Abstract
Solutions of 1,3-diketones and 1,3-ketoester derivatives react with fluorine to give the corresponding 2,2-difluoro-1,3-dicarbonyl derivatives in the presence of quinuclidine. Quinuclidine reacts with fluorine in situ to generate a fluoride ion that facilitates limiting enolization processes, and an electrophilic N–F fluorinating agent that is reactive towards neutral enol species.

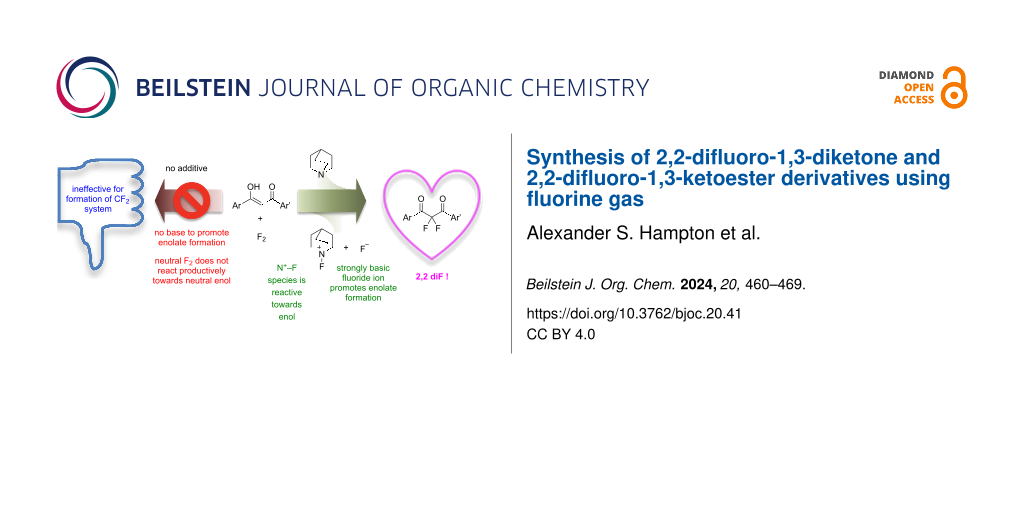
Graphical Abstract
Introduction
Fluorine is present in many agrochemical and pharmaceutical products owing to the beneficial properties imparted such as increased metabolic stability, lipophilicity and bioavailability of the bioactive entity [1-3]. In 2018, 30% of FDA approved drugs contained at least one fluorine atom, with an average of 2.7 fluorine atoms per fluorinated drug, and fluorine is also present in the structures of 50% of marketed agrochemicals [4]. In the context of the research reported here, the incorporation of difluoromethylene (CF2) units into life science products is growing in importance and a number of commercially significant pharmaceuticals [lubiprostone (constipation), maraviroc (HIV), tafluproct (anti-inflamatory), ledipasvir (hepatitis-C)] and agrochemicals [isopyrazam (fungicide), riodipine (calcium channel blocker), primisulfuron-methyl (pesticide)] owe their enhanced bioactivity, in part, to the presence of difluoromethylene units.
To meet the demands of synthetic chemists within the life science discovery and manufacturing arenas, many fluorination methods have been developed over the years to introduce difluoromethylene groups into organic systems. Approaches using nucleophilic fluorination include halogen exchange of gem-dihalo groups to corresponding CF2 derivatives using silver tetrafluoroborate [5] or mercury(II) fluoride [6], deoxyfluorination of carbonyl derivatives using diethylaminosulfur trifluoride (DAST) or related Deoxo-Fluor and Xtalfluor reagents [7,8]. Alternatively, oxidative fluorodesulfurizations of carbonyl derivatives using a combination of sources of halonium and fluoride ions such as 1,3-dibromo-5,5-dimethylhydantoin (DBH) and tetrabutylammonium dihydrogen trifluoride have been achieved [9-11].
The transformation of methylene to difluoromethylene using electrophilic fluorinating agents offers an alternative fluorination route, for example, the reactions of MeCN solutions of 1,3-diketones with electrophilic fluorinating agents such as Selectfluor eventually give the corresponding 2,2-difluoro-1,3-diketone derivatives [12]. Monofluorination of the 1,3-diketone substrates is rapid, but the second fluorination step occurs only after reaction for several days. In the solid phase, mechanical milling of the diketone substrate with solid Selectfluor in the presence of sodium carbonate [13,14], and reaction of ketones with a strong base and an N–F reagent give rise to the corresponding 2,2-difluoroketones [15]. In related kinetic studies concerning the electrophilic 2-fluorination of 1,3-diketones with Selectfluor [16,17], we demonstrated that the rate-determining step for difluorination was enolization of the intermediate 2-fluoro-1,3-diketone. Monofluorination of 1,3-diketones occurs rapidly because the substrates lie predominantly in their enol tautomeric forms. The resulting 2-difluoro-1,3-diketones, on the other hand, are formed in their keto-tautomeric forms. Thus, we found difluorination could only be achieved upon addition of water or a base to accelerate the enolization of the monofluoro-diketone intermediates. In addition, imines and α-diboryl ketone derivatives can also be transformed to 2,2-difluoroketones using an N–F electrophilic fluorinating reagent [18]. Alternatively, building blocks containing CF2 units such as ethyl bromodifluoroacetate and difluoromethylphenyl sulfoxide offer the possibility of transferring difluoromethylene groups directly into organic systems [19-25] and there is now a very extensive literature on carbon–carbon bond-forming reactions using these and other difluoromethylated building blocks [3,26-32].
Since profit margins in the life science industries are always under constant pressure, less expensive methods of introducing fluorine selectively into active intermediates for manufacture on the industrial scale are required and, as a relatively inexpensive strategy, direct fluorination of substrates using fluorine gas has been used successfully for the production of 5-fluorouracil (generic, anticancer) and voriconazole (V-FEND, Pfizer, antifungal) [33]. Methods have been developed for the selective monofluorination of 1,3-dicarbonyl derivatives by fluorine gas using batch and continuous flow techniques [34-36]. Difluorination occurs very slowly in comparison to monofluorination, although some difluorinated by-products are, in general, formed upon fluorination of dicarbonyl substrates and difluorinated products can be readily separated from monofluorinated systems [34]. Direct fluorination of diazo compounds using F2 [37] is the only report of a useful synthetic procedure to selectively prepare a difluoromethylene containing product using F2 but, in these cases, CFCs, now banned under the Montreal protocol, were used as the reaction medium.
