilbenoid natural products, in good overall yields.

Graphical Abstract
Introduction
Oligostilbenoids constitute a family of natural products with various biological functions (Figure 1). Monomeric stilbene units are interconnected in a number of ways to lead to complex structures [1-3]. Despite a long history of isolation and biological studies of these natural products, relatively little attention has been paid by the synthetic community to chemical synthesis of polyphenolic oligostilbenoids. Most synthetic works on these unique natural products have recently appeared in the literature [4-10].
![[1860-5397-12-266-1]](/bjoc/content/figures/1860-5397-12-266-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In connection with our research on benzofurans [11,12], our laboratory has been involved in the synthesis of these benzofuran-containing natural products for the last several years [13-15]. For example, we have reported a concise total synthesis of diptoindonesin G, a potent cytotoxic and immunosuppressant agent [16,17], by using a highly efficient domino cyclodehydration/intramolecular Friedel–Crafts acylation/regioselective demethylation sequence as a key transformation. Very recently, a dual functional role of diptoindonesin G in modulating α and β estrogen receptors (ER) has been discovered, thereby suggesting it as a promising drug lead for the treatment of breast cancer [18]. Our continuing interest in this area led us to design an alternative approach to oligostilbenoids. As shown in Scheme 1, our idea stemmed from recognition of the symmetry element [19] of the target molecules. We expected that the key intermediate (inset box of Scheme 1) could be constructed from the monoiodo compounds 1, 2, or 3 through a sequence involving Sonogashira coupling, iodocyclization [20-26], and Suzuki coupling. As the starting materials (1, 2, and 3) were readily available from the corresponding C2 symmetric precursors via monoiodination, we decided to evaluate this route. In particular, we wondered what functional group as a G moiety would be appropriate for the successful iodine-mediated cyclization. Ester, aldehyde, and alkoxy groups have been used as nucleophiles of iodocyclization for the syntheses of a number of heterocycles, respectively [27-30]. Although Larock’s work on relative reactivity of these functional groups toward alkyne during electrophilic cyclization has been reported [31], the study with the substrates having these nucleophiles on the same aromatic ring has not been disclosed, to the best of our knowledge. Here we wish to describe our results.
![[1860-5397-12-266-i1]](/bjoc/content/inline/1860-5397-12-266-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
By following the known procedures, monoiodination of the commercially available methyl 3,5-dimethoxybenzoate, 3,5-dimethoxybenzaldehyde, and 3,5-dimethoxybenzyl alcohol under the influence of either I2/silver trifluoroacetate or N-iodosuccinimide afforded 1 [32-34], 2 [35], and 7 [36,37], respectively (Scheme 2). The hydroxy group of 7 was protected as an acetate, providing 3 in 96% yield. Sonogashira coupling of the resulting iodides 1, 2, and 3 with alkynylanisole proceeded without any event to give the corresponding alkynes, 4, 5, and 6, setting the stage for iodocyclization.
![[1860-5397-12-266-i2]](/bjoc/content/inline/1860-5397-12-266-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
When 4 was exposed to I2 and NaHCO3 in CH2Cl2, two isolable products were obtained (Scheme 3). Surprisingly, 8 was isolated in 32% yield presumably as a consequence of HI-promoted cyclization even in the presence of excess base. The structure of 8 was confirmed by X-ray crystallographic analysis (Figure 2) [38]. The other major product 9, less polar than 8, resulted from 6-endo-dig iodocyclization. Obviously, the ester moiety in 4 was competitively involved in the iodine-mediated electrophilic cyclization. Only a trace amount of 10 was isolated upon subjection of 5 to the same reaction conditions. We suspected that the aldehyde in 5 also acted as a nucleophile to furnish unstable oxocarbenium species (inset box of Scheme 3) as a major product which decomposed eventually. On the other hand, 6 was successfully converted to the desired 3-iodobenzofuran 11 in good yield. These results led us conclude that either ester or aldehyde groups perform as better nucleophiles than an alkoxy group in the iodocyclization.
