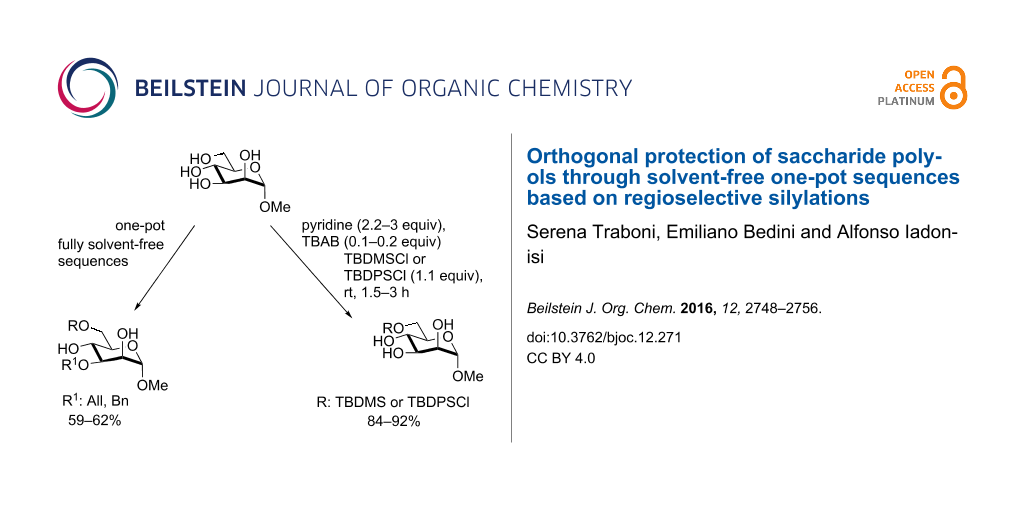
Abstract
tert-Butyldimethylsilyl (TBDMS) and tert-butyldiphenylsilyl (TBDPS) are alcohol protecting groups widely employed in organic synthesis in view of their compatibility with a wide range of conditions. Their regioselective installation on polyols generally requires lengthy reactions and the use of high boiling solvents. In the first part of this paper we demonstrate that regioselective silylation of sugar polyols can be conducted in short times with the requisite silyl chloride and a very limited excess of pyridine (2–3 equivalents). Under these conditions, that can be regarded as solvent-free conditions in view of the insolubility of the polyol substrates, the reactions are faster than in most examples reported in the literature, and can even be further accelerated with a catalytic amount of tetrabutylammonium bromide (TBAB). The strategy proved also useful for either the selective TBDMS protection of secondary alcohols or the fast per-O-trimethylsilylation of saccharide polyols. In the second part of the paper the scope of the silylation approach was significantly extended with the development of unprecedented “one-pot” and “solvent-free” sequences allowing the regioselective silylation/alkylation (or the reverse sequence) of saccharide polyols in short times. The developed methodologies represent a very useful and experimentally simple tool for the straightforward access to saccharide building-blocks useful in organic synthesis.

Graphical Abstract
Introduction
The application of an orthogonal set of protecting groups represents a typical issue in organic synthesis in the elaboration of highly functionalized molecules such as carbohydrates, and lengthy multistep procedures are often needed to this aim [1,2]. Silyl groups are widely applied in organic chemistry in orthogonal protection strategies owing to their stability to a broad range of conditions and feasible removal under conditions compatible with many other used alcohol protecting groups [1-3]. For this reason, silyl protecting groups are often serving as temporary protecting groups with polyol and saccharide substrates. The most robust and adopted silyl protecting groups are featuring the presence of hindered substituents at silicon such as in tert-butyldimethylsilyl (TBDMS) and tert-butyldiphenylsilyl (TBDPS) groups. Their bulkiness allows in many cases regioselective silyl protection of primary alcohols. Commonly, O-silylation is performed by exposing the alcohol to a suitably substituted silyl chloride in the presence of a base, a catalyst (often coinciding with the base) and an aprotic solvent [2-9]. The use of more expensive silyl triflates is also reported, especially when poorly reactive alcohols have to be protected [2,10-13].
A regioselective silylation of polar saccharide polyols is typically performed with the appropriate silyl chloride in the presence of a high boiling solvent such as DMF or pyridine, often in the presence of a nucleophilic catalyst (more frequently imidazole and DMAP) [14-21]. The protection generally takes several hours and the work-up is burdened by necessary removal of the high boiling solvent. A good regioselective control was also reported in an alternative silylation approach based on a dehydrogenative mechanism in which expensive trialkyl silanes were used as silylating agents [22]. Very recently, Vogel and co-workers reported an original strategy based on unusual silylating agents such as silyl methallylsulfinates; this approach proved very high-yielding under neutral conditions, but the preliminary synthesis of the requisite reagent relied on a non-trivial two-step procedure starting from the corresponding silyl chloride [23].
The regioselective silylation of secondary saccharide alcohols can also be achieved either taking advantage of the inherent difference of reactivity among the hydroxy functions [24-32] or exploiting the activation effect of boron complexes [33].
Over the last years, we have addressed our interest towards the development of solvent-free protocols aimed at regioselective protection of highly functionalized saccharide substrates. This effort led to a simple protocol for the selective benzylation of primary saccharide alcohols [34], the first catalytic tin-mediated procedure for regioselective benzylation/allylation of hydroxy groups incorporated into vicinal diols [35], and three alternative acetalation protocols [36]. Besides the avoided use of solvents, these approaches appear advantageous owing to their experimental ease, the reactions being performed under air by simply mixing the requisite reagents.
In this paper, we wish to report the extension of the scope of the solvent-free strategies to the silylation reaction, and the feasible incorporation of this step into unprecedented one-pot, fully solvent-free sequences yielding orthogonally protected saccharide building-blocks under simple experimental conditions and in short times.
Results and Discussion
In preliminary experiments, methyl mannopyranoside (1) was selected as the model substrate and exposed to TBDMSCl in the presence of a slight or moderate excess of several bases (Table 1).
Table 1: Regioselective silylation of 1 under solvent free conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-271-i2.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base (equiv) | Additive (equiv) | Temperature, time | Isolated yield |
|---|---|---|---|---|
| 1 | DIPEA (5) | – | 50 °C, 5 h | <15 |
| 2 | DIPEA (5) | TBAB (0.3) | 50 °C, 5.5 h | 49 |
| 3 | DIPEA (5) | TBAB (0.3), Bu2SnO (0.1) | 50 °C, 5.5 h | 52 |
| 4 | DIPEA (5) | TBAI (0.3), Bu2SnO (0.1) | 50 °C, 5.5 h | 48 |
| 5 | TEA (5) | TBAB (0.3) | 50 °C, 5.5 h | 53 |
| 6 | pyridine (5) | TBAB (0.3) | 50 °C, 1 h | 89 |
| 7 | pyridine (2.5) | TBAB (0.3) | rt, 1 h | 80 |
| 8 | pyridine (2.2) | TBAB (0.1) | rt, 1.5 h | 84 |
| 9 | pyridine (2.2) | – | rt, 1.5 h | 70 |
aGeneral conditions: substrate, base, additive, TBDMSCl (1.2 equiv for entries 1–6, 1.1 equiv for entries 7–9).