Here, we demonstrate that the addition of quinuclidine to direct fluorination reactions of 1,3-diketone and 1,3-ketoester substrates using fluorine gas can give difluorinated products by a simple batch process, offering a potentially valuable route to the synthesis of difluoromethylene compounds that is suitable for inexpensive scale-up.
Results
2-Fluorinations of 1,3-diaryldiketone derivatives such as 1,3-diphenylpropane-1,3-dione (dibenzoylmethane, DBM, 1a) using electrophilic fluorinating reagents such as Selectfluor, NFSI, and NFOBS under a range of conditions have been described extensively [3,12,13,30,38-43]. We confirmed that reaction of compound 1a with Selectfluor in acetonitrile (MeCN) gave high yields of the monofluorinated product 2a with no difluorinated product being observed by 19F NMR analysis of the product mixture after 5 h (Scheme 1).
![[1860-5397-20-41-i1]](/bjoc/content/inline/1860-5397-20-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Monofluorination of 1,3-diphenylpropane-1,3-dione with Selectfluor.
Scheme 1: Monofluorination of 1,3-diphenylpropane-1,3-dione with Selectfluor.
In contrast, attempts to fluorinate 1a with one equivalent of fluorine gas in MeCN gave no noticeable conversion on analysis by 19F NMR spectroscopy, and a large excess of fluorine led to formation of a dark brown tar from which no useful product could be isolated. On the bases of these failed attempts, coupled with our previous experiences with the DBM scaffold [16,17,36], we used the difluorination of 1a with fluorine gas as a model process to assess how direct fluorination reactions could be achieved using reaction additives.
The lack of reactivity of 1a towards one equiv of fluorine gas when compared with strong reactivity towards Selectfluor suggested the use of a cationic, electrophilic reagent to be important. Given the structural similarity of 1,4-diazabicyclo[2.2.2]octane (DABCO) to the Selectfluor system, a 10% v/v mixture of fluorine in nitrogen was passed through a solution of 1a in acetonitrile containing one equivalent of DABCO, using a fluorination apparatus and gas flow controller equipment described previously [35]. Our aim was to form a N–F system in situ and thus mimic the successful monofluorination observed between 1a-enol and Selectfluor. After purging the product mixture with nitrogen, a known quantity of α,α,α-trifluorotoluene was added to the product mixture and the crude yields of fluorinated products were estimated by 19F NMR integration (monofluoro product 2a, δF −189.9 ppm; difluoro product 3a, δF −102.7 ppm) (Table 1, entry 3).
Table 1: Screening conditions for the fluorination of 1,3-diphenylpropane-1,3-dione (1a).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-41-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Base additive | Equiv of additive | Equiv of F2 | Crude yield by 19F NMR spectroscopya,b | ||
| 1a [%] | 2a [%] | 3a [%] | ||||
| 1 | – | – | 1 | 100 | 0 | 0 |
| 2 | – | – | 20 | polyfluorinated tar | ||
| 3 | DABCO | 1 | 1 | 32 | 4 | 20 |
| 4 | DABCO | 1 | 2 | 1 | 1 | 37 |
| 5 | DABCO | 1 | 3 | polyfluorinated tar | ||
| 6 | DABCO | 2 | 2 | many fluorinated products | ||
| 7 | DABCO | 0.1 | 1 | 22 | 28 | 8 |
| 8 | quinuclidine | 1 | 1 | 42 | 10 | 43 |
| 9 | quinuclidine | 1.2 | 1 | 54 | 1 | 43 |
| 10 | Et3N | 1 | 1 | 56 | 25 | 6 |
| 11 | Cs2CO3 | 1 | 1 | 0 | 4 | 14 |
| 12 | NaCl | 1 | 1 | 0 | 33 | 12 |
aConversion levels determined by NMR spectroscopy by comparing the integrals (CF dp at −189.9 ppm, CF2 s at −102.7 ppm) to α,α,α-trifluorotoluene standard. bThe mass balances included mixtures of soluble, unidentified products, and insoluble materials.
Using excess fluorine or DABCO (entries 5 and 6 in Table 1) led to the formation of tars, while 0.1 equiv of DABCO (entry 7) gave only relatively low conversions to 2a and 3a. Other organic nitrogen bases were tested, and we found that quinuclidine (entries 8 and 9, Table 1) gave high conversion to difluorinated product 3a, with very little monofluorinated product 2a being observed. Suspensions of caesium carbonate or sodium chloride (entries 11 and 12 in Table 1) also gave some 2a and 3a, but also unwanted tar.
This set of reactions showed that the basic species we screened all facilitated mono- and difluorination to some degree. The quinuclidine-mediated fluorination of 1a led to the highest conversion to difluorinated product 3a so we next sought to optimize this process at a preparative scale by varying the reaction stoichiometry. We found that 2.3 equiv of fluorine and 1.1 equiv of quinuclidine gave 99% conversion of 1a with 2a and 3a being the only products observed by 19F NMR spectroscopy in a 16:120 ratio (see Supporting Information File 1). To isolate the main difluorinated product 3a, the reaction vessel was purged with nitrogen and the product mixture was partitioned between water and DCM to remove HF and salt by-products. Purification of 3a by column chromatography gave 3a as a white crystalline solid in 65% isolated yield (Scheme 2) and the structure was confirmed by NMR spectroscopy and X-ray crystallography (Figure 1).
![[1860-5397-20-41-i2]](/bjoc/content/inline/1860-5397-20-41-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2,2-difluoro-1,3-diphenylpropane-1,3-dione (3a).
Scheme 2: Synthesis of 2,2-difluoro-1,3-diphenylpropane-1,3-dione (3a).
