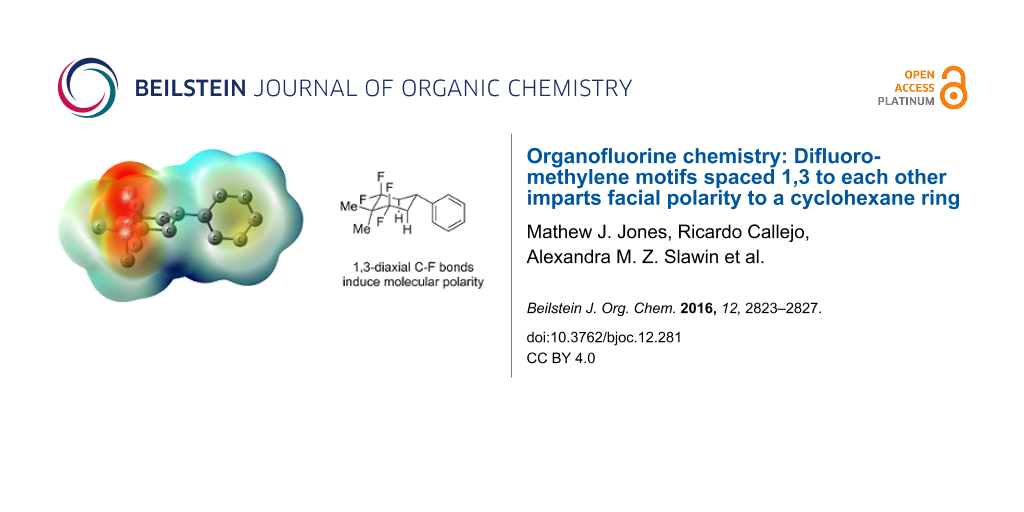
Abstract
2,2-Dimethyl-5-phenyl-1,1,3,3-tetrafluororocyclohexane has been prepared and characterised as an example of a facially polarised cyclohexane containing 1,3 related CF2 groups. The dipolar nature of the ring arises from the axial orientation of two of the C–F bonds pointing in the same direction, and set by the chair conformation of the cyclohexane. This electrostatic profile is revealed experimentally both in the solid-state (X-ray) packing of the rings and by solution (NMR) in different solvents. A computationally derived electrostatic profile of this compound is consistent with a more electronegative and a more electropositive face of the cyclohexane ring. This placing of CF2 groups 1,3 to each other in a cyclohexane ring is introduced as a new design strategy which could be applicable to the preparation of polar hydrophobic cyclohexane motifs.

Graphical Abstract
Introduction
Selective incorporation of fluorine atoms is a powerful strategy for modulating the properties of organic compounds [1-3]. For instance, the replacement of hydrogen by fluorine is commonly practised in medicinal [4,5] and agrochemical [6,7] research programmes. The dipole moment associated with the C–F bond has also rendered fluorine containing compounds important in the development of organic materials such as liquid crystals [8,9]. Strategic fluorination can add polarity to a molecule, however, such compounds do not generally increase in their hydrophilic capacity, thus selective fluorination leads to polar-hydrophobic properties [10]. Although selective mono-fluorination and CF3 incorporation have had wide currency, the difluoromethylene (CF2) motif has received relatively less attention despite possessing unique properties [11-14]. For example CH2F2 shows the highest molecular dipole (1.97 D) of the fluoromethane series, relative to CH3F (1.87 D) or CHF3 (1.65 D) [15,16]. Also (poly)vinylidine fluoride (~CF2CH2~) is the prototypical piezoelectric polymer, when the CF2 group are orientated (poled) in the melt [17].
The search for new and more diverse polar hydrophobic scaffolds is attractive for drug discovery research and new materials [18]. In our own lab we have been exploring the synthesis and properties of differently fluorinated aliphatic structures based on the cyclopentane (1) and cyclohexane (2 and 3) rings, compounds which can show quite large dipole moments depending on the stereochemistry [19,20] (Figure 1). All-syn 1,2,3,4,5,6-hexafluorocyclohexane, which was prepared recently, emerges as among the most polar aliphatic compounds reported in the literature [21]. The specific arrangement with all of the fluorines “up” induces a facial polarisation to the cyclohexane, a unique phenomenon which may have unexplored utility in organic chemistry. For example derivatives of these compounds could be potentially useful as polar hydrophobic substituents in drug discovery that will simultaneously contact both electropositive and electronegative domains within a target protein.