In all cases 6-O-silylated derivative 2 was obtained as the main product, but yields and rates were strongly dependent on the adopted base: pyridine (Table 1, entries 6–9) gave much better results than tertiary amines (Table 1, entries 1–5) that, on the other hand, had previously performed better than pyridine in the tin-catalyzed solvent-free regioselective benzylation or allylation of sugars [35]. The silylation rate was not appreciably influenced by tin catalysis (compare entries 2 and 3 in Table 1), although a previous report described the stoichiometric use of stannylene acetals in the regioselective silylation of saccharide primary alcohols [37]. At this stage, it should be noted that pyridine is frequently employed as the solvent for silylations, but the conditions herein described can be referred to as “solvent-free” because of the very limited amount of pyridine used, that is by far not sufficient for dissolution of the polar polyol substrates. Interestingly, reactions with pyridine were found to give slightly improved yields (within comparable times) on using a catalytic amount of TBAB (compare in Table 1 entries 6–8 with entry 9), which may be accounted for by a possible role of the bromide ion in the activation of the silylating agent as also suggested by literature [38]. Taking into account several parameters such as the used amount of pyridine and TBAB, the reaction yield and its length, we elected conditions of Table 1, entry 8 as the optimized conditions. Notably, the conversion of 1 to 2 under these conditions took a much shorter time than previously reported by using the same silylating agent [14,15,18]. With optimized conditions in hand, the TBDMS regioselective installation was tested on a range of saccharide building-blocks, and good yields were achieved in short times with several polyols (Table 2, entries 1–3, 5 and 8).
Table 2: Regioselective silylation of saccharide primary alcohols under solvent-free conditionsa.
| Entry | Substrate | Pyridine/silylating agent/TBAB (equivalents)b | Time (h) | Product, isolated yield |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-271-i3.svg?max-width=637&scale=1.0)
1 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 1.5 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-271-i4.svg?max-width=637&scale=1.0)
2, 84% |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-271-i5.svg?max-width=637&scale=1.0)
3 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 1.5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-271-i6.svg?max-width=637&scale=1.0)
10, 83% |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-271-i7.svg?max-width=637&scale=1.0)
4 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 1.5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-271-i8.svg?max-width=637&scale=1.0)
11, 86% |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-271-i9.svg?max-width=637&scale=1.0)
5 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 1.5 | low conversion |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-271-i10.svg?max-width=637&scale=1.0)
6 |
pyridine/TBDMSCl/TBAB (3.0:2.0:0.1) | 2.5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-271-i11.svg?max-width=637&scale=1.0)
12, 82% |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-271-i12.svg?max-width=637&scale=1.0)
7 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 1.5 | complex mixture |
| 7 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-271-i13.svg?max-width=637&scale=1.0)
8 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 1.5 | complex mixture |
| 8c |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-271-i14.svg?max-width=637&scale=1.0)
9 |
pyridine/TBDMSCl/TBAB (2.2:1.1:0.1) | 2 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-271-i15.svg?max-width=637&scale=1.0)
13, 73% (α/β 1:2) |
| 9 | 1 | pyridine/TBDPSCl/TBAB (2.2:1.1:0.2) | 3 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-271-i16.svg?max-width=637&scale=1.0)
14, 68% |
| 10 | 1 | pyridine/TBDPSCl/TBAB (3.0:1.1:0.2) | 3 | 14, 92% |
| 11 | 3 | pyridine/TBDPSCl/TBAB (3.0:1.1:0.2) | 3 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-271-i17.svg?max-width=637&scale=1.0)
15, 84% |
| 12 | 4 | pyridine/TBDPSCl/TBAB (3.0:1.1:0.2) | 2 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-271-i18.svg?max-width=637&scale=1.0)
16, 98% |
| 13 | 7 | pyridine/TBDPSCl/TBAB (3.0:1.1:0.2) | 5 | very sluggish |
aGeneral conditions: polyol substrate, pyridine, silylating agent (TBDMSCl or TBDPSCl), and TBAB at rt. See pertinent entries for stoichiometric ratios. bWith respect to the polyol substrate. cThe crude silylation mixture was acetylated in situ (direct addition of pyridine and acetic anhydride), prior to purification of the product.
Interestingly, galacto-configured substrates exhibited a peculiar reactivity with the reaction outcome depending on their anomeric configuration. In accordance with yields obtained by Lee and Taylor with similar substrates under standard conditions [14], only allyl β-galactoside (6) was silylated in a good yield (Table 2, entry 5), whereas the corresponding α-anomer was almost quantitatively recovered (Table 2, entry 4). As will be shown below, partial protection of allyl α-galactoside in itinere can render this substrate much more reactive towards these silylation conditions. High-yielding silylation of allyl β-galactoside (6) required the employment of a higher stoichiometric excess of both pyridine and TBDMSCl (Table 2, entry 5), exhibiting an extent of conversion apparently ruled by an equilibrium-like control.
Among the screened polyols in Table 2, glycals 7 and 8 were the only substrates to exhibit a lower regioselectivity, the silylation rate being comparable at the primary (O-6) and the allylic (O-3) position, to provide a mixture of products (Table 2, entries 6 and 7). A similar competitive reactivity was very recently described also with the above mentioned method based on silyl methallylsulfinates [23]. On the other hand, the method herein proposed was found compatible with a reducing sugar such as D-mannose, which was converted in a good yield into the corresponding 6-O-silylated product 13, isolated after in situ peracetylation (Table 2, entry 8).
The scope of the TBAB-catalyzed silyl protection under solvent-free conditions was next examined for the regioselective attachment of TBDPS (Table 2, entries 9–13), a commonly used silyl protecting group bulkier than TBDMS and more resistant to acidic conditions. Optimization on mannoside 1 indicated that in this case a moderate increase of the pyridine excess (3.0 instead of 2.2 equivalents) is beneficial for the achievement of a higher yield while maintaining the use of a minimal excess of the silylating agent (Table 2, entries 9 and 10). As with TBDMS protection of 6 (Table 2, entry 5), in this case a sort of steady state was observed when adopting less than three equivalents of pyridine, with coexistence of the reagent and the product.
The TBDPS selective protection of polyols 3 and 4 proceeded in high yields (Table 2, entries 11 and 12), and expectedly the reactions took slightly longer times than for TBDMS protection. The only disappointing result observed in attempted TPDPS protections was the poor yield recorded with glucal 7 (Table 2, entry 13), a surprising outcome which is somehow consistent with the very slow rate observed for the same reaction under standard conditions [39,40]. As already observed for the synthesis of 2, the herein described conditions for regioselective TBDMS and TBDPS protections entail shorter reaction times than most of the reported protocols in the literature on monosaccharide polyols [14-18,20,41-53]; comparable silylation rates were indeed be found in a few examples, often involving relatively less polar thioaryl glycosides as the substrates and DMF as the solvent [19,54-59]. Having established the scope of TBAB-catalyzed mono silylations with a minimal excess of pyridine, some effort was devoted to ascertain the feasible exploitation of a similar strategy to either regioselective di-O-silylations or the protection of secondary alcohols in absence of primary ones (Table 3).
Table 3: Solvent-free regioselective silylations committing secondary alcoholsa.
| Entry | Substrate | Time (h) | Product, isolated yield |
|---|---|---|---|
| 1 | 1 | 4 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-271-i19.svg?max-width=637&scale=1.0)
17, 72% |
| 2 | 3 | 4.5 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-271-i20.svg?max-width=637&scale=1.0)
18, 60% |
| 3 | 8 | 5 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-271-i21.svg?max-width=637&scale=1.0)
19, 56% |
| 4 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-271-i22.svg?max-width=637&scale=1.0)
20 |
1 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-271-i23.svg?max-width=637&scale=1.0)
21, 56% |
| 5 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-271-i24.svg?max-width=637&scale=1.0)
22 (α:β ca. 2.5) |
6 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-271-i25.svg?max-width=637&scale=1.0)
23 (α:β ca. 4), 75% |
| 6 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-271-i26.svg?max-width=637&scale=1.0)
24 (α:β ca. 2.5) |
6 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-12-271-i27.svg?max-width=637&scale=1.0)
25 (α:β ca. 4), 50% (95%)b |
aGeneral conditions (entries 1–3): pyridine (5 equiv), TBDMSCl (2.5 equiv), TBAB (0.3 equiv), 50 °C. For entry 4: pyridine (6 equiv), TBDMSCl (3.5 equiv), TBAB (0.3 equiv), rt. For entries 5 and 6: pyridine (3 equiv), TBDMSCl (1.5 equiv), TBAB (0.15 equiv), 50 °C. bIn parenthesis is indicated the conversion yield.