![[1860-5397-20-41-1]](/bjoc/content/figures/1860-5397-20-41-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of 2,2-difluoro-1,3-diphenylpropane-1,3-dione (3a).
Figure 1: Molecular structure of 2,2-difluoro-1,3-diphenylpropane-1,3-dione (3a).
To expand the substrate scope of this difluorination method, a range of DBM derivatives 1b–n was synthesized from para-substituted acetophenones, para-substituted benzoyl chlorides and lithium hexamethyldisilazane following a literature procedure reported by Liu and co-workers (see Supporting Information File 1) [44]. Subsequently, difluorinations of DBM substrates 1b–n were performed under conditions similar to those optimized for the preparation of 3a. The desired difluorinated products 3b–n were synthesized and isolated in good yields (Table 2).
Table 2: Difluorination of dibenzoylmethane derivatives 3a–n using fluorine gas and quinuclidine.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-41-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | 1,3-Diketone | Product | Structure | Isolated yield [%] |
| 1 | 1a | 3a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-41-i7.svg?max-width=637&scale=1.0)
|
65 |
| 2 | 1b | 3b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-41-i8.svg?max-width=637&scale=1.0)
|
41a
10a (7a) 12a (Ar–F) |
| 3 | 1c | 3c |
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-41-i9.svg?max-width=637&scale=1.0)
|
31a
16a (Ar–F) |
| 4 | 1d | 3d |
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-41-i10.svg?max-width=637&scale=1.0)
|
60 |
| 5 | 1e | 3e |
![[Graphic 7]](/bjoc/content/inline/1860-5397-20-41-i11.svg?max-width=637&scale=1.0)
|
59 |
| 6 | 1f | 3f |
![[Graphic 8]](/bjoc/content/inline/1860-5397-20-41-i12.svg?max-width=637&scale=1.0)
|
50 |
| 7 | 1g | 3g |
![[Graphic 9]](/bjoc/content/inline/1860-5397-20-41-i13.svg?max-width=637&scale=1.0)
|
72 |
| 8 | 1h | 3h |
![[Graphic 10]](/bjoc/content/inline/1860-5397-20-41-i14.svg?max-width=637&scale=1.0)
|
76 |
| 9 | 1i | 3i |
![[Graphic 11]](/bjoc/content/inline/1860-5397-20-41-i15.svg?max-width=637&scale=1.0)
|
77 |
aConversion estimated by NMR spectroscopy.
Unfortunately, substrates bearing electron-donating groups 1b (–Me) and 1c (–OMe) reacted with fluorine to give tarry materials and products arising from fluorination of both the desired enolic sites and the aryl rings. No products could be isolated from these complex mixtures and yields were estimated by 19F NMR spectroscopy.
In contrast, substrates bearing electron-withdrawing groups deactivated the aryl rings sufficiently to suppress competing ring fluorination and difluorinated products 3d–i could be isolated in high yields. Again, purification by column chromatography gave the products 3 as white crystalline solids and the structures of compounds 3f and 3i were confirmed by X-ray crystallography (Figure 2 and Supporting Information File 1). Molecules 3a, f, and i all exist in the solid state with the dicarbonyl moiety rotated to maximize the distances between the lone pairs of the electron-rich fluorine and oxygen atoms. Usually, one of the fluorine atoms lies in a syn orientation to an oxygen (e.g., 3f has an F–C–C–O dihedral angle of 15.6°) creating a dipole. This dipole appears to aid crystal packing by forming weak intermolecular interactions with an aryl ring in an adjacent molecule. The two aryl rings within the molecule are near-perpendicular to each other and this conformation leads to enhanced, orthogonal π-stacking interactions.
![[1860-5397-20-41-2]](/bjoc/content/figures/1860-5397-20-41-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal packing structure of 3f as determined by SXRC.
Figure 2: Crystal packing structure of 3f as determined by SXRC.
We next turned our attention to difluorination of related 2-ketoester substrates. Monofluorination of 2-ketoesters using fluorine gas has been scaled up to the manufacturing level [33], whereas preparative methods for the synthesis of 2,2-difluoro-3-ketoesters using fluorine gas have not been realized. Ethyl benzoylacetate (4a) was used as a model system for the development of conditions for selective difluorination using fluorine gas. After screening basic additives as mediating agents and subsequent optimization (see Supporting Information File 1), we found that reaction of ethyl benzoylacetate (4a), quinuclidine (1.5 equiv), and fluorine (3 equiv) in acetonitrile gave the desired difluorinated product 5a in 85% isolated yield. Purification of 5a was achieved very readily by eluting the reaction mixture through a small quantity of silica gel with chloroform and evaporating the residual solvent to leave the crude product which could be further purified by recrystallization. Subsequently, a range of ethyl benzoylacetate derivatives was prepared (see Supporting Information File 1) [45,46] and successfully subjected to difluorination conditions (Table 3).
![[Graphic 12]](/bjoc/content/inline/1860-5397-20-41-i16.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-20-41-i17.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-20-41-i18.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-20-41-i19.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-20-41-i20.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-20-41-i21.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-20-41-i22.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-20-41-i23.svg?max-width=637&scale=1.0)
Purification by column chromatography using the minimum amount of silica gel with chloroform as the eluent yielded 5c–g in high yields. As was observed in attempted fluorination reaction of 1c towards difluorodiketone 3c, methoxy ketoester derivative 4b gave substantial amounts of product arising from competing fluorination of the aromatic ring. Structures of difluorinated ketoesters 5a–h were confirmed by NMR spectroscopy. The 13C{1H} NMR spectra contained signals supporting the presence of ketone (e.g., δC = 185.6 ppm for 5a) and ester (δC = 161.9 ppm for 5a) functionalities. Difluoroketoester products were found to hydrate readily to give gem-diol derivatives during aqueous work-up [39], thus reducing the efficiency of extraction. Indeed, attempts to grow a single crystal of 5e from a mixture of EtOH and water led to the isolation of the corresponding gem-diol (Figure 3). There are very few examples of organic structures containing a C(OH)2–CF2–C fragment in the CCDC and only three acyclic examples (CSD 5.43 (Nov. 2021); ref codes IZICEA [47], XOPZEK and XOPZIO [48]) are known. Interestingly, in contrast to the previously described acyclic structures no OH···O(H) hydrogen bonds are present in structure 5e – the molecules are linked by OH···O(NO2) interactions.