![[1860-5397-12-281-1]](/bjoc/content/figures/1860-5397-12-281-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected fluorinated polar alicyclic scaffolds.
Figure 1: Selected fluorinated polar alicyclic scaffolds.
In this paper, we now report the design, synthesis and characterisation of a novel tetrafluorocyclohexane structure 4, rendered polar by placing two CF2 groups located 1,3 from each other (Scheme 1). This forces two C–F bonds to lie 1,3-diaxial to each other while the cyclohexane adopts the classic chair conformation. To the best of our knowledge, 2,2,6,6-tetrafluorocyclohexanol is the only derivative containing this feature reported so far. It was prepared in an eight step synthesis [22]. At the outset we explored a more direct synthesis of this arrangement based on deoxofluorination of 1,3-diketones 5 with DAST or Deoxo-Fluor® [23] (Scheme 1). An alternative route envisaged a fluorodesulfurisation using 1,3-bis-dithianes 6 as substrates [24] (Scheme 1). Both synthetic approaches have been widely used for the introduction of the CF2 group in open-chain and (macro)cyclic systems [23,24], but not in a 1,3 relationship.
![[1860-5397-12-281-i1]](/bjoc/content/inline/1860-5397-12-281-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic plan to the preparation of 1,1,3,3-tetrafluorocyclohexane structures.
Scheme 1: Retrosynthetic plan to the preparation of 1,1,3,3-tetrafluorocyclohexane structures.
Results and Discussion
The preparation of diketones 5a–c and bis-dithianes 6a–c was carried out by previously described methods. The dimethyl-diketone 5c was prepared by double methylation of 5b [25] (Scheme 2). Two different O-alkylated products 7 and 8 were also obtained as significant byproducts.
![[1860-5397-12-281-i2]](/bjoc/content/inline/1860-5397-12-281-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of starting materials 5c and 6a–c.
Scheme 2: Preparation of starting materials 5c and 6a–c.
The reaction of ketones 5a–c with 1,2-ethanedithiol in the presence of catalytic BF3·OEt2 afforded the corresponding bis-dithianes 6a–c in moderate to good yields [26] (Scheme 2). The reaction of diketone 5c also afforded the monoderivatised ketone 9 as a minor product.
Direct fluorination of diketones 5a–c was explored with DAST or Deoxo-Fluor® (Scheme 3). When diketones 5a and 5b were treated with DAST or Deoxo-Fluor®, either neat or in DCM as a solvent, only complex and intractable products were obtained. These highly enolisable diketones [27,28] were incompatible with deoxyfluorination. In order to block enolisation, diketone 5c containing two methyl groups α to the ketones was subject to fluorination with DAST and Deoxo-Fluor®. In this case the reaction was successful and the desired tetrafluorocyclohexane 4c could be obtained, most preferably with DAST although in a low overall yield (Scheme 3). The presence of the gem-dimethyl motif renders the carbonyl groups independent of each other and this facilitates the difluorination process relative to 5a and 5b. Nevertheless, other unidentified byproducts were also obtained, which made purification of 4c by column chromatography difficult and compromised the yield.
![[1860-5397-12-281-i3]](/bjoc/content/inline/1860-5397-12-281-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deoxofluorination of diketones 5. Reagents and conditions: a) DAST, DCM, rt, overnight, 4a (traces), 4b (traces), 4c (14%); b) Deoxo-Fluor®, DCM, rt, overnight, 4a (traces), 4b (traces), 4c (10%).
Scheme 3: Deoxofluorination of diketones 5. Reagents and conditions: a) DAST, DCM, rt, overnight, 4a (traces)...
Treatment of bis-dithianes 6 with NIS and HF·Py as previously described for linear systems [28] proved unsuccessful (Scheme 4) and did not offer a way forward.
