Abstract
An efficient synthesis of a series of γ-ketoallylphosphonates through a direct conversion of both primary and secondary Morita–Baylis–Hillman (MBH) alcohols by trialkyl phosphites with or without DMAP, used as additive, and under solvent-free conditions, is described herein for the first time. Subsequently, a highly regioselective Luche reduction of the primary phosphonate 2a (R = H) gave the corresponding γ-hydroxyallylphosphonate 5 that further reacted with tosylamines in the presence of diiodine (15 mol %) as a catalyst, affording the corresponding SN2-type products 6a–d in 63 to 70% isolated yields. Alternatively, the alcohol 5 produced the corresponding acetate 7 which, mediated by Ce(III), was successfully converted into the corresponding γ-aminoallylphosphonates 8a–d.

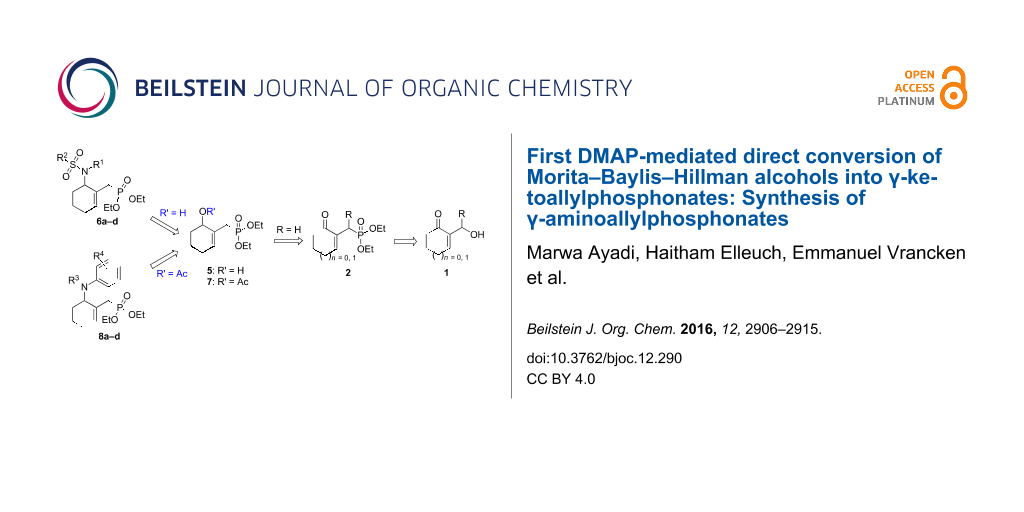
Graphical Abstract
Introduction
Phosphonates and their derivatives are an important class of substances that have a wide range of applications in numerous areas such as medicinal [1-3] and agricultural chemistry [4,5]. Among them, multifunctional derivatives have been exploited as valuable building blocks in natural product syntheses, e.g., calyculins A and B as potent serine-threonine protein phosphatase inhibitors [6], as well as ligands in enantioselective reactions [7]. Furthermore, allylphosphonates are important bioactive compounds that exhibit interesting antimicrobial and antimalarial properties [8,9], as well as useful substrates for the synthesis of valuable organic compounds [10-12].
Historically, the Michaelis−Arbuzov rearrangement [13] is the most widely and generally high yielding strategy for phosphonate synthesis. The current three step protocol involves firstly the mesylation of corresponding alcohols and then the conversion of the intermediate mesylates into their halides. Further an Arbuzov reaction of alkyl phosphites with such halides affords the phosphonates.
Interestingly, a direct conversion of common allyl alcohols into the corresponding phosphonates, with or without catalyst, was previously reported. Accordingly, Bodalski and co-workers [14] and then Swamy’s research group [15] described a direct route to allylphosphonates by treatment of Morita–Baylis–Hillman alcohols with chlorophosphites without any additive, followed by thermal Arbuzov rearrangement of the intermediate allyl phosphites (Scheme 1, reaction 1).
![[1860-5397-12-290-i1]](/bjoc/content/inline/1860-5397-12-290-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of allylphosphonates from acyclic MBH adducts.
Scheme 1: Synthesis of allylphosphonates from acyclic MBH adducts.