![[1860-5397-20-41-3]](/bjoc/content/figures/1860-5397-20-41-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure and crystal packing of 5e as determined by SXRC.
Figure 3: Molecular structure and crystal packing of 5e as determined by SXRC.
Discussion
Keto–enol tautomer studies have shown that DBM 1a and related systems 1b–i exist almost entirely (ca. 90%) in their enolic forms in MeCN [17]. Our initial experiments showed 1a to be unreactive towards 1 equiv of fluorine gas, suggesting that the neutral enol group and neutral, elemental fluorine do not react to give the desired 2-fluoro-1,3-diketone 2a. Supplementation of the reaction mixture with either a tertiary amine or inorganic base led to varying mixtures of mono- and difluoro products 2a and 3a, respectively, with the tertiary amines proving most effective. Inorganic bases offer the possibility of deprotonating 1a-enol to form a more reactive enolate 1a-enolate. Nitrogen-centered bases react with fluorine gas to form N-fluoroammonium fluorides and fluoride ion [49]. Thus, on addition of tertiary amines, fluorine can react to generate basic fluoride ions and deliver reactive, electrophilic N–F species. Given that Selectfluor is sufficiently electrophilic to react with the neutral enol forms of dicarbonyls 1a–i, we believe that N-fluoroammonium ion 6 (Scheme 3) reacts with 1a–i-enol, whereas fluorine does not appear to react with neutral 1a–i-enol to give 2-fluoro products 2a–i. Conversely, fluorine could react directly with the anionic 1a–i-enolate species in parallel with N-fluoroammonium ion 6. Fluoride ions formed through the reactions between fluorine and quinuclidine or fluorine and enolate species, may deprotonate 1a–i-enol, to form enolates of 1a–i that are reactive towards both fluorine and N-fluoroammonium ion 6. The fluorination of 1a–i affords monofluoro products 2a–i in their keto tautomeric forms. For difluorodiketones 3a–i to be formed, enolization of 2a–i-keto must occur through deprotonation at the 2-position, and this process is a key limiting factor [17]. The challenge posed by enolization of 2a–i-keto may be estimated from pKa differences between acidic species and potential base species. The pKa(MeCN) for dibenzoylmethane (1a) can be estimated from pKa(DMSO) [50], where pKa(MeCN) = pKa(DMSO) + 12.9 = 13.4 + 12.9 = 26.3. Mayr and co-workers have shown the 2-fluoro-substituted species to be only slightly less acidic than their non-fluorinated homologues owing to the dominant π-donor effect of the 2-fluoro group [51,52]. On this basis, quinuclidine with pKaH(MeCN) ≈ 18.0–19.5 (estimated using pKaH(water) = 11.0 and pKaH(DMSO) = 9.8), is not predicted to be sufficiently basic to offer significant acceleration of the enolization processes of residual 1a–i-keto or, more critically, the 2-fluoro-keto intermediates 2a–i-keto that are formed after monofluorination [50,53,54]. Consequently, we believe a stronger base must be formed during the fluorination process in the presence of quinuclidine, and it is this base that accelerates enolization of 2a–i-keto to allow difluorination to occur. The fluoride ion is a relatively strong base (pKa(MeCN) of HF is ≈25 based on pKa(DMSO) [55,56]), especially when formed in situ under anhydrous conditions, where solvation of fluoride ion is not possible. Since the pKa(MeCN) of 1a-keto is ≈26.3, and we expect a pKa(MeCN) of 2a-keto to be similar in value [51,52], we suggest fluoride ion may be sufficiently basic to cause significant acceleration of the deprotonation of 2a–i-keto and allow formation of 2a–i-enolates, which then react rapidly with fluorine gas, or N-fluoroammonium ion 6, to form difluoroketones 3a–i. Quinuclidine hydrofluoride has independently been shown to be an effective form of soluble fluoride ion in a variety of carbon–fluorine bond-forming processes [57,58]. Enols are, in general, significantly more acidic than their isomeric keto forms, for example, the pKa(DMSO) of acetone is ≈26.5, whereas the pKa(DMSO) of acetone enol is ≈18.2 [59]. Thus, assuming a similar difference in pKa values between 1a-keto and 1a-enol, we expect pKa(MeCN) of 1a-enol to be ≈18. On this basis, quinuclidine with pKaH(MeCN) ≈ 18.0–19.5, could also be an effective base to facilitate the formation of 1a-enolate from 1a-enol and thus facilitate the initial monofluorination step by either fluorine or N-fluoroammonium ion 6.
![[1860-5397-20-41-i3]](/bjoc/content/inline/1860-5397-20-41-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of the quinuclidine-mediated difluorination of 1,3-dicarbonyl substrates.
Scheme 3: Proposed mechanism of the quinuclidine-mediated difluorination of 1,3-dicarbonyl substrates.
Carbonate ions are also expected to be highly basic in MeCN, however, their limited solubility is likely to inhibit their ability to act as an effective base for the formation of enolates of 1a and 2a, and this is reflected in the modest levels of formation of 3a (Scheme 4). Chloride ion, on the other hand, is less basic (pKa(MeCN) of HCl is 10.30 [60]), however, its greater solubility seemingly allows some level of deprotonation of 1a-enol to occur, where the enolate of 1a can react with fluorine to afford 2a and fluoride ion (Scheme 4). The resulting fluoride ion can then act as an additional, stronger base catalyst to facilitate further enolization processes and thus form 3a. Similar arguments are also applicable to the fluorinations of ethyl benzoylacetate derivatives 4a–g.
![[1860-5397-20-41-i4]](/bjoc/content/inline/1860-5397-20-41-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanisms of carbonate and chloride ion-mediated difluorination of 1,3-dicarbonyl substrates.