![[1860-5397-12-266-i3]](/bjoc/content/inline/1860-5397-12-266-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-266-2]](/bjoc/content/figures/1860-5397-12-266-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Having secured gram quantities of 3-iodobenzofuran 11 in hand, our next task was to elaborate the conversion of 11 to 2,3-diarylbenzofurans (Scheme 4). To this end, 11 was first transformed to aldehyde 13 via 12 through a two-step sequence consisting of deacetylation and Dess–Martin oxidation [39]. To our delight, the subsequent Suzuki cross-coupling of 13 with 3,5-dimethoxyphenylboronic acid under the reaction conditions analogous as described before [26] proceeded well and furnished 14 in 88% yield. This intermediate was used for our previous syntheses of permethylated analogues of viniferifuran, malibatiol A, and shoreaphenol [13]. Under similar reaction conditions, several other arylboronic acids reacted with 13 to give the corresponding products in good yields, demonstrating the general usefulness of this route for the synthesis of a range of structural analogues at a late stage.
![[1860-5397-12-266-i4]](/bjoc/content/inline/1860-5397-12-266-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The direct Friedel–Crafts type intramolecular cyclization of 14 induced by BCl3 was attempted but a complex mixture was observed. Thus, oxidation of the aldehyde in 14 to carboxylic acid was carried out. Pinnick oxidation [40-42] of 14 went smoothly to give 18, an intermediate previously employed for the synthesis of diptoindonesin G (Scheme 5) [43,44].
![[1860-5397-12-266-i5]](/bjoc/content/inline/1860-5397-12-266-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have established a highly scalable and flexible synthetic route to several benzofuran-containing oligostilbenoid natural products by relying on a symmetry-breaking strategy from 3,5-dimethoxybenzyl alcohol. The relative reactivity of ester, aldehyde, and methoxy moieties toward the neighboring alkyne in the course of iodocyclization was investigated, revealing that neither ester nor aldehyde was compatible under these conditions to reach the desired product. The versatile key intermediates for the syntheses of several oligostilbenoids such as viniferifuran, shoreaphenol, malibatol A, and diptoindonesin G were rapidly accessed in a highly efficient manner, allowing for large-scale preparations of the target natural products as well as unnatural analogues.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, and 1H and 13C NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 2.1 MB | Download |
| Supporting Information File 2: Chemical information file of compound 8. | ||
| Format: CIF | Size: 661.2 KB | Download |
References
-
Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Angew. Chem., Int. Ed. 2011, 50, 586. doi:10.1002/anie.201000044
Return to citation in text: [1] -
Keyler, M. H.; Matsuura, B. S.; Stephenson, C. R. J. Chem. Rev. 2015, 115, 8976. doi:10.1021/cr500689b