Recently, an efficient protocol for the conversion of common allyl and benzyl alcohols into the corresponding phosphonates through their treatment with triethyl phosphite and ZnI2, was described [16]. Similarly, Das and co-workers [17] have directly converted acyclic Morita–Baylis–Hillman (MBH) alcohols into the corresponding allylphosphonates upon their treatment with trialkyl phosphite in the presence of FeCl3 (Scheme 1, reaction 2).
On the other hand, treatment of MBH acetates with triethyl phosphite, without any additive, provided, after thermal Arbuzov rearrangement, a variety of diethyl allylphosphonates (Scheme 1, reaction 3) [18]. Moreover, the asymmetric allylic substitution of MBH carbonates with diphenyl phosphonate using chiral thiourea phosphite as catalyst, afforded the related allylphosphonates (Scheme 1, reaction 4) [19].
We have previously reported a direct nucleophilic allylic substitution of cyclic MBH alcohols by β-dicarbonyl compounds [20]. In addition, in our recent work [21], we have described an efficient protocol for the synthesis of a new series of allylphosphonates in high yields and selectivity, using cyclic MBH acetates as starting materials, in the presence of DMAP or imidazole as additives (Scheme 2, reaction 6).
![[1860-5397-12-290-i2]](/bjoc/content/inline/1860-5397-12-290-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of γ-ketoallylphosphonates from cyclic MBH adducts.
Scheme 2: Synthesis of γ-ketoallylphosphonates from cyclic MBH adducts.
Moreover, it has been demonstrated that the presence of a nitrogen atom and a phosphonate moiety in multifunctional compounds may improve their synthetic and biological potentials. Aminophosphonates and their derivatives [22-26] are thus recognized as promising compounds and a new class of drugs that are widely used in a variety of commercial applications. Indeed, they are known to influence various biochemical processes in plants, modifying or inhibiting them, or to have biological activities as for example antibiotics, antibacterial, anti-cancer or antithrombotic agents [22-26].
In this context, α-aminophosphonates have particularly attracted considerable attention owing to their biological activities [27-33] since they are considered as important surrogates for α-amino carboxylic acids, peptide mimics as well as versatile intermediates for the design of potential anticancer agents.
β-Aminophosphonates present also an important place among the various compounds containing both a P–C bond and an amino group. Indeed, they are known for their important role in medicinal chemistry [34-36] as anti-HIV agents, enzyme inhibitors and antibacterial agents. In addition, α- and β-aminophosphonates have been widely described in literature. However, γ-aminophosphonates have received much less attention. These derivatives were originally isolated from microorganisms [37,38], e.g., the fosmidomycin [39] is as an antimalarial drug that was isolated from broths of bacteria of the genus Streptomyces [40]. The γ-aminophosphonates were prepared through numerous synthetic methods [41-45]. For instance, they were prepared by reductive amination of γ-aminophosphonyl ketones using sodium borohydride [41], or by conjugate addition of diethyl methylphosphonite to 2-cyclohexenone followed by Bucherer–Bergs amino acid synthesis [42]. Another synthetic approach for a series of α-fluorinated-γ-aminophosphonates has been reported through a palladium-catalyzed hydrogenation of α-fluorovinylphosphonates [43].
In continuation of our interest [21] in the construction of carbon–phosphorus bonds using both cyclic and acyclic MBH adducts, we report herein, for the first time, a direct and facile access to γ-ketoallylphosphonates from primary and secondary MBH alcohols, with or without DMAP used as additive, and under solvent-free conditions (Scheme 1, reaction 5 and Scheme 2, reaction 7). Taking into consideration the importance of aminophosphonates (vide infra), we developed in the second part of this study an efficient conversion of these γ-ketoallylphosphonates into related γ-aminoallylphosphonates.
Results and Discussion
In our first attempt, a mixture of cyclic MBH alcohol 1a and triethyl phosphite was reacted in toluene without any additive. After stirring the reaction mixture at 110 °C for 72 h, the starting materials were completely recovered (Table 1, entry 1).