Scheme 4: Proposed mechanisms of carbonate and chloride ion-mediated difluorination of 1,3-dicarbonyl substra...
Conclusion
From our experiments, we conclude that quinuclidine is the most effective mediating agent for the difluorination of 1,3-dicarbonyl species using fluorine. Difluorinations of 1,3-diketones 1 and 1,3-ketoesters 4 were achieved by the addition of two equivalents of quinuclidine. We propose that the fluoride ion, generated in situ, deprotonates enolic forms of 1,3-dicarbonyls and accelerates the rate-limiting enolization of 2-fluoro-1,3-dicarbonyl intermediates. The resulting enolates are nucleophilic and could react with fluorine or in situ-generated N-fluoroammonium ion 7 to form 2-fluoro- and 2,2-difluoro-1,3-dicarbonyl products.
Supporting Information
Associated CDCC submission numbers: 2288841–2288848.
| Supporting Information File 1: Experimental procedures, characterization data, and copies of 1H, 19F and 13C{1H} NMR spectra. | ||
| Format: PDF | Size: 2.2 MB | Download |
Acknowledgements
We thank Dr Dmitry S. Yufit for conducting X-ray crystallographic studies. Parts of this work have already been published in Alexander Hampton’s Ph.D. thesis [61].
Data Availability Statement
All data that supports the findings of this study are available in the published article and/or the supporting information to this article.
References
-
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] [2] [3] -
de la Torre, B. G.; Albericio, F. Molecules 2019, 24, 809. doi:10.3390/molecules24040809
Return to citation in text: [1] -
Bloodworth, A. J.; Bowyer, K. J.; Mitchell, J. C. Tetrahedron Lett. 1987, 28, 5347–5350. doi:10.1016/s0040-4039(00)96726-1
Return to citation in text: [1] -
Modarai, B.; Khoshdel, E. J. Org. Chem. 1977, 42, 3527–3531. doi:10.1021/jo00442a017
Return to citation in text: [1] -
Fäh, C.; Mathys, R.; Hardegger, L. A.; Meyer, S.; Bur, D.; Diederich, F. Eur. J. Org. Chem. 2010, 4617–4629. doi:10.1002/ejoc.201000712
Return to citation in text: [1] -
Markovskij, L. N.; Pashinnik, V. E.; Kirsanov, A. V. Synthesis 1973, 787–789. doi:10.1055/s-1973-22302
Return to citation in text: [1] -
Middleton, W. J.; Bingham, E. M. J. Org. Chem. 1980, 45, 2883–2887. doi:10.1021/jo01302a025
Return to citation in text: [1] -
Singh, R. P.; Majumder, U.; Shreeve, J. M. J. Org. Chem. 2001, 66, 6263–6267. doi:10.1021/jo0157674
Return to citation in text: [1] -
Singh, R. P.; Shreeve, J. M. Org. Lett. 2001, 3, 2713–2715. doi:10.1021/ol016319l
Return to citation in text: [1] -
Banks, R. E.; Lawrence, N. J.; Popplewell, A. L. J. Chem. Soc., Chem. Commun. 1994, 343–344. doi:10.1039/c39940000343
Return to citation in text: [1] [2] -
Howard, J. L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D. L. Green Chem. 2017, 19, 2798–2802. doi:10.1039/c6gc03139k
Return to citation in text: [1] [2] -
Howard, J. L.; Sagatov, Y.; Browne, D. L. Tetrahedron 2018, 74, 3118–3123. doi:10.1016/j.tet.2017.11.066
Return to citation in text: [1] -
Differding, E.; Rüegg, G. M.; Lang, R. W. Tetrahedron Lett. 1991, 32, 1779–1782. doi:10.1016/s0040-4039(00)74328-0
Return to citation in text: [1] -
Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b
Return to citation in text: [1] [2] -
Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k
Return to citation in text: [1] [2] [3] [4] -
Iacono, C. E.; Stephens, T. C.; Rajan, T. S.; Pattison, G. J. Am. Chem. Soc. 2018, 140, 2036–2040. doi:10.1021/jacs.7b12941
Return to citation in text: [1] -
Prakash, G. K. S.; Hu, J.; Wang, Y.; Olah, G. A. Org. Lett. 2004, 6, 4315–4317. doi:10.1021/ol048166i
Return to citation in text: [1] -
Zhu, L.; Li, Y.; Ni, C.; Hu, J.; Beier, P.; Wang, Y.; Prakash, G. K. S.; Olah, G. A. J. Fluorine Chem. 2007, 128, 1241–1247. doi:10.1016/j.jfluchem.2007.05.003
Return to citation in text: [1] -
Furuta, S.; Kuroboshi, M.; Hiyama, T. Bull. Chem. Soc. Jpn. 1998, 71, 2687–2694. doi:10.1246/bcsj.71.2687
Return to citation in text: [1] -
Hallinan, E. A.; Fried, J. Tetrahedron Lett. 1984, 25, 2301–2302. doi:10.1016/s0040-4039(01)80239-2
Return to citation in text: [1] -
Curran, T. T. J. Org. Chem. 1993, 58, 6360–6363. doi:10.1021/jo00075a033
Return to citation in text: [1] -
Iseki, K.; Kuroki, Y.; Asada, D.; Kobayashi, Y. Tetrahedron Lett. 1997, 38, 1447–1448. doi:10.1016/s0040-4039(97)00044-0
Return to citation in text: [1] -
Sato, K.; Omote, M.; Ando, A.; Kumadaki, I. J. Fluorine Chem. 2004, 125, 509–515. doi:10.1016/j.jfluchem.2003.11.023
Return to citation in text: [1] -
Hiyama, T. In Organofluorine Compounds: Chemistry and Applications; Yamamoto, H., Ed.; Springer: Berlin, Heidelberg, Germany, 2000. doi:10.1007/978-3-662-04164-2
Return to citation in text: [1] -
Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612–633. doi:10.1021/cr500366b
Return to citation in text: [1] -
Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a