As a matter of fact, upon doubling the stoichiometric amount of TBAB, pyridine and TBDMSCl, the regioselective synthesis of di-O-TBDMS derivatives was achieved at 50 °C in satisfying yields from glycosides 1 and 3 (Table 3, entries 1 and 2), and glycal 8 (entry 3).
Double silylation at primary positions of the disaccharide lactoside 20 [60] also proved feasible at room temperature within short times (Table 3, entry 4). Model substrates devoid of a primary alcohol were also examined; thioethyl rhamnoside 22 (as an anomeric mixture) was 3-O-silylated in a good yield, and the anomeric composition of the products revealed the higher reactivity of the α-anomer (Table 3, entry 5). The same regioselectivity was also found starting from the corresponding 4-O-benzoylated precursor 24 [61] (Table 3, entry 6) and a satisfying yield and an excellent conversion were observed in spite of the increased hindrance and deactivation of the O-3 hydroxy group due to the adjacent electron-withdrawing benzoyl group. As already described above in Table 1 for mono-silylations, tin catalysis did not affect the double silylation processes as evidenced by the reaction of Table 3, entry 1 that in the presence of 0.1 equiv of Bu2SnO gave 17 in the same yield within the same time. Not unexpectedly, secondary alcohols exhibited a recalcitrant reactivity towards TBDPSCl under these solvent-free conditions, and the di-O-silylation process resulted in limited synthetic usefulness (data not shown).
The scope of the solvent-free conditions was further examined in the synthesis of per-O-trimethylsilylated derivatives (Scheme 1), widely used precursors in one-pot strategies for orthogonal protection of carbohydrates [62-67].
![[1860-5397-12-271-i1]](/bjoc/content/inline/1860-5397-12-271-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Multiple O-trimethylsilylations of saccharide compounds.
Scheme 1: Multiple O-trimethylsilylations of saccharide compounds.
Application of the TBAB-catalyzed protocol on glucoside 3 gave product 26 in high yield within a few minutes (Scheme 1, reaction 1). The method proved also applicable to glucosamine hydrochloride, although in this case a higher excess of pyridine was needed for the conversion to occur (Scheme 1, reaction 2). Consistent with previous literature reports [68-72], silylation left unaltered the amino functionality which could be protected in situ with a Troc group without isolation of 27 [69], as shown in the one-pot, two-step sequence in Scheme 1, reaction 3.
Owing to the importance of the one-pot functional diversification of carbohydrates in modern organic synthesis [62-67,73-76], the scope of the solvent-free silylation approaches herein introduced was next extended to the development of fully solvent-free one-pot sequences leading to the sequential alkylation/silylation of saccharide polyols with high regiocontrol. For this purpose, we tried to combine the previously reported tin-catalyzed procedure for benzylation/allylation of saccharide secondary alcohols [35] with the present protocol for silylation of primary alcohols. Some experiments were carried out on methyl mannoside (1) in order to establish which order of steps (alkylation/silylation or the reverse sequence) might be higher yielding. Initial experiments indicated that the silylation step was not apparently effective when performed after the tin-catalyzed 3-O-benzylation of mannoside (1, Table 4, entry 1). A competitive reaction of residual benzyl bromide from the first step with pyridine may account for this result. Indeed, on repeating the experiment suitably increasing the amount of pyridine in the second step (from 2.2 to 5 equivalents), the desired product 29 was obtained in a satisfying 61% yield within a few hours (Table 4, entry 2). It should be outlined that this result is especially relevant taking into account that the solvent-free 3-O-benzylation alone (the first step of the sequence) occurs in a comparable yield under tin catalysis [35]. The reverse protection sequence (6-O-silylation/3-O-benzylation) was found to be less effective in terms of yields, apparently because of the partial loss of the TBDMS group in the second step where relatively forced thermal conditions are needed (Table 4, entry 3). This latter one-pot sequence was instead much more rewarding when the more robust TBDPS group was installed first (Table 4, entries 5 and 6). Interestingly, this latter group was satisfyingly stable under the especially forced conditions (a reaction temperature of 90 °C) required to carry out the tin-mediated 2-O-benzylation of a gluco-substrate (Table 4, entry 6). In addition, the TBDPS installation also proceeded in satisfying yields after a preliminary allylation (Table 4, entry 4) or benzylation step (Table 4, entry 8).
Table 4: One-pot, regioselective protection of sugar polyols with silyl and alkyl groupsa.
| Entry | Substrate | First step conditions (equiv) | Second step conditions (equiv) | Product, isolated yield |
|---|---|---|---|---|
| 1 | 1 | DIPEA (2.5), Bu2SnO (0.1), TBAB (0.3), BnBr (4), 70 °C, 3.5 h | pyridine (2.2), TBDMSCl (2.0), rt, 3 h | low silylation yields |
| 2 | 1 | DIPEA (2.5), Bu2SnO (0.1), TBAB (0.3), BnBr (4), 70 °C, 3.5 h | pyridine (5.0), TBDMSCl (2.0), rt, 3 h |
![[Graphic 27]](/bjoc/content/inline/1860-5397-12-271-i28.svg?max-width=637&scale=1.0)
29, 61% |
| 3 | 1 | pyridine (2.2), TBDMSCl (1.1), TBAB (0.3), rt, 1.5 h | DIPEA (3.5), Bu2SnO (0.1), BnBr (5.5), 70 °C, 4.5 h | 29, 39% |
| 4 | 1 | DIPEA (4), Bu2SnO (0.1), TBAB (0.3), AllBr (8), 90 °C, 3.5 h | pyridine (5.0), TBDPSCl (1.5), rt, 3.5 h |
![[Graphic 28]](/bjoc/content/inline/1860-5397-12-271-i29.svg?max-width=637&scale=1.0)
30, 59% |
| 5 | 1 | pyridine (3.0), TBDPSCl (1.1), TBAB (0.3), rt, 3 h | DIPEA (2.5), Bu2SnO (0.1), BnBr (6), 80 °C, 4 h |
![[Graphic 29]](/bjoc/content/inline/1860-5397-12-271-i30.svg?max-width=637&scale=1.0)
31, 62% |
| 6 | 3 | pyridine (3.0), TBDPSCl (1.1), TBAB (0.3), rt, 3 h | DIPEA (2.5), Bu2SnO (0.2), BnBr (7), 90 °C, 4 h |
![[Graphic 30]](/bjoc/content/inline/1860-5397-12-271-i31.svg?max-width=637&scale=1.0)
32, 42% |
| 7 | 8 | DIPEA (2.5), Bu2SnO (0.1), BnBr (2), TBAB (0.3) 70 °C, 2.5 h | pyridine (3.5), TBDMSCl (2.0), TBAB (0.3), rt, 1 h |
![[Graphic 31]](/bjoc/content/inline/1860-5397-12-271-i32.svg?max-width=637&scale=1.0)
33, 69% |
| 8 | 8 | DIPEA (2.5), Bu2SnO (0.1), BnBr (2), TBAB (0.3), 70 °C, 2.5 h | pyridine (3.5), TBDPSCl (1.5), rt, 2.5 h |
![[Graphic 32]](/bjoc/content/inline/1860-5397-12-271-i33.svg?max-width=637&scale=1.0)
34, 75% |
| 9 | 5 | DIPEA (2.5), Bu2SnO (0.1), BnBr (4), TBAB (0.3), 70 °C, 2 h | pyridine (6), TBDMSCl (2), rt, 1.5 h |
![[Graphic 33]](/bjoc/content/inline/1860-5397-12-271-i34.svg?max-width=637&scale=1.0)
35, 57% |
aGeneral conditions: upon completion of the first step (see times in pertinent entries), the temperature was modified according to conditions of the second step, and the requisite reagents added.
In the case of glycal 8, the application of a benzylation/silylation sequence resulted in very good yields with both silylating agents (Table 4, entries 7 and 8), although the 6-O-silylation alone had been low yielding in the initial set of experiments (Table 2, entries 6, 7 and 13). Comparison of these data seems to indicate that the silylation step is much more effective on less polar, partially protected polyols. As a further evidence of this trend, the 3-O-benzylation/6-O-silylation sequence was succesfully applied to access α-galactoside 35 (Table 4, entry 9), although, as shown earlier in Table 2, entry 4, direct silylation of the same precursor was quite unfruitful.