Return to citation in text: [1] -
Wang, X.-F.; Yao, C.-S. J. Asian Nat. Prod. Res. 2016, 18, 376. doi:10.1080/10286020.2015.1094464
Return to citation in text: [1] -
Li, W.; Li, H.; Li, Y.; Hou, Z. Angew. Chem., Int. Ed. 2006, 45, 7609. doi:10.1002/anie.200603097
Return to citation in text: [1] -
Snyder, S. A.; Breazzano, S. P.; Ross, A. G.; Lin, Y.; Zografos, A. L. J. Am. Chem. Soc. 2009, 131, 1753. doi:10.1021/ja806183r
Return to citation in text: [1] -
Nicolaou, K. C.; Wu, T. R.; Kang, Q.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2009, 48, 3440. doi:10.1002/anie.200900438
Return to citation in text: [1] -
Jeffrey, J. L.; Sarpong, R. Org. Lett. 2009, 11, 5450. doi:10.1021/ol902141z
Return to citation in text: [1] -
Snyder, S. A.; Gollner, A.; Chiriac, M. I. Nature 2011, 474, 461. doi:10.1038/nature10197
Return to citation in text: [1] -
Jepsen, T. H.; Thomas, S. B.; Lin, Y.; Stathakis, C. I.; de Miguel, I.; Snyder, S. A. Angew. Chem., Int. Ed. 2014, 53, 6747. doi:10.1002/anie.201402858
Return to citation in text: [1] -
Matsuura, B. S.; Keylor, M. H.; Li, B.; Lin, Y.; Allison, S.; Pratt, D. A.; Stephenson, C. R. J. Angew. Chem., Int. Ed. 2015, 54, 3754. doi:10.1002/anie.201409773
Return to citation in text: [1] -
Kim, I.; Lee, S.-H.; Lee, S. Tetrahedron Lett. 2008, 49, 6579. doi:10.1016/j.tetlet.2008.09.034
Return to citation in text: [1] -
Kim, I.; Kim, K.; Choi, J. J. Org. Chem. 2009, 74, 8492. doi:10.1021/jo901937u
Return to citation in text: [1] -
Kim, I.; Choi, J. Org. Biomol. Chem. 2009, 7, 2788. doi:10.1039/b901911a
Return to citation in text: [1] [2] -
Kim, K.; Kim, I. Org. Lett. 2010, 12, 5314. doi:10.1021/ol102322g
Return to citation in text: [1] -
Lee, J. H.; Kim, M.; Kim, I. J. Org. Chem. 2014, 79, 6153. doi:10.1021/jo500885w
Return to citation in text: [1] -
Juliawaty, L. D.; Sahidin, H.; akim, E. H.; Achmad, S. A.; Syah, Y. M.; Latip, J.; Said, I. M. Nat. Prod. Commun. 2009, 4, 947.
Return to citation in text: [1] -
Ge, H. M.; Yang, W. H.; Shen, Y.; Jiang, N.; Guo, Z. K.; Luo, Q.; Xu, Q.; Ma, J.; Tan, R. X. Chem. – Eur. J. 2010, 16, 6338. doi:10.1002/chem.201000230
Return to citation in text: [1] -
Zhao, J.; Wang, L.; James, T.; Jung, Y.; Kim, I.; Tan, R.; Hoffmann, F. M.; Xu, W. Chem. Biol. 2015, 22, 1608. doi:10.1016/j.chembiol.2015.10.011
Return to citation in text: [1] -
Ho, T.-L. Symmetry: A Basis for Synthesis Design; John Wiley & Sons, Inc.: New York, 1995.
Return to citation in text: [1] -
Kim, I.; Choi, J.; Won, H. K.; Lee, G. H. Tetrahedron Lett. 2007, 48, 6863. doi:10.1016/j.tetlet.2007.07.180
Return to citation in text: [1] -
Kim, I.; Won, H. K.; Choi, J.; Lee, G. H. Tetrahedron 2007, 63, 12954. doi:10.1016/j.tet.2007.10.037
Return to citation in text: [1] -
Kim, I.; Kim, S. G.; Kim, J. Y.; Lee, G. H. Tetrahedron Lett. 2007, 48, 8976. doi:10.1016/j.tetlet.2007.10.101
Return to citation in text: [1] -