Return to citation in text: [1] -
Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683–730. doi:10.1021/cr400473a
Return to citation in text: [1] -
Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b
Return to citation in text: [1] [2] -
Pattison, G. Eur. J. Org. Chem. 2018, 3520–3540. doi:10.1002/ejoc.201800532
Return to citation in text: [1] -
Adler, P.; Teskey, C. J.; Kaiser, D.; Holy, M.; Sitte, H. H.; Maulide, N. Nat. Chem. 2019, 11, 329–334. doi:10.1038/s41557-019-0215-z
Return to citation in text: [1] -
Butters, M.; Ebbs, J.; Green, S. P.; MacRae, J.; Morland, M. C.; Murtiashaw, C. W.; Pettman, A. J. Org. Process Res. Dev. 2001, 5, 28–36. doi:10.1021/op0000879
Return to citation in text: [1] [2] -
Chambers, R. D.; Greenhall, M. P.; Hutchinson, J. J. Chem. Soc., Chem. Commun. 1995, 21–22. doi:10.1039/c39950000021
Return to citation in text: [1] [2] -
Harsanyi, A.; Sandford, G. Green Chem. 2015, 17, 3000–3009. doi:10.1039/c5gc00402k
Return to citation in text: [1] [2] -
Lisse, E.; Sandford, G. J. Fluorine Chem. 2018, 206, 117–124. doi:10.1016/j.jfluchem.2017.12.012
Return to citation in text: [1] [2] -
Patrick, T. B.; Scheibel, J. J.; Cantrell, G. L. J. Org. Chem. 1981, 46, 3917–3918. doi:10.1021/jo00332a034
Return to citation in text: [1] -
Stavber, G.; Zupan, M.; Jereb, M.; Stavber, S. Org. Lett. 2004, 6, 4973–4976. doi:10.1021/ol047867c
Return to citation in text: [1] -
Stavber, G.; Stavber, S. Adv. Synth. Catal. 2010, 352, 2838–2846. doi:10.1002/adsc.201000477
Return to citation in text: [1] [2] -
Stavber, G.; Zupan, M.; Stavber, S. Tetrahedron Lett. 2007, 48, 2671–2673. doi:10.1016/j.tetlet.2007.02.077
Return to citation in text: [1] -
Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730–4737. doi:10.1021/jo00120a014
Return to citation in text: [1] -
Xiao, J.-C.; Shreeve, J. M. J. Fluorine Chem. 2005, 126, 473–476. doi:10.1016/j.jfluchem.2004.10.043
Return to citation in text: [1] -
Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023
Return to citation in text: [1] -
Yang, N.-Y.; Li, Z.-L.; Ye, L.; Tan, B.; Liu, X.-Y. Chem. Commun. 2016, 52, 9052–9055. doi:10.1039/c6cc00364h
Return to citation in text: [1] -
Clay, R. J.; Collom, T. A.; Karrick, G. L.; Wemple, J. Synthesis 1993, 290–292. doi:10.1055/s-1993-25849
Return to citation in text: [1] -
Venkat Ragavan, R.; Vijayakumar, V.; Rajesh, K.; Palakshi Reddy, B.; Karthikeyan, S.; Suchetha Kumari, N. Bioorg. Med. Chem. Lett. 2012, 22, 4193–4197. doi:10.1016/j.bmcl.2012.04.008
Return to citation in text: [1] -
Han, C.; Kim, E. H.; Colby, D. A. J. Am. Chem. Soc. 2011, 133, 5802–5805. doi:10.1021/ja202213f
Return to citation in text: [1] -
Khatri, H. R.; Han, C.; Luong, E.; Pan, X.; Adam, A. T.; Alshammari, M. D.; Shao, Y.; Colby, D. A. J. Org. Chem. 2019, 84, 11665–11675. doi:10.1021/acs.joc.9b01595
Return to citation in text: [1] -
Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595
Return to citation in text: [1] -
Olmstead, W. N.; Bordwell, F. G. J. Org. Chem. 1980, 45, 3299–3305. doi:10.1021/jo01304a033
Return to citation in text: [1] [2] -
Zhang, Z.; Puente, Á.; Wang, F.; Rahm, M.; Mei, Y.; Mayr, H.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2016, 55, 12845–12849. doi:10.1002/anie.201605616
Return to citation in text: [1] [2] -
Zhang, Z.; Puente, Á.; Wang, F.; Rahm, M.; Mei, Y.; Mayr, H.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2016, 55, 14494. doi:10.1002/anie.201609842
Return to citation in text: [1] [2] -
Coetzee, J. F.; Padmanabhan, G. R. J. Am. Chem. Soc. 1965, 87, 5005–5010. doi:10.1021/ja00950a006
Return to citation in text: [1] -
Cox, B. G. Acids and Bases: Solvent Effects on Acid-Base Strength; Oxford University Press: Oxford, UK, 2013. doi:10.1093/acprof:oso/9780199670512.001.0001
Return to citation in text: [1] -
Bordwell, F. G. Acc. Chem. Res. 1988, 21, 456–463. doi:10.1021/ar00156a004
Return to citation in text: [1] -
Ashworth, I. W.; Frodsham, L.; Moore, P.; Ronson, T. O. J. Org. Chem. 2022, 87, 2111–2119. doi:10.1021/acs.joc.1c01768
Return to citation in text: [1] -
Chambers, R. D.; Holmes, T. F.; Korn, S. R.; Sandford, G. J. Chem. Soc., Chem. Commun. 1993, 855–856. doi:10.1039/c39930000855
Return to citation in text: [1] -
Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. doi:10.1021/ja508369z
Return to citation in text: [1] -
Bordwell, F. G.; Zhang, S.; Eventova, I.; Rappoport, Z. J. Org. Chem. 1997, 62, 5371–5373. doi:10.1021/jo970404i
Return to citation in text: [1] -
Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kütt, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; Pihl, V.; Koppel, I. A.; Leito, I. J. Phys. Org. Chem. 2013, 26, 162–170. doi:10.1002/poc.2946
Return to citation in text: [1] -
Hampton, A. S. Fluorine Gas as a Selective Difluorinating Reagent. Ph.D. Thesis, Durham University, Durham, U.K., 2020.