![[1860-5397-12-281-i4]](/bjoc/content/inline/1860-5397-12-281-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Fluorodesulfurisation of bis-dithianes 6. Reagents and conditions: a) NIS, HF·Py, DCM, −78 °C to rt, overnight.
Scheme 4: Fluorodesulfurisation of bis-dithianes 6. Reagents and conditions: a) NIS, HF·Py, DCM, −78 °C to rt...
Tetrafluorocyclohexane 4c was a crystalline solid (mp 70 ºC). The structure of this compound was established by NMR, showing a characteristic AB system in 19F NMR for axial/equatorial fluorines, and a triplet (1JCF = 250 Hz) in 13C NMR for the CF2 group. In addition, single crystal X-ray diffraction data were obtained for 4c which confirmed the structure (Figure 2). The C–CF2–C angles in the cyclohexane ring are considerably wider (≈116°) than that of the C–CH2–C angles (≈112°), which is now recognised as a general phenomenon of the CF2 group in an aliphatic chain/ring [10-13]. The two axial fluorines flex away from each other with widened right angles of 96.3°, distorting a co-parallel orientation. This distortion is also observed for the 1,3-diaxial C–F bonds in the solid state structures of 2 [20] and 3 [21] consistent with electrostatic dipolar repulsion between these C–F bonds and indicative of the origin of the polarity of these molecules. In terms of intermolecular packing in the solid state there are interactions between the non-fluorine face of the cyclohexyl ring and the phenyl group of a neighbouring molecule (Figure 2). These C-H···π interactions are a feature of the solid state packing found in phenyl derivatives of 2 [29].
![[1860-5397-12-281-2]](/bjoc/content/figures/1860-5397-12-281-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structure of compound 4c. The image shows two molecules stacked with the non-fluorine face pointing to an adjacent phenyl ring consistent with an electrostatic ordering. The structure has been deposited in the Cambridge Crystallographic Data Centre (CCDC) 1502954.
Figure 2: X-ray structure of compound 4c. The image shows two molecules stacked with the non-fluorine face po...
This electrostatic ordering was reinforced by an NMR shift experiment in different solvents. Selected 1H NMR signals of 4c showed a very clear upfield shift in [2H8]-toluene when compared with the spectrum obtained in chloroform (CDCl3) (Figure 3). In particular, the two axial protons H-3 and the axial methyl group at C-4 display higher upfield shifts in toluene (Δδ ≈ 0.5 ppm and Δδ ≈ 0.3 ppm, respectively), whereas that of the equatorial protons H-2, the equatorial methyl group at C-5 and the axial proton H-1 do not experience shifts of the same magnitude (Δδ ≈ 0.2 ppm, Δδ ≈ −0.1 ppm and Δδ ≈ 0.1 ppm, respectively). These observations are consistent with the anisotropic influence of the electronegative aromatic solvent (toluene) interacting with the more electropositive (non-fluorine) face of the cyclohexane ring (Figure 3).
![[1860-5397-12-281-3]](/bjoc/content/figures/1860-5397-12-281-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra of 4c. A) shows the spectrum in [2H8]-toluene, and B) shows the spectrum in chloroform (CDCl3). There are significant upfield-shifted proton signals in toluene, consistent with an electrostatic interaction between toluene and the electropositive hydrogen face of 1,1,3,3-tetrafluorocyclohexyl 4c.
Figure 3: 1H NMR spectra of 4c. A) shows the spectrum in [2H8]-toluene, and B) shows the spectrum in chlorofo...
To extend these experimental observations, a DFT computational study, at the B3LYP-D3/6-311+G(d,p) level, was carried out in order to obtain an electrostatic potential map of cyclohexane 4c. The profile confirms the presence of an electronegative face (in red) determined by the two 1,3-diaxial C–F bonds and an electropositive face (dark blue), which is most intense at the hydrogens of the diaxial C–H bonds pointing antiperiplanar to the diaxial C–F bonds (Figure 4). A molecular dipole moment for compound 4c was calculated at 3.1 D and can be compared for example to 1,2,4,5-tetrafluorocyclohexane (2, 5.2 D) [18].