Conclusion
In the first part of this paper we have introduced a very simple approach to carry out the selective TBDMS or TBDPS protection of carbohydrate polyols taking advantage of reactions performed in the presence of a very limited amount of pyridine (3 equivalents or less for the hydroxy group to be protected). The method can indeed be regarded as a solvent-free approach because of the limited stoichiometric amount of the base that is not sufficient to dissolve the highly polar saccharide substrates. Under these conditions, a catalytic role played by TBAB was also evidenced. Besides the limited amount of the used base, the proposed method is endowed with further practical advantages such as the experimental ease (all reaction herein reported were conducted in air by simply mixing the reagents), and high reaction rates, often comparing favourably with literature examples, in spite of the poor solubility of the starting substrates in the reaction medium. The scope of the silylation approach was also extended to secondary alcohols and to the per-O-trimethylsilylation of saccharides.
In the second part of the paper, we have explored the feasible combination of the silylation methodology here introduced with a previously developed tin-catalyzed methodology for regioselective alkyl protection of carbohydrates. The merging of both methods led to unprecedented one-pot and fully solvent-free synthetic sequences, providing a straightforward and experimentally simple access to saccharide building-blocks orthogonally protected with a silyl at the primary position and a benzyl (or an allyl) group at well defined and predicatable secondary positions.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 7.2 MB | Download |
References
-
Ágoston, K.; Streicher, H.; Fügedi, P. Tetrahedron: Asymmetry 2016, 27, 707–728. doi:10.1016/j.tetasy.2016.06.010
See for a very recent review.
Return to citation in text: [1] [2] -
Wuts, P. G. M. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. Greene’s Protective Groups in Organic Synthesis; Wiley: 2014; pp 201–270.
Return to citation in text: [1] [2] [3] [4] -
Corey, E. J.; Venkateswarlu, A. J. Am. Chem. Soc. 1972, 94, 6190–6191. doi:10.1021/ja00772a043
Return to citation in text: [1] [2] -
Bartoszewicz, A.; Kalek, M.; Stawinski, J. Tetrahedron 2008, 64, 8843–8850. doi:10.1016/j.tet.2008.06.070
See for use of iodine as the catalyst (also ref [5]).
Return to citation in text: [1] -
Bartoszewicz, A.; Kalek, M.; Nilsson, J.; Hiresova, R.; Stawinski, J. Synlett 2008, 37–40. doi:10.1055/s-2007-992379
Return to citation in text: [1] [2] -
Yoshida, K.; Fujino, Y.; Itatsu, Y.; Inoue, H.; Kanoko, Y.; Takao, K.-i. Tetrahedron Lett. 2016, 57, 627–631. doi:10.1016/j.tetlet.2015.12.114
See for use of an N-oxide catalyst.
Return to citation in text: [1] -
Lombardo, L. Tetrahedron Lett. 1984, 25, 227–228. doi:10.1016/S0040-4039(00)99846-0
See for use of DIPEA as a catalyst.
Return to citation in text: [1] -
Aizpurua, J. M.; Palomo, C. Tetrahedron Lett. 1985, 26, 475–476. doi:10.1016/S0040-4039(00)61915-9
See for use of DBU as the base.
Return to citation in text: [1] -
Kim, S.; Chang, H. Synth. Commun. 1984, 14, 899–904. doi:10.1080/00397918408063758
See for use of tetramethylguanidine as the catalyst.
Return to citation in text: [1] -
Corey, E. J.; Cho, H.; Rücker, C.; Hua, D. H. Tetrahedron Lett. 1981, 22, 3455–3458. doi:10.1016/S0040-4039(01)81930-4
Return to citation in text: [1] -
Mander, L. N.; Sehti, S. P. Tetrahedron Lett. 1984, 25, 5953–5956. doi:10.1016/S0040-4039(01)81731-7
Return to citation in text: [1] -
Boschelli, D.; Takemasa, T.; Nishitani, Y.; Masamune, S. Tetrahedron Lett. 1985, 26, 5239–5242. doi:10.1016/S0040-4039(00)95004-4
Return to citation in text: [1] -
Gassman, P. G.; Haberman, L. M. J. Org. Chem. 1986, 51, 5010–5013. doi:10.1021/jo00375a050
Return to citation in text: [1] -
Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. doi:10.1021/ja110332r
Return to citation in text: [1] [2] [3] [4] -
El-Badri, M. H.; Willenbring, D.; Tantillo, D. J.; Gervay-Hague, J. J. Org. Chem. 2007, 72, 4663–4672. doi:10.1021/jo070229y
Return to citation in text: [1] [2] [3] -
Chambers, D. J.; Evans, G. R.; Fairbanks, A. J. Tetrahedron 2005, 61, 7184–7192. doi:10.1016/j.tet.2005.05.032
Return to citation in text: [1] [2] -
Mikkelsen, L. M.; Krintel, S. L.; Jiménez-Barbero, J.; Skrydstrup, T. J. Org. Chem. 2002, 67, 6297–6308. doi:10.1021/jo020339z
Return to citation in text: [1] [2] -
Belaissaoui, A.; Saez, I. M.; Cowling, S. J.; Zeng, X.; Goodby, J. W. Chem. – Eur. J. 2012, 18, 2366–2373. doi:10.1002/chem.201102193
Return to citation in text: [1] [2] [3] -
Lam, S. N.; Gervay-Hague, J. Carbohydr. Res. 2002, 337, 1953–1965. doi:10.1016/S0008-6215(02)00227-6
Return to citation in text: [1] [2] -
Suriano, F.; Coulembier, O.; Dubois, P. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3271–3280. doi:10.1002/pola.24109
Return to citation in text: [1] [2] -
Nakagawa, Y.; Doi, T.; Taketani, T.; Takegoshi, K.; Igarashi, Y.; Ito, Y. Chem. – Eur. J. 2013, 19, 10516–10525. doi:10.1002/chem.201301368
Return to citation in text: [1] -
Chung, M.-K.; Orlova, G.; Goddard, J. D.; Schlaf, M.; Harris, R.; Beveridge, T. J.; White, G.; Hallett, F. R. J. Am. Chem. Soc. 2002, 124, 10508–10518. doi:10.1021/ja026723v
Return to citation in text: [1] -
Marković, D.; Tchawou, W. A.; Novosjolova, I.; Laclef, S.; Stepanovs, D.; Turksm, M.; Vogel, P. Chem. – Eur. J. 2016, 22, 4196–4205. doi:10.1002/chem.201504380
Return to citation in text: [1] [2] -
Halmos, T.; Montserret, R.; Filippi, J.; Antonakis, K. Carbohydr. Res. 1987, 170, 57–69. doi:10.1016/0008-6215(87)85005-X
Return to citation in text: [1] -
Arias-Pérez, M. S.; Santos, M. J. Tetrahedron 1996, 52, 10785–10798. doi:10.1016/0040-4020(96)00600-X
Return to citation in text: [1] -
Ikeda, Y.; Furukawa, K.; Yamada, H. Carbohydr. Res. 2002, 337, 1499–1501. doi:10.1016/S0008-6215(02)00168-4
Return to citation in text: [1] -
Chung, M.-K.; Schlaf, M. J. Am. Chem. Soc. 2005, 127, 18085–18092. doi:10.1021/ja056283i
Return to citation in text: [1] -
Teumelsan, N.; Huang, X. J. Org. Chem. 2007, 72, 8976–8979. doi:10.1021/jo7013824
Return to citation in text: [1] -
Pastore, A.; Adinolfi, M.; Iadonisi, A.; Valerio, S. Carbohydr. Res. 2010, 345, 1316–1323. doi:10.1016/j.carres.2010.02.025
Return to citation in text: [1] -
Heuckendorff, M.; Pedersen, C. M.; Bols, M. J. Org. Chem. 2013, 78, 7234–7248. doi:10.1021/jo4012464
Return to citation in text: [1] -
Zhang, Y.; Chen, C.; Jin, L.; Tan, H.; Wang, F.; Cao, H. Carbohydr. Res. 2015, 401, 109–114. doi:10.1016/j.carres.2014.09.010
Return to citation in text: [1] -
Garro, H. A.; Garcia, C.; Martin, V. S.; Tonn, C. E.; Pungitore, C. R. Bioorg. Med. Chem. Lett. 2015, 25, 914–918. doi:10.1016/j.bmcl.2014.12.052
Return to citation in text: [1] -
Lee, D.; Taylor, M. S. Org. Biomol. Chem. 2013, 11, 5409–5412. doi:10.1039/c3ob40981c
Return to citation in text: [1] -
Giordano, M.; Iadonisi, A. Tetrahedron Lett. 2013, 54, 1550–1552. doi:10.1016/j.tetlet.2013.01.023
Return to citation in text: [1] -
Giordano, M.; Iadonisi, A. J. Org. Chem. 2014, 79, 213–222. doi:10.1021/jo402399n
Return to citation in text: [1] [2] [3] [4] -
Traboni, S.; Bedini, E.; Giordano, M.; Iadonisi, A. Adv. Synth. Catal. 2015, 357, 3562–3572. doi:10.1002/adsc.201500745
Return to citation in text: [1] -
Bredenkamp, M. W. S. Afr. J. Chem. 1995, 48, 154–156.