Table 1: Optimization of the reaction conditions for the γ-ketophosphonate 2a from 1aa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-290-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry |
DMAP
(equiv) |
Solvent | T (°C) | t (h) | Yield 2a (%)b |
|---|---|---|---|---|---|
| 1 | 0 | toluene | 110 | 72 | nonec |
| 2 | 0.5 | toluene | 110 | 72 | 30 |
| 3 | 1 | toluene | 110 | 120 | 45 |
| 4 | 1 | THF | 66 | 120 | trace |
| 5 | 0 | none | rt/80 | 8 | nonec |
| 6 | 0.5 | none | 80 | 4 | 35 |
| 7 | 1 | none | 80 | 1 | 75 |
aReaction conditions: MBH adduct 1a (1 mmol), triethyl phosphite (2 mmol), DMAP (0–1 equiv) and solvent (5 mL) at the indicated temperature. bYields of isolated pure compounds after column chromatography. cNo reaction detected based on TLC and 1H NMR analysis.
Under the previous conditions and in the presence of 0.5 equiv of DMAP, a sluggish reaction occurred, affording the desired phosphonate 2a within 72 h in a 30% yield (Table 1, entry 2). Using 1 equiv of DMAP in refluxing toluene required a longer reaction time of 120 h to completely convert the allyl alcohol 1a into the phosphonate 2a in 45% yield (Table 1, entry 3). The nucleophilic substitution of alcohol 1a with triethyl phosphite in THF at reflux was significantly less effective (Table 1, entry 4). The TLC and 1H NMR analyses of the crude reaction mixture conducted without any additive and under solvent-free conditions, at room temperature or at 80 °C, indicated that no reaction occurred and the starting materials were completely recovered (Table 1, entry 5).
However, when 0.5 equiv of DMAP was employed under the previous conditions, the phosphonate 2a was isolated within 4 h in 35% yield (Table 1, entry 6). After screening several amounts of DMAP, under solvent-free conditions at 80 °C, we observed that the best yield (75%) was obtained using 1 equiv of DMAP (Table 1, entry 7). Under these reaction conditions, the allylic phosphonate 2a was produced as the sole product within 1 h and its structure was confirmed by 1H, 13C and 31P NMR analysis [21].
A series of γ-ketoallylphosphonates 2a–f was prepared from different cyclic MBH alcohols and triethyl phosphite using the optimized reaction conditions (Table 2).
Table 2: Direct conversion of cyclic MBH alcohols 1a–f into γ-ketoallylphosphonates 2a–fa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-290-i9.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-290-i10.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-290-i11.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-290-i12.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-290-i13.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-290-i14.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-290-i15.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-290-i16.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-290-i17.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-290-i18.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-290-i19.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-290-i20.svg?max-width=637&scale=1.0)
aReaction conditions: MBH alcohols (1 mmol), trialkyl/triaryl phosphite (2 mmol) and DMAP (1 equiv) at 80 °C. bYields of isolated pure compounds after column chromatography.
These adducts were obtained from primary (1a and 1f) or secondary (1b–e) MBH alcohols (R = linear/branched alkyl or aryl). The total conversion of alcohols 1a–f into phosphonates 2a–f was complete within 1–4 h in 58–84% yields. Our results suggested that this reaction worked well with six-membered cyclic MBH alcohols 1a–e (Table 2, entries 1–5), as well as five-membered cyclic MBH alcohol 1f (Table 2, entry 6).
A putative reaction pathway could start from a first β-conjugate addition of DMAP onto the allylic alcohol 1a (i), followed by the elimination of the hydroxide ion that would afford the intermediate I (ii). Similarly, a second β’-conjugate addition of triethyl phosphite onto I (iii), followed by the release of DMAP would provide the phosphonium intermediate II (iv). Finally, the hydroxide ion is expected to react with II via an Arbuzov rearrangement to provide the desired γ-ketoallylphosphonate 2a (v) (Scheme 3).
![[1860-5397-12-290-i3]](/bjoc/content/inline/1860-5397-12-290-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for DMAP-mediated direct nucleophilic α-substitution of MBH alcohol 1a.
Scheme 3: Proposed mechanism for DMAP-mediated direct nucleophilic α-substitution of MBH alcohol 1a.
We next investigated the scope and the limitations of this synthetic method. We have thus examined, the behavior of the acyclic MBH alcohol 3a [46,47] towards triethyl phosphite under the above conditions (1 equiv of DMAP, solvent-free conditions, 80 °C, Scheme 4).
![[1860-5397-12-290-i4]](/bjoc/content/inline/1860-5397-12-290-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Direct conversion of acyclic MBH alcohols 3a–c into γ-ketoallylphosphonates 4a–f.