![[1860-5397-12-281-4]](/bjoc/content/figures/1860-5397-12-281-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Electrostatic potential map for 4c calculated at the B3LYP/6-311G(d,p) level for an optimised structure calculated at the B3LYP-D3/6-311+G(d,p) level. The image is plotted on a colour scale from −0.003 a.u. (red) to +0.003 a.u. (blue) and mapped onto an isodensity surface (ρ = 4 × 10−4 a.u.); hydrogen atoms are omitted for clarity.
Figure 4: Electrostatic potential map for 4c calculated at the B3LYP/6-311G(d,p) level for an optimised struc...
Conclusion
In this study we have prepared a novel polar aliphatic structure 4c based on the 1,1,3,3-tetrafluorocyclohexyl motif. The compound was prepared by direct difluorination of the corresponding α,α-dimethylated 1,3-diketone with DAST. The presence of the two 1,3-diaxial fluorine atoms in structure 4c is responsible for the facial polarisation on the cyclohexane ring. We have demonstrated the intermolecular electrostatic interaction of the positive non-fluorous face with the π-system of an aromatic ring by single crystal X-ray diffraction and NMR experiments. Additionally, the polarity of 4c has been calculated by computational optimisations. It follows that functionalised cyclohexanes containing the 1,3-di-CF2 motif would offer new possibilities for the design of performance molecules extending from organic materials research to medicinal chemistry.
Supporting Information
| Supporting Information File 1: Experimental part and NMR spectra of new synthesised compounds. | ||
| Format: PDF | Size: 1.4 MB | Download |
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) and the European Research Council (ERC). The authors acknowledge the EPSRC National Mass Spectrometry Facility (Swansea) and Mrs Caroline Horsburgh (University of St Andrews) for analytical assistance. D.O’H. thanks the Royal Society for a Wolfson Research Merit Award.
References
-
Liu, W.; Huang, X.; Cheng, M.-J.; Neilson, R. J.; Goddard, W. A., III; Groves, J. T. Science 2012, 337, 1322–1325. doi:10.1126/science.1222327
Return to citation in text: [1] -
O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/B711844A
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2004. doi:10.1002/352760393X
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/B610213C
Return to citation in text: [1] -
Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Oxford, 2009.
Return to citation in text: [1] -
Boiko, V. N. Beilstein J. Org. Chem. 2010, 6, 880–921. doi:10.3762/bjoc.6.88
Return to citation in text: [1] -
Leroux, F.; Jeschke, P.; Schlosser, M. Chem. Rev. 2005, 105, 827–856. doi:10.1021/cr040075b
Return to citation in text: [1] -
Kirsch, P.; Bremer, M. ChemPhysChem 2010, 11, 357–360. doi:10.1002/cphc.200900745
Return to citation in text: [1] -
Kirsch, P.; Binder, W.; Hahn, A.; Jährling, K.; Lenges, M.; Lietzau, L.; Maillard, D.; Meyer, V.; Poetsch, E.; Ruhl, A.; Unger, G.; Fröhlich, R. Eur. J. Org. Chem. 2008, 3479–3487. doi:10.1002/ejoc.200800149
Return to citation in text: [1] -
Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910
Return to citation in text: [1] [2] -
Callejo, R.; Corr, M. J.; Yang, M.; Wang, M.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2016, 22, 8137–8151. doi:10.1002/chem.201600519
Return to citation in text: [1] [2] -