Return to citation in text: [1] -
Amantini, D.; Fringuelli, F.; Pizzo, F.; Vaccaro, L. J. Org. Chem. 2001, 66, 6734–6737. doi:10.1021/jo015814s
Here TBAB was reported to catalyze the trimethylsilylation of alcohols in the presence of trimethylsilyl azide.
Return to citation in text: [1] -
Saidhareddy, P.; Ajay, S.; Shaw, A.-K. RSC Adv. 2014, 4, 4253–4259. doi:10.1039/C3RA45530K
Return to citation in text: [1] -
Fei, X.; Holmes, T.; Diddle, J.; Hintz, L.; Delaney, D.; Stock, A.; Renner, D.; McDevitt, M.; Berkowitz, D. B.; Soukup, J. K. ACS Chem. Biol. 2014, 9, 2875–2882. doi:10.1021/cb500458f
Return to citation in text: [1] -
Dohi, H.; Nishida, Y.; Takeda, T.; Kobayashi, K. Carbohydr. Res. 2002, 337, 983–989. doi:10.1016/S0008-6215(02)00093-9
Return to citation in text: [1] -
Despras, G.; Robert, R.; Sendid, B.; Machez, E.; Poulain, D.; Mallet, J.-M. Bioorg. Med. Chem. 2012, 20, 1817–1831. doi:10.1016/j.bmc.2011.12.048
Return to citation in text: [1] -
Peng, P.; Xiong, D.-C.; Ye, X.-S. Carbohydr. Res. 2014, 384, 1–8. doi:10.1016/j.carres.2013.11.009
Return to citation in text: [1] -
Tam, P. H.; Lowary, T. L. Org. Biomol. Chem. 2010, 8, 181–192. doi:10.1039/B916580K
Return to citation in text: [1] -
Cai, C.; Dickinson, D. M.; Li, L.; Masuko, S.; Suflita, M.; Schultz, V.; Nelson, S. D.; Bhaskar, U.; Liu, J.; Linhardt, R. J. Org. Lett. 2014, 16, 2240–2243. doi:10.1021/ol500738g
Return to citation in text: [1] -
Huang, X.; Huang, L.; Wang, H.; Ye, X.-S. Angew. Chem., Int. Ed. 2004, 43, 5221–5224. doi:10.1002/anie.200460176
Return to citation in text: [1] -
Guillemineau, M.; Auzanneau, F.-I. J. Org. Chem. 2012, 77, 8864–8878. doi:10.1021/jo301644w
Return to citation in text: [1] -
Lemanski, G.; Ziegler, T. Helv. Chim. Acta 2000, 83, 2655–2675. doi:10.1002/1522-2675(20001004)83:10<2655::AID-HLCA2655>3.0.CO;2-U
Return to citation in text: [1] -
Keliris, A.; Ziegler, T.; Mishra, R.; Pohmann, R.; Sauer, M. G.; Ugurbil, K.; Engelmann, J. Bioorg. Med. Chem. 2011, 19, 2529–2540. doi:10.1016/j.bmc.2011.03.023
Return to citation in text: [1] -
McGavin, R. S.; Gagne, R. A.; Chervenak, M. C.; Bundle, D. R. Org. Biomol. Chem. 2005, 3, 2723–2732. doi:10.1039/b416105j
Return to citation in text: [1] -
Srivastava, J.; Khare, A.; Khare, N. K. ARKIVOC 2009, vii, 180–188.
Return to citation in text: [1] -
Xiong, J.; Lu, Z.; Ding, N.; Ren, S.; Li, Y. Eur. J. Org. Chem. 2013, 6158–6166. doi:10.1002/ejoc.201300575
Return to citation in text: [1] -
Pozsgay, V. J. Org. Chem. 1999, 64, 7277–7280. doi:10.1021/jo990811p
Return to citation in text: [1] -
McGavin, R.; Bundle, D. R. Org. Biomol. Chem. 2005, 3, 2733–2740. doi:10.1039/b416106h
Return to citation in text: [1] -
Tanaka, Y.; Nakahara, Y.; Hojo, H.; Nakahara, Y. Tetrahedron 2003, 59, 4059–4067. doi:10.1016/S0040-4020(03)00615-X
Return to citation in text: [1] -
Chang, S.-M.; Tu, Z.; Jan, H.-M.; Pan, J.-F.; Lin, C.-H. Chem. Commun. 2013, 49, 4265–4267. doi:10.1039/C2CC37099A
Return to citation in text: [1] -
Roy, S.; Roy, N. J. Carbohydr. Chem. 2003, 22, 521–535. doi:10.1081/CAR-120026456
Return to citation in text: [1] -
Boto, A.; Hernandez, D.; Hernández, R.; Suárez, E. J. Org. Chem. 2006, 71, 1938–1948. doi:10.1021/jo052313o
Return to citation in text: [1] -
Krylov, V. B.; Argunov, D. A.; Vinnitskiy, D. Z.; Verkhnyatskaya, S. A.; Gerbst, A. G.; Ustyuzhanina, N. E.; Dmitrenok, A. S.; Huebner, J.; Holst, O.; Siebert, H.-C.; Nifantiev, N. E. Chem. – Eur. J. 2014, 20, 16516–16522. doi:10.1002/chem.201405083
Return to citation in text: [1] -
Pastore, A.; Adinolfi, M.; Iadonisi, A. Eur. J. Org. Chem. 2008, 6206–6212. doi:10.1002/ejoc.200800914
Return to citation in text: [1] -
Bedini, E.; Carabellese, A.; Comegna, D.; De Castro, C.; Parrilli, M. Tetrahedron 2006, 62, 8474–8483. doi:10.1016/j.tet.2006.06.084
Return to citation in text: [1] -
Wang, C.-C.; Lee, J.-C.; Luo, S.-Y.; Kulkarni, S. S.; Huang, Y.-W.; Lee, C.-C.; Chang, K.-L.; Hung, S.-C. Nature 2007, 446, 896–899. doi:10.1038/nature05730
Return to citation in text: [1] [2] -
Français, A.; Urban, D.; Beau, J.-M. Angew. Chem., Int. Ed. 2007, 46, 8662–8665. doi:10.1002/anie.200703437
Return to citation in text: [1] [2] -
Wang, C.-C.; Kulkarni, S. S.; Lee, J.-C.; Luo, S.-Y.; Hung, S.-C. Nat. Protoc. 2008, 3, 97–113. doi:10.1038/nprot.2007.493
Return to citation in text: [1] [2] -
Bourdreux, Y.; Lemétais, A.; Urban, D.; Beau, J.-M. Chem. Commun. 2011, 47, 2146–2148. doi:10.1039/c0cc04398b
Return to citation in text: [1] [2] -
Patil, P. S.; Lee, C.-C.; Huang, Y.-W.; Zulueta, M. M.; Hung, S.-C. Org. Biomol. Chem. 2013, 11, 2605–2612. doi:10.1039/c3ob40079d
Return to citation in text: [1] [2] -
Joseph, A. A.; Verma, V. P.; Liu, X.-Y.; Wu, C.-H.; Dhurandhare, V. M.; Wang, C.-C. Eur. J. Org. Chem. 2012, 744–753. doi:10.1002/ejoc.201101267
Return to citation in text: [1] [2] -
Irmak, M.; Groschner, A.; Boysen, M. M. K. Chem. Commun. 2007, 177–179. doi:10.1039/B612986B
Return to citation in text: [1] -