Return to citation in text: [1]
| 49. | Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595 |
| 17. | Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k |
| 50. | Olmstead, W. N.; Bordwell, F. G. J. Org. Chem. 1980, 45, 3299–3305. doi:10.1021/jo01304a033 |
| 1. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 2. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 3. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 7. | Fäh, C.; Mathys, R.; Hardegger, L. A.; Meyer, S.; Bur, D.; Diederich, F. Eur. J. Org. Chem. 2010, 4617–4629. doi:10.1002/ejoc.201000712 |
| 8. | Markovskij, L. N.; Pashinnik, V. E.; Kirsanov, A. V. Synthesis 1973, 787–789. doi:10.1055/s-1973-22302 |
| 34. | Chambers, R. D.; Greenhall, M. P.; Hutchinson, J. J. Chem. Soc., Chem. Commun. 1995, 21–22. doi:10.1039/c39950000021 |
| 35. | Harsanyi, A.; Sandford, G. Green Chem. 2015, 17, 3000–3009. doi:10.1039/c5gc00402k |
| 36. | Lisse, E.; Sandford, G. J. Fluorine Chem. 2018, 206, 117–124. doi:10.1016/j.jfluchem.2017.12.012 |
| 60. | Raamat, E.; Kaupmees, K.; Ovsjannikov, G.; Trummal, A.; Kütt, A.; Saame, J.; Koppel, I.; Kaljurand, I.; Lipping, L.; Rodima, T.; Pihl, V.; Koppel, I. A.; Leito, I. J. Phys. Org. Chem. 2013, 26, 162–170. doi:10.1002/poc.2946 |
| 6. | Modarai, B.; Khoshdel, E. J. Org. Chem. 1977, 42, 3527–3531. doi:10.1021/jo00442a017 |
| 34. | Chambers, R. D.; Greenhall, M. P.; Hutchinson, J. J. Chem. Soc., Chem. Commun. 1995, 21–22. doi:10.1039/c39950000021 |
| 61. | Hampton, A. S. Fluorine Gas as a Selective Difluorinating Reagent. Ph.D. Thesis, Durham University, Durham, U.K., 2020. |
| 5. | Bloodworth, A. J.; Bowyer, K. J.; Mitchell, J. C. Tetrahedron Lett. 1987, 28, 5347–5350. doi:10.1016/s0040-4039(00)96726-1 |
| 3. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 26. | Hiyama, T. In Organofluorine Compounds: Chemistry and Applications; Yamamoto, H., Ed.; Springer: Berlin, Heidelberg, Germany, 2000. doi:10.1007/978-3-662-04164-2 |
| 27. | Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612–633. doi:10.1021/cr500366b |
| 28. | Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a |
| 29. | Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683–730. doi:10.1021/cr400473a |
| 30. | Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b |
| 31. | Pattison, G. Eur. J. Org. Chem. 2018, 3520–3540. doi:10.1002/ejoc.201800532 |
| 32. | Adler, P.; Teskey, C. J.; Kaiser, D.; Holy, M.; Sitte, H. H.; Maulide, N. Nat. Chem. 2019, 11, 329–334. doi:10.1038/s41557-019-0215-z |
| 57. | Chambers, R. D.; Holmes, T. F.; Korn, S. R.; Sandford, G. J. Chem. Soc., Chem. Commun. 1993, 855–856. doi:10.1039/c39930000855 |
| 58. | Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. doi:10.1021/ja508369z |
| 4. | de la Torre, B. G.; Albericio, F. Molecules 2019, 24, 809. doi:10.3390/molecules24040809 |
| 33. | Butters, M.; Ebbs, J.; Green, S. P.; MacRae, J.; Morland, M. C.; Murtiashaw, C. W.; Pettman, A. J. Org. Process Res. Dev. 2001, 5, 28–36. doi:10.1021/op0000879 |
| 59. | Bordwell, F. G.; Zhang, S.; Eventova, I.; Rappoport, Z. J. Org. Chem. 1997, 62, 5371–5373. doi:10.1021/jo970404i |
| 15. | Differding, E.; Rüegg, G. M.; Lang, R. W. Tetrahedron Lett. 1991, 32, 1779–1782. doi:10.1016/s0040-4039(00)74328-0 |
| 18. | Iacono, C. E.; Stephens, T. C.; Rajan, T. S.; Pattison, G. J. Am. Chem. Soc. 2018, 140, 2036–2040. doi:10.1021/jacs.7b12941 |
| 55. | Bordwell, F. G. Acc. Chem. Res. 1988, 21, 456–463. doi:10.1021/ar00156a004 |
| 56. | Ashworth, I. W.; Frodsham, L.; Moore, P.; Ronson, T. O. J. Org. Chem. 2022, 87, 2111–2119. doi:10.1021/acs.joc.1c01768 |
| 13. | Howard, J. L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D. L. Green Chem. 2017, 19, 2798–2802. doi:10.1039/c6gc03139k |
| 14. | Howard, J. L.; Sagatov, Y.; Browne, D. L. Tetrahedron 2018, 74, 3118–3123. doi:10.1016/j.tet.2017.11.066 |
| 19. | Prakash, G. K. S.; Hu, J.; Wang, Y.; Olah, G. A. Org. Lett. 2004, 6, 4315–4317. doi:10.1021/ol048166i |
| 20. | Zhu, L.; Li, Y.; Ni, C.; Hu, J.; Beier, P.; Wang, Y.; Prakash, G. K. S.; Olah, G. A. J. Fluorine Chem. 2007, 128, 1241–1247. doi:10.1016/j.jfluchem.2007.05.003 |
| 21. | Furuta, S.; Kuroboshi, M.; Hiyama, T. Bull. Chem. Soc. Jpn. 1998, 71, 2687–2694. doi:10.1246/bcsj.71.2687 |
| 22. | Hallinan, E. A.; Fried, J. Tetrahedron Lett. 1984, 25, 2301–2302. doi:10.1016/s0040-4039(01)80239-2 |
| 23. | Curran, T. T. J. Org. Chem. 1993, 58, 6360–6363. doi:10.1021/jo00075a033 |