Scheme 4: Direct conversion of acyclic MBH alcohols 3a–c into γ-ketoallylphosphonates 4a–f.
We were pleased to note that the nucleophilic substitution reaction worked also well and gave the allylphosphonate 4a in 90% yield within 1 h (Table 3, entry 1). More interestingly, repeating this reaction without DMAP and under solvent-free conditions at 80 °C gave, in only 30 min, the phosphonate 4a in 92% yield (Table 3, entry 2). Encouraged by these results, the behavior of a variety of acyclic MBH alcohols 3a–c towards trialkyl/triaryl phosphites was examined to determine the substrate scope of the present procedure (Table 3).
Table 3: Direct conversion of acyclic MBH alcohols 3a–c into γ-ketoallylphosphonates 4a–fa.
| Entry | MBH alcohol | Phosphite reagent/DMAP (equiv) | γ- Ketoallylphosphonate | t (min) | Yield (%)b |
Ratio
Z/Ec |
|---|---|---|---|---|---|---|
| 1 | 3a | P(OEt)3/1 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-290-i21.svg?max-width=637&scale=1.0)
4a [18-48] |
60 | 90 | N/Ad |
| 2 | 3a | P(OEt)3/0 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-290-i22.svg?max-width=637&scale=1.0)
4a [18-48] |
30 | 92 | N/Ad |
| 3 | 3a | P(OMe)3/0 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-290-i23.svg?max-width=637&scale=1.0)
4b [18-48] |
30 | 60 | N/Ad |
| 4 | 3a | P(OPh)3/0 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-290-i24.svg?max-width=637&scale=1.0)
4c |
30 | 62 | N/Ad |
| 5 | 3b | P(OEt)3/0 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-290-i25.svg?max-width=637&scale=1.0)
4d [18-48] |
30 | 92 | 87:13 |
| 6 | 3b | P(OMe)3/0 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-290-i26.svg?max-width=637&scale=1.0)
4e [18-48] |
30 | 56 | 94:06 |
| 7 | 3c | P(OEt)3/0 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-290-i27.svg?max-width=637&scale=1.0)
4f [18-48] |
30 | 82 | 100:0 |
aReaction conditions: MBH alcohols (1 mmol), trialkyl/triaryl phosphite (2 mmol) at 80 °C. bYields of isolated pure compounds after column chromatography. cEstimated from the integrated intensities of the 31P NMR signal of the crude products. dMeans not applicable.
As shown in entries 3 and 4 (Table 3), the substitution of the primary alcohol 3a with a trialkyl/triaryl phosphites gave the corresponding phosphonates 4b and 4c in 60–62% yields.
The reaction of secondary alcohols 3b and 3c with trialkyl phosphites was also surveyed. Under the previous reaction conditions (solvent-free, 80 °C, without any additive) an SN2’-type reaction followed by an Arbuzov rearrangement gave the corresponding primary allylphosphonates 4d–f with a high Z-stereoselectivity (Z/E = 87–100/13–0) and in 56–92% yields (Table 2, entries 5–7). It seems that the Z/E ratios depend on the nature of the R allylic group. Indeed, when it is an alkyl group, i.e., R1 = Me, the phosphonates 4d and 4e were obtained in high Z-diastereoselectivities (de = 74–88%), whereas the phosphonate 4f was provided with an excellent Z-diastereoselectivity (de = 100%) when R1 = Ph. In this work, the observed high Z-diastereoselectities are in good agreement with a previous report by Basavaiah and co-workers on the nucleophilic addition of triethyl phosphite onto Morita–Baylis–Hillman acetates [18-48].
We envisaged in the second part of this study a further functionalization of these allylphosphonates as the literature survey revealed that the aminophosphonates are useful molecules in organic synthesis and in biology (vide supra).
The synthesis of γ-tosylaminophosphonates was achieved in a two-step sequence. First, the γ-hydroxyphosphonate 5 was prepared and isolated in a good yield (88%) via a highly selective Luche reduction [49,50] of the γ-ketophosphonate 2a using NaBH4 in the presence of CeCl3·6H2O in methanol at 0 °C. The TLC of the reaction mixture and analysis of 1H NMR and 31P NMR spectra showed the exclusive formation of the 1,2-adduct 5 (Scheme 5). The literature survey revealed that γ-hydroxyphosphonates may display interesting biological activities [51,52].