Joseph, A.; Dhurandhare, V. M.; Chang, C. W.; Verma, V. P.; Mishra, G. P.; Ku, C. C.; Lin, C.-C.; Wang, C.-C. Chem. Commun. 2015, 51, 104–106. doi:10.1039/C4CC06645F
Return to citation in text: [1] [2] -
Lim, J.; Grove, B. C.; Roth, A.; Breaker, R. R. Angew. Chem., Int. Ed. 2006, 45, 6689–6693. doi:10.1002/anie.200602534
Return to citation in text: [1] -
George, J.; Reddy, B. V. S. Org. Biomol. Chem. 2012, 10, 4731–4738. doi:10.1039/c2ob25315a
Return to citation in text: [1] -
Minuth, T.; Irmak, M.; Groschner, A.; Lehnert, T.; Boysen, M. M. K. Eur. J. Org. Chem. 2009, 997–1008. doi:10.1002/ejoc.200801035
Return to citation in text: [1] -
Tran, A.-T.; Jones, R. A.; Pastor, J.; Boisson, J.; Smith, N.; Galan, M. C. Adv. Synth. Catal. 2011, 353, 2593–2598. doi:10.1002/adsc.201100228
Return to citation in text: [1] -
Tatina, M.; Yousuf, S. K.; Mukherjee, D. Org. Biomol. Chem. 2012, 10, 5357–5360. doi:10.1039/c2ob25452b
Return to citation in text: [1] -
Xu, H.; Ren, B.; Zhao, W.; Xin, X.; Lu, Y.; Pei, Y.; Dong, H.; Pei, Z. Tetrahedron 2016, 72, 3490–3499. doi:10.1016/j.tet.2016.04.076
Return to citation in text: [1] -
Zulueta, M. M. L.; Janreddy, D.; Hung, S.-C. Isr. J. Chem. 2015, 55, 347–359. doi:10.1002/ijch.201400171
See for a recent review.
Return to citation in text: [1]
| 35. | Giordano, M.; Iadonisi, A. J. Org. Chem. 2014, 79, 213–222. doi:10.1021/jo402399n |
| 35. | Giordano, M.; Iadonisi, A. J. Org. Chem. 2014, 79, 213–222. doi:10.1021/jo402399n |
| 5. | Bartoszewicz, A.; Kalek, M.; Nilsson, J.; Hiresova, R.; Stawinski, J. Synlett 2008, 37–40. doi:10.1055/s-2007-992379 |
| 1. |
Ágoston, K.; Streicher, H.; Fügedi, P. Tetrahedron: Asymmetry 2016, 27, 707–728. doi:10.1016/j.tetasy.2016.06.010
See for a very recent review. |
| 2. | Wuts, P. G. M. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. Greene’s Protective Groups in Organic Synthesis; Wiley: 2014; pp 201–270. |
| 14. | Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. doi:10.1021/ja110332r |
| 15. | El-Badri, M. H.; Willenbring, D.; Tantillo, D. J.; Gervay-Hague, J. J. Org. Chem. 2007, 72, 4663–4672. doi:10.1021/jo070229y |
| 16. | Chambers, D. J.; Evans, G. R.; Fairbanks, A. J. Tetrahedron 2005, 61, 7184–7192. doi:10.1016/j.tet.2005.05.032 |
| 17. | Mikkelsen, L. M.; Krintel, S. L.; Jiménez-Barbero, J.; Skrydstrup, T. J. Org. Chem. 2002, 67, 6297–6308. doi:10.1021/jo020339z |
| 18. | Belaissaoui, A.; Saez, I. M.; Cowling, S. J.; Zeng, X.; Goodby, J. W. Chem. – Eur. J. 2012, 18, 2366–2373. doi:10.1002/chem.201102193 |
| 19. | Lam, S. N.; Gervay-Hague, J. Carbohydr. Res. 2002, 337, 1953–1965. doi:10.1016/S0008-6215(02)00227-6 |
| 20. | Suriano, F.; Coulembier, O.; Dubois, P. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3271–3280. doi:10.1002/pola.24109 |
| 21. | Nakagawa, Y.; Doi, T.; Taketani, T.; Takegoshi, K.; Igarashi, Y.; Ito, Y. Chem. – Eur. J. 2013, 19, 10516–10525. doi:10.1002/chem.201301368 |
| 38. |
Amantini, D.; Fringuelli, F.; Pizzo, F.; Vaccaro, L. J. Org. Chem. 2001, 66, 6734–6737. doi:10.1021/jo015814s
Here TBAB was reported to catalyze the trimethylsilylation of alcohols in the presence of trimethylsilyl azide. |
| 2. | Wuts, P. G. M. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. Greene’s Protective Groups in Organic Synthesis; Wiley: 2014; pp 201–270. |
| 10. | Corey, E. J.; Cho, H.; Rücker, C.; Hua, D. H. Tetrahedron Lett. 1981, 22, 3455–3458. doi:10.1016/S0040-4039(01)81930-4 |
| 11. | Mander, L. N.; Sehti, S. P. Tetrahedron Lett. 1984, 25, 5953–5956. doi:10.1016/S0040-4039(01)81731-7 |
| 12. | Boschelli, D.; Takemasa, T.; Nishitani, Y.; Masamune, S. Tetrahedron Lett. 1985, 26, 5239–5242. doi:10.1016/S0040-4039(00)95004-4 |
| 13. | Gassman, P. G.; Haberman, L. M. J. Org. Chem. 1986, 51, 5010–5013. doi:10.1021/jo00375a050 |
| 14. | Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. doi:10.1021/ja110332r |
| 15. | El-Badri, M. H.; Willenbring, D.; Tantillo, D. J.; Gervay-Hague, J. J. Org. Chem. 2007, 72, 4663–4672. doi:10.1021/jo070229y |
| 18. | Belaissaoui, A.; Saez, I. M.; Cowling, S. J.; Zeng, X.; Goodby, J. W. Chem. – Eur. J. 2012, 18, 2366–2373. doi:10.1002/chem.201102193 |
| 2. | Wuts, P. G. M. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. Greene’s Protective Groups in Organic Synthesis; Wiley: 2014; pp 201–270. |
| 3. | Corey, E. J.; Venkateswarlu, A. J. Am. Chem. Soc. 1972, 94, 6190–6191. doi:10.1021/ja00772a043 |
| 4. |
Bartoszewicz, A.; Kalek, M.; Stawinski, J. Tetrahedron 2008, 64, 8843–8850. doi:10.1016/j.tet.2008.06.070
See for use of iodine as the catalyst (also ref [5]). |
| 5. | Bartoszewicz, A.; Kalek, M.; Nilsson, J.; Hiresova, R.; Stawinski, J. Synlett 2008, 37–40. doi:10.1055/s-2007-992379 |
| 6. |
Yoshida, K.; Fujino, Y.; Itatsu, Y.; Inoue, H.; Kanoko, Y.; Takao, K.-i. Tetrahedron Lett. 2016, 57, 627–631. doi:10.1016/j.tetlet.2015.12.114
See for use of an N-oxide catalyst. |
| 7. |
Lombardo, L. Tetrahedron Lett. 1984, 25, 227–228. doi:10.1016/S0040-4039(00)99846-0