| 24. | Iseki, K.; Kuroki, Y.; Asada, D.; Kobayashi, Y. Tetrahedron Lett. 1997, 38, 1447–1448. doi:10.1016/s0040-4039(97)00044-0 |
| 25. | Sato, K.; Omote, M.; Ando, A.; Kumadaki, I. J. Fluorine Chem. 2004, 125, 509–515. doi:10.1016/j.jfluchem.2003.11.023 |
| 51. | Zhang, Z.; Puente, Á.; Wang, F.; Rahm, M.; Mei, Y.; Mayr, H.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2016, 55, 12845–12849. doi:10.1002/anie.201605616 |
| 52. | Zhang, Z.; Puente, Á.; Wang, F.; Rahm, M.; Mei, Y.; Mayr, H.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2016, 55, 14494. doi:10.1002/anie.201609842 |
| 12. | Banks, R. E.; Lawrence, N. J.; Popplewell, A. L. J. Chem. Soc., Chem. Commun. 1994, 343–344. doi:10.1039/c39940000343 |
| 51. | Zhang, Z.; Puente, Á.; Wang, F.; Rahm, M.; Mei, Y.; Mayr, H.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2016, 55, 12845–12849. doi:10.1002/anie.201605616 |
| 52. | Zhang, Z.; Puente, Á.; Wang, F.; Rahm, M.; Mei, Y.; Mayr, H.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2016, 55, 14494. doi:10.1002/anie.201609842 |
| 9. | Middleton, W. J.; Bingham, E. M. J. Org. Chem. 1980, 45, 2883–2887. doi:10.1021/jo01302a025 |
| 10. | Singh, R. P.; Majumder, U.; Shreeve, J. M. J. Org. Chem. 2001, 66, 6263–6267. doi:10.1021/jo0157674 |
| 11. | Singh, R. P.; Shreeve, J. M. Org. Lett. 2001, 3, 2713–2715. doi:10.1021/ol016319l |
| 16. | Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b |
| 17. | Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k |
| 50. | Olmstead, W. N.; Bordwell, F. G. J. Org. Chem. 1980, 45, 3299–3305. doi:10.1021/jo01304a033 |
| 53. | Coetzee, J. F.; Padmanabhan, G. R. J. Am. Chem. Soc. 1965, 87, 5005–5010. doi:10.1021/ja00950a006 |
| 54. | Cox, B. G. Acids and Bases: Solvent Effects on Acid-Base Strength; Oxford University Press: Oxford, UK, 2013. doi:10.1093/acprof:oso/9780199670512.001.0001 |
| 16. | Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b |
| 17. | Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k |
| 36. | Lisse, E.; Sandford, G. J. Fluorine Chem. 2018, 206, 117–124. doi:10.1016/j.jfluchem.2017.12.012 |
| 37. | Patrick, T. B.; Scheibel, J. J.; Cantrell, G. L. J. Org. Chem. 1981, 46, 3917–3918. doi:10.1021/jo00332a034 |
| 3. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 12. | Banks, R. E.; Lawrence, N. J.; Popplewell, A. L. J. Chem. Soc., Chem. Commun. 1994, 343–344. doi:10.1039/c39940000343 |
| 13. | Howard, J. L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D. L. Green Chem. 2017, 19, 2798–2802. doi:10.1039/c6gc03139k |
| 30. | Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b |
| 38. | Stavber, G.; Zupan, M.; Jereb, M.; Stavber, S. Org. Lett. 2004, 6, 4973–4976. doi:10.1021/ol047867c |
| 39. | Stavber, G.; Stavber, S. Adv. Synth. Catal. 2010, 352, 2838–2846. doi:10.1002/adsc.201000477 |
| 40. | Stavber, G.; Zupan, M.; Stavber, S. Tetrahedron Lett. 2007, 48, 2671–2673. doi:10.1016/j.tetlet.2007.02.077 |
| 41. | Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730–4737. doi:10.1021/jo00120a014 |
| 42. | Xiao, J.-C.; Shreeve, J. M. J. Fluorine Chem. 2005, 126, 473–476. doi:10.1016/j.jfluchem.2004.10.043 |
| 43. | Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023 |
| 48. | Khatri, H. R.; Han, C.; Luong, E.; Pan, X.; Adam, A. T.; Alshammari, M. D.; Shao, Y.; Colby, D. A. J. Org. Chem. 2019, 84, 11665–11675. doi:10.1021/acs.joc.9b01595 |
| 17. | Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k |
| 39. | Stavber, G.; Stavber, S. Adv. Synth. Catal. 2010, 352, 2838–2846. doi:10.1002/adsc.201000477 |
| 47. | Han, C.; Kim, E. H.; Colby, D. A. J. Am. Chem. Soc. 2011, 133, 5802–5805. doi:10.1021/ja202213f |
| 33. | Butters, M.; Ebbs, J.; Green, S. P.; MacRae, J.; Morland, M. C.; Murtiashaw, C. W.; Pettman, A. J. Org. Process Res. Dev. 2001, 5, 28–36. doi:10.1021/op0000879 |
| 45. | Clay, R. J.; Collom, T. A.; Karrick, G. L.; Wemple, J. Synthesis 1993, 290–292. doi:10.1055/s-1993-25849 |
| 46. | Venkat Ragavan, R.; Vijayakumar, V.; Rajesh, K.; Palakshi Reddy, B.; Karthikeyan, S.; Suchetha Kumari, N. Bioorg. Med. Chem. Lett. 2012, 22, 4193–4197. doi:10.1016/j.bmcl.2012.04.008 |
| 35. | Harsanyi, A.; Sandford, G. Green Chem. 2015, 17, 3000–3009. doi:10.1039/c5gc00402k |
| 44. | Yang, N.-Y.; Li, Z.-L.; Ye, L.; Tan, B.; Liu, X.-Y. Chem. Commun. 2016, 52, 9052–9055. doi:10.1039/c6cc00364h |
© 2024 Hampton et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.