Wang, Y.; Callejo, R.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2014, 10, 18–25. doi:10.3762/bjoc.10.4
Return to citation in text: [1] [2] -
Urbina-Blanco, C. A.; Skibinski, M.; O’Hagan, D.; Nolan, S. P. Chem. Commun. 2013, 49, 7201–7203. doi:10.1039/c3cc44312d
Return to citation in text: [1] [2] -
O’Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26
Return to citation in text: [1] -
Papoušek, D.; Papoušková, Z.; Chong, D. P. J. Phys. Chem. 1995, 99, 15387–15395. doi:10.1021/j100042a010
Return to citation in text: [1] -
Fox, G. L.; Schlegel, H. B. J. Chem. Phys. 1990, 92, 4351–4357. doi:10.1063/1.457742
Return to citation in text: [1] -
Kawai, H. Jpn. J. Appl. Phys. 1969, 8, 975–976. doi:10.1143/JJAP.8.975
Return to citation in text: [1] -
Wudarczyk, J.; Papamokos, G.; Margaritis, V.; Schollmeyer, D.; Hinkel, F.; Baumgarten, M.; Floudas, G.; Müllen, K. Angew. Chem., Int. Ed. 2016, 55, 3220–3223. doi:10.1002/anie.201508249
Return to citation in text: [1] [2] -
Fang, Z.; Al-Maharik, N.; Slawin, A. M. Z.; Bühl, M.; O’Hagan, D. Chem. Commun. 2016, 52, 5116–5119. doi:10.1039/C6CC01348A
Return to citation in text: [1] -
Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f
Return to citation in text: [1] [2] -
Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O'Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232
Return to citation in text: [1] [2] -
Duhamel, P.; Leblond, B.; Poirer, J.-M. J. Chem. Soc., Chem. Commun. 1993, 476–477. doi:10.1039/C39930000476
Return to citation in text: [1] -
Singh, R. P.; Shreeve, J. M. Synthesis 2002, 2561–2578. doi:10.1055/s-2002-35626
Return to citation in text: [1] [2] -
Sondej, S. C.; Katzenellenbogen, J. A. J. Org. Chem. 1986, 51, 3508–3513. doi:10.1021/jo00368a022
Return to citation in text: [1] [2] -
Eddy, N. A.; Kelly, C. B.; Mercadante, M. A.; Leadbeater, N. E.; Fenteany, G. Org. Lett. 2012, 14, 498–501. doi:10.1021/ol2030873
Return to citation in text: [1] -
Smissman, E. E.; Diebold, J. L. J. Org. Chem. 1968, 33, 1466–1468. doi:10.1021/jo01268a033
Return to citation in text: [1] -
Singh, R. P.; Majumder, U.; Shreeve, J. M. J. Org. Chem. 2001, 66, 6263–6267. doi:10.1021/jo0157674
Return to citation in text: [1] -
Asato, A. E.; Liu, R. S. H. Tetrahedron Lett. 1986, 27, 3337–3340. doi:10.1016/S0040-4039(00)84789-9
Return to citation in text: [1] [2] -
Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354
Return to citation in text: [1]
| 28. | Asato, A. E.; Liu, R. S. H. Tetrahedron Lett. 1986, 27, 3337–3340. doi:10.1016/S0040-4039(00)84789-9 |
| 26. | Smissman, E. E.; Diebold, J. L. J. Org. Chem. 1968, 33, 1466–1468. doi:10.1021/jo01268a033 |
| 27. | Singh, R. P.; Majumder, U.; Shreeve, J. M. J. Org. Chem. 2001, 66, 6263–6267. doi:10.1021/jo0157674 |
| 28. | Asato, A. E.; Liu, R. S. H. Tetrahedron Lett. 1986, 27, 3337–3340. doi:10.1016/S0040-4039(00)84789-9 |
| 1. | Liu, W.; Huang, X.; Cheng, M.-J.; Neilson, R. J.; Goddard, W. A., III; Groves, J. T. Science 2012, 337, 1322–1325. doi:10.1126/science.1222327 |
| 2. | O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/B711844A |
| 3. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, 2004. doi:10.1002/352760393X |
| 10. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 23. | Singh, R. P.; Shreeve, J. M. Synthesis 2002, 2561–2578. doi:10.1055/s-2002-35626 |
| 24. | Sondej, S. C.; Katzenellenbogen, J. A. J. Org. Chem. 1986, 51, 3508–3513. doi:10.1021/jo00368a022 |