See for use of DIPEA as a catalyst. |
| 8. |
Aizpurua, J. M.; Palomo, C. Tetrahedron Lett. 1985, 26, 475–476. doi:10.1016/S0040-4039(00)61915-9
See for use of DBU as the base. |
| 9. |
Kim, S.; Chang, H. Synth. Commun. 1984, 14, 899–904. doi:10.1080/00397918408063758
See for use of tetramethylguanidine as the catalyst. |
| 35. | Giordano, M.; Iadonisi, A. J. Org. Chem. 2014, 79, 213–222. doi:10.1021/jo402399n |
| 1. |
Ágoston, K.; Streicher, H.; Fügedi, P. Tetrahedron: Asymmetry 2016, 27, 707–728. doi:10.1016/j.tetasy.2016.06.010
See for a very recent review. |
| 2. | Wuts, P. G. M. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. Greene’s Protective Groups in Organic Synthesis; Wiley: 2014; pp 201–270. |
| 3. | Corey, E. J.; Venkateswarlu, A. J. Am. Chem. Soc. 1972, 94, 6190–6191. doi:10.1021/ja00772a043 |
| 33. | Lee, D.; Taylor, M. S. Org. Biomol. Chem. 2013, 11, 5409–5412. doi:10.1039/c3ob40981c |
| 35. | Giordano, M.; Iadonisi, A. J. Org. Chem. 2014, 79, 213–222. doi:10.1021/jo402399n |
| 24. | Halmos, T.; Montserret, R.; Filippi, J.; Antonakis, K. Carbohydr. Res. 1987, 170, 57–69. doi:10.1016/0008-6215(87)85005-X |
| 25. | Arias-Pérez, M. S.; Santos, M. J. Tetrahedron 1996, 52, 10785–10798. doi:10.1016/0040-4020(96)00600-X |
| 26. | Ikeda, Y.; Furukawa, K.; Yamada, H. Carbohydr. Res. 2002, 337, 1499–1501. doi:10.1016/S0008-6215(02)00168-4 |
| 27. | Chung, M.-K.; Schlaf, M. J. Am. Chem. Soc. 2005, 127, 18085–18092. doi:10.1021/ja056283i |
| 28. | Teumelsan, N.; Huang, X. J. Org. Chem. 2007, 72, 8976–8979. doi:10.1021/jo7013824 |
| 29. | Pastore, A.; Adinolfi, M.; Iadonisi, A.; Valerio, S. Carbohydr. Res. 2010, 345, 1316–1323. doi:10.1016/j.carres.2010.02.025 |
| 30. | Heuckendorff, M.; Pedersen, C. M.; Bols, M. J. Org. Chem. 2013, 78, 7234–7248. doi:10.1021/jo4012464 |
| 31. | Zhang, Y.; Chen, C.; Jin, L.; Tan, H.; Wang, F.; Cao, H. Carbohydr. Res. 2015, 401, 109–114. doi:10.1016/j.carres.2014.09.010 |
| 32. | Garro, H. A.; Garcia, C.; Martin, V. S.; Tonn, C. E.; Pungitore, C. R. Bioorg. Med. Chem. Lett. 2015, 25, 914–918. doi:10.1016/j.bmcl.2014.12.052 |
| 36. | Traboni, S.; Bedini, E.; Giordano, M.; Iadonisi, A. Adv. Synth. Catal. 2015, 357, 3562–3572. doi:10.1002/adsc.201500745 |
| 23. | Marković, D.; Tchawou, W. A.; Novosjolova, I.; Laclef, S.; Stepanovs, D.; Turksm, M.; Vogel, P. Chem. – Eur. J. 2016, 22, 4196–4205. doi:10.1002/chem.201504380 |
| 22. | Chung, M.-K.; Orlova, G.; Goddard, J. D.; Schlaf, M.; Harris, R.; Beveridge, T. J.; White, G.; Hallett, F. R. J. Am. Chem. Soc. 2002, 124, 10508–10518. doi:10.1021/ja026723v |
| 34. | Giordano, M.; Iadonisi, A. Tetrahedron Lett. 2013, 54, 1550–1552. doi:10.1016/j.tetlet.2013.01.023 |
| 39. | Saidhareddy, P.; Ajay, S.; Shaw, A.-K. RSC Adv. 2014, 4, 4253–4259. doi:10.1039/C3RA45530K |
| 40. | Fei, X.; Holmes, T.; Diddle, J.; Hintz, L.; Delaney, D.; Stock, A.; Renner, D.; McDevitt, M.; Berkowitz, D. B.; Soukup, J. K. ACS Chem. Biol. 2014, 9, 2875–2882. doi:10.1021/cb500458f |
| 14. | Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. doi:10.1021/ja110332r |
| 23. | Marković, D.; Tchawou, W. A.; Novosjolova, I.; Laclef, S.; Stepanovs, D.; Turksm, M.; Vogel, P. Chem. – Eur. J. 2016, 22, 4196–4205. doi:10.1002/chem.201504380 |
| 69. | Joseph, A.; Dhurandhare, V. M.; Chang, C. W.; Verma, V. P.; Mishra, G. P.; Ku, C. C.; Lin, C.-C.; Wang, C.-C. Chem. Commun. 2015, 51, 104–106. doi:10.1039/C4CC06645F |
| 62. | Wang, C.-C.; Lee, J.-C.; Luo, S.-Y.; Kulkarni, S. S.; Huang, Y.-W.; Lee, C.-C.; Chang, K.-L.; Hung, S.-C. Nature 2007, 446, 896–899. doi:10.1038/nature05730 |
| 63. | Français, A.; Urban, D.; Beau, J.-M. Angew. Chem., Int. Ed. 2007, 46, 8662–8665. doi:10.1002/anie.200703437 |
| 64. | Wang, C.-C.; Kulkarni, S. S.; Lee, J.-C.; Luo, S.-Y.; Hung, S.-C. Nat. Protoc. 2008, 3, 97–113. doi:10.1038/nprot.2007.493 |
| 65. | Bourdreux, Y.; Lemétais, A.; Urban, D.; Beau, J.-M. Chem. Commun. 2011, 47, 2146–2148. doi:10.1039/c0cc04398b |
| 66. | Patil, P. S.; Lee, C.-C.; Huang, Y.-W.; Zulueta, M. M.; Hung, S.-C. Org. Biomol. Chem. 2013, 11, 2605–2612. doi:10.1039/c3ob40079d |
| 67. | Joseph, A. A.; Verma, V. P.; Liu, X.-Y.; Wu, C.-H.; Dhurandhare, V. M.; Wang, C.-C. Eur. J. Org. Chem. 2012, 744–753. doi:10.1002/ejoc.201101267 |
| 73. | Tran, A.-T.; Jones, R. A.; Pastor, J.; Boisson, J.; Smith, N.; Galan, M. C. Adv. Synth. Catal. 2011, 353, 2593–2598. doi:10.1002/adsc.201100228 |
| 74. | Tatina, M.; Yousuf, S. K.; Mukherjee, D. Org. Biomol. Chem. 2012, 10, 5357–5360. doi:10.1039/c2ob25452b |
| 75. | Xu, H.; Ren, B.; Zhao, W.; Xin, X.; Lu, Y.; Pei, Y.; Dong, H.; Pei, Z. Tetrahedron 2016, 72, 3490–3499. doi:10.1016/j.tet.2016.04.076 |
| 76. |
Zulueta, M. M. L.; Janreddy, D.; Hung, S.-C. Isr. J. Chem. 2015, 55, 347–359. doi:10.1002/ijch.201400171
See for a recent review. |
| 62. | Wang, C.-C.; Lee, J.-C.; Luo, S.-Y.; Kulkarni, S. S.; Huang, Y.-W.; Lee, C.-C.; Chang, K.-L.; Hung, S.-C. Nature 2007, 446, 896–899. doi:10.1038/nature05730 |