![[1860-5397-12-290-i5]](/bjoc/content/inline/1860-5397-12-290-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: I2-Catalyzed direct synthesis of γ-tosylaminophosphonates 6 from alcohol 5.
Scheme 5: I2-Catalyzed direct synthesis of γ-tosylaminophosphonates 6 from alcohol 5.
The next step was to realize the nucleophilic substitution of the γ-hydroxyphosphonate 5a with a variety of differently substituted tosylamines catalyzed by 15 mol % of iodine [53] in refluxing methylene chloride. Under these optimized conditions, the γ-tosylaminophosphonates 6a–d were obtained in good yields (63–70%) (Table 4, entries 1–4).
Table 4: Synthesis of γ-tosylaminophosphonates 6a–d from alcohol 5.
| Entry | Tosylamine | γ-Tosylaminophosphonate | t (h) | Yield (%)a |
|---|---|---|---|---|
| 1 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-290-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-290-i29.svg?max-width=637&scale=1.0)
6a |
25 | 69 |
| 2 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-290-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-290-i31.svg?max-width=637&scale=1.0)
6b |
12 | 63 |
| 3 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-290-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-12-290-i33.svg?max-width=637&scale=1.0)
6c |
20 | 68 |
| 4 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-12-290-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-12-290-i35.svg?max-width=637&scale=1.0)
6d |
4 | 70 |
aYields of isolated pure compounds after column chromatography.
Scheme 6 illustrates a possible mechanism for the I2-catalyzed substitution reaction of γ-hydroxyallylphosphonate 5 with tosylamines. This could first involve an activation of the hydroxy moiety of adduct 5 through a coordination of the iodine catalyst with the hydroxy group to give an iodine-coordinated intermediate I that would subsequently undergo a nucleophilic attack by the tosylamine. The release of diiodine and elimination of a molecule of water is then expected to afford the desired γ-tosylaminoallylphosphonates 6.
![[1860-5397-12-290-i6]](/bjoc/content/inline/1860-5397-12-290-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed mechanism for I2-catalyzed direct nucleophilic substitution of γ-hydroxyallylphosphonate 5 with tosylamines.
Scheme 6: Proposed mechanism for I2-catalyzed direct nucleophilic substitution of γ-hydroxyallylphosphonate 5...
However, this reaction failed using primary, secondary amines or aniline as nucleophiles. Therefore, an alternative synthesis of γ-aminophosphonates 8 has been developed. Following our previous report [54], we have first converted the MBH alcohol 5 into its acetate 7 in 90% yields using a mixture of Ac2O and DMAP as reagents (Scheme 7).
![[1860-5397-12-290-i7]](/bjoc/content/inline/1860-5397-12-290-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Ce(III)-mediated conversion of acetate 7 into γ-aminophosphonates 8a–d.
Scheme 7: Ce(III)-mediated conversion of acetate 7 into γ-aminophosphonates 8a–d.
The MBH acetate 7 was further converted into the γ-aminophosphonates 8a–c within short reactions times of 1–2 h and in 60–81% isolated yields using anilines, and 1 equiv of cerium(III) chloride hexahydrate in refluxing toluene (Table 5, entries 1–3). The nucleophilic allylic substitution of the acetate 7 worked also with β-naphthylamine and gave the corresponding γ-aminophosphonate 8d in only a modest yield (Table 5, entry 4).
![[Graphic 29]](/bjoc/content/inline/1860-5397-12-290-i36.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-12-290-i37.svg?max-width=637&scale=1.0)
![[Graphic 31]](/bjoc/content/inline/1860-5397-12-290-i38.svg?max-width=637&scale=1.0)
![[Graphic 32]](/bjoc/content/inline/1860-5397-12-290-i39.svg?max-width=637&scale=1.0)
![[Graphic 33]](/bjoc/content/inline/1860-5397-12-290-i40.svg?max-width=637&scale=1.0)
![[Graphic 34]](/bjoc/content/inline/1860-5397-12-290-i41.svg?max-width=637&scale=1.0)
![[Graphic 35]](/bjoc/content/inline/1860-5397-12-290-i42.svg?max-width=637&scale=1.0)
![[Graphic 36]](/bjoc/content/inline/1860-5397-12-290-i43.svg?max-width=637&scale=1.0)
Conclusion
We have developed a mild, direct and convenient procedure for the conversion of both cyclic and acyclic MBH alcohols, with trialkyl and triaryl phosphites, into γ-ketoallylphosphonates under solvent-free conditions, in the presence or absence of the promoter DMAP. The corresponding products have been further involved in two alternative efficient synthetic routes for γ-amino- and γ-tosylaminophosphonates that were readily obtained in high yields and selectivities.