Choi, J.; Lee, G. H.; Kim, I. Synlett 2008, 1243. doi:10.1055/s-2008-1072721
Return to citation in text: [1] -
Kim, K.; Kim, I. J. Comb. Chem. 2010, 12, 379. doi:10.1021/cc100015k
Return to citation in text: [1] -
Jung, Y.; Kim, I. Tetrahedron 2012, 68, 8198. doi:10.1016/j.tet.2012.07.068
Return to citation in text: [1] -
Jung, Y.; Kim, I. Asian J. Org. Chem. 2016, 5, 147. doi:10.1002/ajoc.201500423
Return to citation in text: [1] [2] -
Biagetti, M.; Bellina, F.; Carpita, A.; Stabile, P.; Rossi, R. Tetrahedron 2002, 58, 5023. doi:10.1016/S0040-4020(02)00469-6
Return to citation in text: [1] -
Yao, T.; Larock, R. C. J. Org. Chem. 2003, 68, 5936. doi:10.1021/jo034308v
Return to citation in text: [1] -
Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 10292. doi:10.1021/jo051299c
Return to citation in text: [1] -
Verma, A. K.; Aggarwal, T.; Rustagi, V.; Larock, R. C. Chem. Commun. 2010, 46, 4064. doi:10.1039/b927185f
Return to citation in text: [1] -
Mehta, S.; Waldo, J. P.; Larock, R. C. J. Org. Chem. 2009, 74, 1141. doi:10.1021/jo802196r
Return to citation in text: [1] -
Manzo, E.; Ciavatta, M. L. Tetrahedron 2012, 68, 4107. doi:10.1016/j.tet.2012.04.010
Return to citation in text: [1] -
Leermann, T.; Broutin, P.-E.; Leroux, F. R.; Colobert, F. Org. Biomol. Chem. 2012, 10, 4095. doi:10.1039/c2ob25373a
Return to citation in text: [1] -
Ehrlich, M.; Carell, T. Eur. J. Org. Chem. 2013, 77. doi:10.1002/ejoc.201201256
Return to citation in text: [1] -
Harrowven, D. C.; Nunn, M. I. T.; Fenwick, D. R. Tetrahedron Lett. 2002, 43, 7345. doi:10.1016/S0040-4039(02)01720-3
Return to citation in text: [1] -
Swindell, C. S.; Fan, W. J. Org. Chem. 1996, 61, 1109. doi:10.1021/jo9519367
Return to citation in text: [1] -
Murata, C.; Ogura, T.; Narita, S.; Kondo, A. P.; Iwasaki, N.; Saito, T.; Usuki, T. Bioorg. Med. Chem. Lett. 2016, 26, 1428. doi:10.1016/j.bmcl.2016.01.067
Return to citation in text: [1] -
CCDC 1503940 contains the supplementary crystallographic data for compound 8. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155. doi:10.1021/jo00170a070
Return to citation in text: [1] -
Lindgren, B. O.; Nilsson, T. Acta Chem. Scand. 1973, 27, 888. doi:10.3891/acta.chem.scand.27-0888
Return to citation in text: [1] -
Kraus, G. A.; Taschner, M. J. J. Org. Chem. 1980, 45, 1175. doi:10.1021/jo01294a058
Return to citation in text: [1] -
Bal, B. S.; Childers, W. E., Jr.; Pinnick, H. W. Tetrahedron 1981, 37, 2091. doi:10.1016/S0040-4020(01)97963-3
Return to citation in text: [1] -
Conversion of 18 to diptoindonesin G was reported via intramolecular Friedel–Crafts acylation/global demethylation in 72% overall yield. See ref. [14].
Return to citation in text: [1] -
Liu, J.-t.; Do, T. J.; Simmons, C. J.; Lynch, J. C.; Gu, W.; Ma, Z.-X.; Xu, W.; Tang, W. Org. Biomol. Chem. 2016, 14, 8927. doi:10.1039/C6OB01657J
See for another total synthesis of diptoindonesin G and its analogues which was published during the preparation of this article.