| 63. | Français, A.; Urban, D.; Beau, J.-M. Angew. Chem., Int. Ed. 2007, 46, 8662–8665. doi:10.1002/anie.200703437 |
| 64. | Wang, C.-C.; Kulkarni, S. S.; Lee, J.-C.; Luo, S.-Y.; Hung, S.-C. Nat. Protoc. 2008, 3, 97–113. doi:10.1038/nprot.2007.493 |
| 65. | Bourdreux, Y.; Lemétais, A.; Urban, D.; Beau, J.-M. Chem. Commun. 2011, 47, 2146–2148. doi:10.1039/c0cc04398b |
| 66. | Patil, P. S.; Lee, C.-C.; Huang, Y.-W.; Zulueta, M. M.; Hung, S.-C. Org. Biomol. Chem. 2013, 11, 2605–2612. doi:10.1039/c3ob40079d |
| 67. | Joseph, A. A.; Verma, V. P.; Liu, X.-Y.; Wu, C.-H.; Dhurandhare, V. M.; Wang, C.-C. Eur. J. Org. Chem. 2012, 744–753. doi:10.1002/ejoc.201101267 |
| 68. | Irmak, M.; Groschner, A.; Boysen, M. M. K. Chem. Commun. 2007, 177–179. doi:10.1039/B612986B |
| 69. | Joseph, A.; Dhurandhare, V. M.; Chang, C. W.; Verma, V. P.; Mishra, G. P.; Ku, C. C.; Lin, C.-C.; Wang, C.-C. Chem. Commun. 2015, 51, 104–106. doi:10.1039/C4CC06645F |
| 70. | Lim, J.; Grove, B. C.; Roth, A.; Breaker, R. R. Angew. Chem., Int. Ed. 2006, 45, 6689–6693. doi:10.1002/anie.200602534 |
| 71. | George, J.; Reddy, B. V. S. Org. Biomol. Chem. 2012, 10, 4731–4738. doi:10.1039/c2ob25315a |
| 72. | Minuth, T.; Irmak, M.; Groschner, A.; Lehnert, T.; Boysen, M. M. K. Eur. J. Org. Chem. 2009, 997–1008. doi:10.1002/ejoc.200801035 |
| 60. | Pastore, A.; Adinolfi, M.; Iadonisi, A. Eur. J. Org. Chem. 2008, 6206–6212. doi:10.1002/ejoc.200800914 |
| 61. | Bedini, E.; Carabellese, A.; Comegna, D.; De Castro, C.; Parrilli, M. Tetrahedron 2006, 62, 8474–8483. doi:10.1016/j.tet.2006.06.084 |
| 14. | Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. doi:10.1021/ja110332r |
| 15. | El-Badri, M. H.; Willenbring, D.; Tantillo, D. J.; Gervay-Hague, J. J. Org. Chem. 2007, 72, 4663–4672. doi:10.1021/jo070229y |
| 16. | Chambers, D. J.; Evans, G. R.; Fairbanks, A. J. Tetrahedron 2005, 61, 7184–7192. doi:10.1016/j.tet.2005.05.032 |
| 17. | Mikkelsen, L. M.; Krintel, S. L.; Jiménez-Barbero, J.; Skrydstrup, T. J. Org. Chem. 2002, 67, 6297–6308. doi:10.1021/jo020339z |
| 18. | Belaissaoui, A.; Saez, I. M.; Cowling, S. J.; Zeng, X.; Goodby, J. W. Chem. – Eur. J. 2012, 18, 2366–2373. doi:10.1002/chem.201102193 |
| 20. | Suriano, F.; Coulembier, O.; Dubois, P. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3271–3280. doi:10.1002/pola.24109 |
| 41. | Dohi, H.; Nishida, Y.; Takeda, T.; Kobayashi, K. Carbohydr. Res. 2002, 337, 983–989. doi:10.1016/S0008-6215(02)00093-9 |
| 42. | Despras, G.; Robert, R.; Sendid, B.; Machez, E.; Poulain, D.; Mallet, J.-M. Bioorg. Med. Chem. 2012, 20, 1817–1831. doi:10.1016/j.bmc.2011.12.048 |
| 43. | Peng, P.; Xiong, D.-C.; Ye, X.-S. Carbohydr. Res. 2014, 384, 1–8. doi:10.1016/j.carres.2013.11.009 |
| 44. | Tam, P. H.; Lowary, T. L. Org. Biomol. Chem. 2010, 8, 181–192. doi:10.1039/B916580K |
| 45. | Cai, C.; Dickinson, D. M.; Li, L.; Masuko, S.; Suflita, M.; Schultz, V.; Nelson, S. D.; Bhaskar, U.; Liu, J.; Linhardt, R. J. Org. Lett. 2014, 16, 2240–2243. doi:10.1021/ol500738g |
| 46. | Huang, X.; Huang, L.; Wang, H.; Ye, X.-S. Angew. Chem., Int. Ed. 2004, 43, 5221–5224. doi:10.1002/anie.200460176 |
| 47. | Guillemineau, M.; Auzanneau, F.-I. J. Org. Chem. 2012, 77, 8864–8878. doi:10.1021/jo301644w |
| 48. | Lemanski, G.; Ziegler, T. Helv. Chim. Acta 2000, 83, 2655–2675. doi:10.1002/1522-2675(20001004)83:10<2655::AID-HLCA2655>3.0.CO;2-U |
| 49. | Keliris, A.; Ziegler, T.; Mishra, R.; Pohmann, R.; Sauer, M. G.; Ugurbil, K.; Engelmann, J. Bioorg. Med. Chem. 2011, 19, 2529–2540. doi:10.1016/j.bmc.2011.03.023 |
| 50. | McGavin, R. S.; Gagne, R. A.; Chervenak, M. C.; Bundle, D. R. Org. Biomol. Chem. 2005, 3, 2723–2732. doi:10.1039/b416105j |
| 51. | Srivastava, J.; Khare, A.; Khare, N. K. ARKIVOC 2009, vii, 180–188. |
| 52. | Xiong, J.; Lu, Z.; Ding, N.; Ren, S.; Li, Y. Eur. J. Org. Chem. 2013, 6158–6166. doi:10.1002/ejoc.201300575 |
| 53. | Pozsgay, V. J. Org. Chem. 1999, 64, 7277–7280. doi:10.1021/jo990811p |
| 19. | Lam, S. N.; Gervay-Hague, J. Carbohydr. Res. 2002, 337, 1953–1965. doi:10.1016/S0008-6215(02)00227-6 |
| 54. | McGavin, R.; Bundle, D. R. Org. Biomol. Chem. 2005, 3, 2733–2740. doi:10.1039/b416106h |
| 55. | Tanaka, Y.; Nakahara, Y.; Hojo, H.; Nakahara, Y. Tetrahedron 2003, 59, 4059–4067. doi:10.1016/S0040-4020(03)00615-X |
| 56. | Chang, S.-M.; Tu, Z.; Jan, H.-M.; Pan, J.-F.; Lin, C.-H. Chem. Commun. 2013, 49, 4265–4267. doi:10.1039/C2CC37099A |
| 57. | Roy, S.; Roy, N. J. Carbohydr. Chem. 2003, 22, 521–535. doi:10.1081/CAR-120026456 |
| 58. | Boto, A.; Hernandez, D.; Hernández, R.; Suárez, E. J. Org. Chem. 2006, 71, 1938–1948. doi:10.1021/jo052313o |
| 59. | Krylov, V. B.; Argunov, D. A.; Vinnitskiy, D. Z.; Verkhnyatskaya, S. A.; Gerbst, A. G.; Ustyuzhanina, N. E.; Dmitrenok, A. S.; Huebner, J.; Holst, O.; Siebert, H.-C.; Nifantiev, N. E. Chem. – Eur. J. 2014, 20, 16516–16522. doi:10.1002/chem.201405083 |
© 2016 Traboni et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)