References
-
Zhang, N.; Casida, J. E. Bioorg. Med. Chem. 2002, 10, 1281–1290. doi:10.1016/S0968-0896(01)00391-1
Return to citation in text: [1] -
Wu, D.; Niu, J.-Q.; Ding, Y.-H.; Wu, X.-Y.; Zhong, B.-H.; Feng, X.-W. Med. Chem. Res. 2012, 21, 1179–1187. doi:10.1007/s00044-011-9616-2
Return to citation in text: [1] -
Romanenko, V. D.; Kukhar, V. P. Beilstein J. Org. Chem. 2013, 9, 991–1001. doi:10.3762/bjoc.9.114
Return to citation in text: [1] -
Nandula, V. K. Glyphosphate Resistance in Corps and Weeds: History, Development, and Management; John Wiley & Sons, Inc., 2010. doi:10.1002/9780470634394.index
Return to citation in text: [1] -
Jackson, E. R.; Dowd, C. S. Curr. Top. Med. Chem. 2012, 12, 706–728. doi:10.2174/156802612799984599
Return to citation in text: [1] -
Touré, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439–4486. doi:10.1021/cr800296p
Return to citation in text: [1] -
Albrecht, L.; Albrecht, A.; Krawczyk, H.; Jørgensen, K. A. Chem. – Eur. J. 2010, 16, 28–48. doi:10.1002/chem.200902634
Return to citation in text: [1] -
Kerst, A. F.; Douros, J. D., Jr. Diethyl allyl phosphonates as an antimicrobial agent. U. S. Patent 3783160, 1974.
Return to citation in text: [1] -
Ettari, R.; Nizi, E.; Di Francesco, M. E.; Micale, N.; Grasso, S.; Zappala, M.; Vičík, R.; Schirmeister, T. ChemMedChem 2008, 3, 1030–1033. doi:10.1002/cmdc.200800050
Return to citation in text: [1] -
Janecki, T.; Bodalski, R. Synthesis 1989, 506–510. doi:10.1055/s-1989-27301
Return to citation in text: [1] -
Attolinis, M.; Peiffer, G.; Maffei, M. Tetrahedron 2000, 56, 2693–2697. doi:10.1016/S0040-4020(00)00172-1
Return to citation in text: [1] -
Bałczewski, P.; Bodzioch, A. Phosphorus, Sulfur Silicon Relat. Elem. 2009, 184, 1076–1090. doi:10.1080/10426500902737380
Return to citation in text: [1] -
Demmer, C. S.; Krogsgaard-Larsen, N.; Bunch, L. Chem. Rev. 2011, 111, 7981–8006. doi:10.1021/cr2002646
Return to citation in text: [1] -
Janecki, T.; Bodalski, R. Synthesis 1990, 799–801. doi:10.1055/s-1990-27019
Return to citation in text: [1] -
Muthiah, C.; Senthil Kumar, K.; Vittal, J. J.; Kumara Swamy, K. C. Synlett 2002, 1787–1790. doi:10.1055/s-2002-34899
Return to citation in text: [1] -
Barney, R. J.; Richardson, R. M.; Wiemer, D. F. J. Org. Chem. 2011, 76, 2875–2879. doi:10.1021/jo200137k
Return to citation in text: [1] -
Das, B.; Bhunia, N.; Damodar, K. Synth. Commun. 2012, 42, 2479–2489. doi:10.1080/00397911.2011.561396
Return to citation in text: [1] -
Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] -
Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Luche, J. L. J. Am. Chem. Soc. 1978, 100, 2226–2227. doi:10.1021/ja00475a040
Return to citation in text: [1] -
Gemal, A. L.; Luche, J. L. J. Am. Chem. Soc. 1981, 103, 5454–5459. doi:10.1021/ja00408a029
Return to citation in text: [1] -
Koh, Y.-h.; Shim, J. H.; Wu, J. Z.; Zhong, W.; Hong, Z.; Girardet, J.-L. J. Med. Chem. 2005, 48, 2867–2875. doi:10.1021/jm049029u
Return to citation in text: [1] -
Kobayashi, Y.; William, A. D.; Tokoro, Y. J. Org. Chem. 2001, 66, 7903–7906. doi:10.1021/jo010701u