| 8. | Kirsch, P.; Bremer, M. ChemPhysChem 2010, 11, 357–360. doi:10.1002/cphc.200900745 |
| 9. | Kirsch, P.; Binder, W.; Hahn, A.; Jährling, K.; Lenges, M.; Lietzau, L.; Maillard, D.; Meyer, V.; Poetsch, E.; Ruhl, A.; Unger, G.; Fröhlich, R. Eur. J. Org. Chem. 2008, 3479–3487. doi:10.1002/ejoc.200800149 |
| 25. | Eddy, N. A.; Kelly, C. B.; Mercadante, M. A.; Leadbeater, N. E.; Fenteany, G. Org. Lett. 2012, 14, 498–501. doi:10.1021/ol2030873 |
| 6. | Boiko, V. N. Beilstein J. Org. Chem. 2010, 6, 880–921. doi:10.3762/bjoc.6.88 |
| 7. | Leroux, F.; Jeschke, P.; Schlosser, M. Chem. Rev. 2005, 105, 827–856. doi:10.1021/cr040075b |
| 23. | Singh, R. P.; Shreeve, J. M. Synthesis 2002, 2561–2578. doi:10.1055/s-2002-35626 |
| 18. | Wudarczyk, J.; Papamokos, G.; Margaritis, V.; Schollmeyer, D.; Hinkel, F.; Baumgarten, M.; Floudas, G.; Müllen, K. Angew. Chem., Int. Ed. 2016, 55, 3220–3223. doi:10.1002/anie.201508249 |
| 4. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/B610213C |
| 5. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Oxford, 2009. |
| 24. | Sondej, S. C.; Katzenellenbogen, J. A. J. Org. Chem. 1986, 51, 3508–3513. doi:10.1021/jo00368a022 |
| 18. | Wudarczyk, J.; Papamokos, G.; Margaritis, V.; Schollmeyer, D.; Hinkel, F.; Baumgarten, M.; Floudas, G.; Müllen, K. Angew. Chem., Int. Ed. 2016, 55, 3220–3223. doi:10.1002/anie.201508249 |
| 21. | Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O'Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232 |
| 21. | Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O'Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232 |
| 22. | Duhamel, P.; Leblond, B.; Poirer, J.-M. J. Chem. Soc., Chem. Commun. 1993, 476–477. doi:10.1039/C39930000476 |
| 29. | Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354 |
| 15. | Papoušek, D.; Papoušková, Z.; Chong, D. P. J. Phys. Chem. 1995, 99, 15387–15395. doi:10.1021/j100042a010 |
| 16. | Fox, G. L.; Schlegel, H. B. J. Chem. Phys. 1990, 92, 4351–4357. doi:10.1063/1.457742 |
| 10. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 11. | Callejo, R.; Corr, M. J.; Yang, M.; Wang, M.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2016, 22, 8137–8151. doi:10.1002/chem.201600519 |
| 12. | Wang, Y.; Callejo, R.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2014, 10, 18–25. doi:10.3762/bjoc.10.4 |
| 13. | Urbina-Blanco, C. A.; Skibinski, M.; O’Hagan, D.; Nolan, S. P. Chem. Commun. 2013, 49, 7201–7203. doi:10.1039/c3cc44312d |
| 11. | Callejo, R.; Corr, M. J.; Yang, M.; Wang, M.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2016, 22, 8137–8151. doi:10.1002/chem.201600519 |
| 12. | Wang, Y.; Callejo, R.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2014, 10, 18–25. doi:10.3762/bjoc.10.4 |
| 13. | Urbina-Blanco, C. A.; Skibinski, M.; O’Hagan, D.; Nolan, S. P. Chem. Commun. 2013, 49, 7201–7203. doi:10.1039/c3cc44312d |
| 14. | O’Hagan, D.; Wang, Y.; Skibinski, M.; Slawin, A. M. Z. Pure Appl. Chem. 2012, 84, 1587–1595. doi:10.1351/PAC-CON-11-09-26 |
| 19. | Fang, Z.; Al-Maharik, N.; Slawin, A. M. Z.; Bühl, M.; O’Hagan, D. Chem. Commun. 2016, 52, 5116–5119. doi:10.1039/C6CC01348A |
| 20. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f |
| 20. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f |
© 2016 Jones et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)