Return to citation in text: [1]
| 43. | Conversion of 18 to diptoindonesin G was reported via intramolecular Friedel–Crafts acylation/global demethylation in 72% overall yield. See ref. [14]. |
| 44. |
Liu, J.-t.; Do, T. J.; Simmons, C. J.; Lynch, J. C.; Gu, W.; Ma, Z.-X.; Xu, W.; Tang, W. Org. Biomol. Chem. 2016, 14, 8927. doi:10.1039/C6OB01657J
See for another total synthesis of diptoindonesin G and its analogues which was published during the preparation of this article. |
| 40. | Lindgren, B. O.; Nilsson, T. Acta Chem. Scand. 1973, 27, 888. doi:10.3891/acta.chem.scand.27-0888 |
| 41. | Kraus, G. A.; Taschner, M. J. J. Org. Chem. 1980, 45, 1175. doi:10.1021/jo01294a058 |
| 42. | Bal, B. S.; Childers, W. E., Jr.; Pinnick, H. W. Tetrahedron 1981, 37, 2091. doi:10.1016/S0040-4020(01)97963-3 |
| 1. | Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Angew. Chem., Int. Ed. 2011, 50, 586. doi:10.1002/anie.201000044 |
| 2. | Keyler, M. H.; Matsuura, B. S.; Stephenson, C. R. J. Chem. Rev. 2015, 115, 8976. doi:10.1021/cr500689b |
| 3. | Wang, X.-F.; Yao, C.-S. J. Asian Nat. Prod. Res. 2016, 18, 376. doi:10.1080/10286020.2015.1094464 |
| 16. | Juliawaty, L. D.; Sahidin, H.; akim, E. H.; Achmad, S. A.; Syah, Y. M.; Latip, J.; Said, I. M. Nat. Prod. Commun. 2009, 4, 947. |
| 17. | Ge, H. M.; Yang, W. H.; Shen, Y.; Jiang, N.; Guo, Z. K.; Luo, Q.; Xu, Q.; Ma, J.; Tan, R. X. Chem. – Eur. J. 2010, 16, 6338. doi:10.1002/chem.201000230 |
| 39. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155. doi:10.1021/jo00170a070 |
| 13. | Kim, I.; Choi, J. Org. Biomol. Chem. 2009, 7, 2788. doi:10.1039/b901911a |
| 14. | Kim, K.; Kim, I. Org. Lett. 2010, 12, 5314. doi:10.1021/ol102322g |
| 15. | Lee, J. H.; Kim, M.; Kim, I. J. Org. Chem. 2014, 79, 6153. doi:10.1021/jo500885w |
| 26. | Jung, Y.; Kim, I. Asian J. Org. Chem. 2016, 5, 147. doi:10.1002/ajoc.201500423 |
| 11. | Kim, I.; Lee, S.-H.; Lee, S. Tetrahedron Lett. 2008, 49, 6579. doi:10.1016/j.tetlet.2008.09.034 |
| 12. | Kim, I.; Kim, K.; Choi, J. J. Org. Chem. 2009, 74, 8492. doi:10.1021/jo901937u |
| 36. | Swindell, C. S.; Fan, W. J. Org. Chem. 1996, 61, 1109. doi:10.1021/jo9519367 |
| 37. | Murata, C.; Ogura, T.; Narita, S.; Kondo, A. P.; Iwasaki, N.; Saito, T.; Usuki, T. Bioorg. Med. Chem. Lett. 2016, 26, 1428. doi:10.1016/j.bmcl.2016.01.067 |
| 4. | Li, W.; Li, H.; Li, Y.; Hou, Z. Angew. Chem., Int. Ed. 2006, 45, 7609. doi:10.1002/anie.200603097 |
| 5. | Snyder, S. A.; Breazzano, S. P.; Ross, A. G.; Lin, Y.; Zografos, A. L. J. Am. Chem. Soc. 2009, 131, 1753. doi:10.1021/ja806183r |
| 6. | Nicolaou, K. C.; Wu, T. R.; Kang, Q.; Chen, D. Y.-K. Angew. Chem., Int. Ed. 2009, 48, 3440. doi:10.1002/anie.200900438 |
| 7. | Jeffrey, J. L.; Sarpong, R. Org. Lett. 2009, 11, 5450. doi:10.1021/ol902141z |
| 8. | Snyder, S. A.; Gollner, A.; Chiriac, M. I. Nature 2011, 474, 461. doi:10.1038/nature10197 |
| 9. | Jepsen, T. H.; Thomas, S. B.; Lin, Y.; Stathakis, C. I.; de Miguel, I.; Snyder, S. A. Angew. Chem., Int. Ed. 2014, 53, 6747. doi:10.1002/anie.201402858 |
| 10. | Matsuura, B. S.; Keylor, M. H.; Li, B.; Lin, Y.; Allison, S.; Pratt, D. A.; Stephenson, C. R. J. Angew. Chem., Int. Ed. 2015, 54, 3754. doi:10.1002/anie.201409773 |
| 38. | CCDC 1503940 contains the supplementary crystallographic data for compound 8. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 27. | Biagetti, M.; Bellina, F.; Carpita, A.; Stabile, P.; Rossi, R. Tetrahedron 2002, 58, 5023. doi:10.1016/S0040-4020(02)00469-6 |
| 28. | Yao, T.; Larock, R. C. J. Org. Chem. 2003, 68, 5936. doi:10.1021/jo034308v |
| 29. | Yue, D.; Yao, T.; Larock, R. C. J. Org. Chem. 2005, 70, 10292. doi:10.1021/jo051299c |
| 30. | Verma, A. K.; Aggarwal, T.; Rustagi, V.; Larock, R. C. Chem. Commun. 2010, 46, 4064. doi:10.1039/b927185f |
| 32. | Manzo, E.; Ciavatta, M. L. Tetrahedron 2012, 68, 4107. doi:10.1016/j.tet.2012.04.010 |
| 33. | Leermann, T.; Broutin, P.-E.; Leroux, F. R.; Colobert, F. Org. Biomol. Chem. 2012, 10, 4095. doi:10.1039/c2ob25373a |
| 34. | Ehrlich, M.; Carell, T. Eur. J. Org. Chem. 2013, 77. doi:10.1002/ejoc.201201256 |
| 20. | Kim, I.; Choi, J.; Won, H. K.; Lee, G. H. Tetrahedron Lett. 2007, 48, 6863. doi:10.1016/j.tetlet.2007.07.180 |
| 21. | Kim, I.; Won, H. K.; Choi, J.; Lee, G. H. Tetrahedron 2007, 63, 12954. doi:10.1016/j.tet.2007.10.037 |
| 22. | Kim, I.; Kim, S. G.; Kim, J. Y.; Lee, G. H. Tetrahedron Lett. 2007, 48, 8976. doi:10.1016/j.tetlet.2007.10.101 |
| 23. | Choi, J.; Lee, G. H.; Kim, I. Synlett 2008, 1243. doi:10.1055/s-2008-1072721 |
| 24. | Kim, K.; Kim, I. J. Comb. Chem. 2010, 12, 379. doi:10.1021/cc100015k |
| 25. | Jung, Y.; Kim, I. Tetrahedron 2012, 68, 8198. doi:10.1016/j.tet.2012.07.068 |
| 26. | Jung, Y.; Kim, I. Asian J. Org. Chem. 2016, 5, 147. doi:10.1002/ajoc.201500423 |
| 35. | Harrowven, D. C.; Nunn, M. I. T.; Fenwick, D. R. Tetrahedron Lett. 2002, 43, 7345. doi:10.1016/S0040-4039(02)01720-3 |
| 19. | Ho, T.-L. Symmetry: A Basis for Synthesis Design; John Wiley & Sons, Inc.: New York, 1995. |
| 18. | Zhao, J.; Wang, L.; James, T.; Jung, Y.; Kim, I.; Tan, R.; Hoffmann, F. M.; Xu, W. Chem. Biol. 2015, 22, 1608. doi:10.1016/j.chembiol.2015.10.011 |
| 31. | Mehta, S.; Waldo, J. P.; Larock, R. C. J. Org. Chem. 2009, 74, 1141. doi:10.1021/jo802196r |
© 2016 Jung et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)