Return to citation in text: [1] -
Wu, W.; Rao, W.; Er, Y. Q.; Loh, J. K.; Poh, C. Y.; Chan, P. W. H. Tetrahedron Lett. 2008, 49, 2620–2624. doi:10.1016/j.tetlet.2008.02.079
Return to citation in text: [1] -
Rezgui, F.; El Gaïed, M. M. J. Chem. Res., Synop. 1999, 510–511. doi:10.1039/A902012H
Return to citation in text: [1]
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 1. | Zhang, N.; Casida, J. E. Bioorg. Med. Chem. 2002, 10, 1281–1290. doi:10.1016/S0968-0896(01)00391-1 |
| 2. | Wu, D.; Niu, J.-Q.; Ding, Y.-H.; Wu, X.-Y.; Zhong, B.-H.; Feng, X.-W. Med. Chem. Res. 2012, 21, 1179–1187. doi:10.1007/s00044-011-9616-2 |
| 3. | Romanenko, V. D.; Kukhar, V. P. Beilstein J. Org. Chem. 2013, 9, 991–1001. doi:10.3762/bjoc.9.114 |
| 8. | Kerst, A. F.; Douros, J. D., Jr. Diethyl allyl phosphonates as an antimicrobial agent. U. S. Patent 3783160, 1974. |
| 9. | Ettari, R.; Nizi, E.; Di Francesco, M. E.; Micale, N.; Grasso, S.; Zappala, M.; Vičík, R.; Schirmeister, T. ChemMedChem 2008, 3, 1030–1033. doi:10.1002/cmdc.200800050 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 7. | Albrecht, L.; Albrecht, A.; Krawczyk, H.; Jørgensen, K. A. Chem. – Eur. J. 2010, 16, 28–48. doi:10.1002/chem.200902634 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 6. | Touré, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439–4486. doi:10.1021/cr800296p |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 4. | Nandula, V. K. Glyphosphate Resistance in Corps and Weeds: History, Development, and Management; John Wiley & Sons, Inc., 2010. doi:10.1002/9780470634394.index |
| 5. | Jackson, E. R.; Dowd, C. S. Curr. Top. Med. Chem. 2012, 12, 706–728. doi:10.2174/156802612799984599 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 15. | Muthiah, C.; Senthil Kumar, K.; Vittal, J. J.; Kumara Swamy, K. C. Synlett 2002, 1787–1790. doi:10.1055/s-2002-34899 |
| 17. | Das, B.; Bhunia, N.; Damodar, K. Synth. Commun. 2012, 42, 2479–2489. doi:10.1080/00397911.2011.561396 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 13. | Demmer, C. S.; Krogsgaard-Larsen, N.; Bunch, L. Chem. Rev. 2011, 111, 7981–8006. doi:10.1021/cr2002646 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 10. | Janecki, T.; Bodalski, R. Synthesis 1989, 506–510. doi:10.1055/s-1989-27301 |
| 11. | Attolinis, M.; Peiffer, G.; Maffei, M. Tetrahedron 2000, 56, 2693–2697. doi:10.1016/S0040-4020(00)00172-1 |
| 12. | Bałczewski, P.; Bodzioch, A. Phosphorus, Sulfur Silicon Relat. Elem. 2009, 184, 1076–1090. doi:10.1080/10426500902737380 |
| 16. | Barney, R. J.; Richardson, R. M.; Wiemer, D. F. J. Org. Chem. 2011, 76, 2875–2879. doi:10.1021/jo200137k |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 54. | Rezgui, F.; El Gaïed, M. M. J. Chem. Res., Synop. 1999, 510–511. doi:10.1039/A902012H |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 51. | Koh, Y.-h.; Shim, J. H.; Wu, J. Z.; Zhong, W.; Hong, Z.; Girardet, J.-L. J. Med. Chem. 2005, 48, 2867–2875. doi:10.1021/jm049029u |
| 52. | Kobayashi, Y.; William, A. D.; Tokoro, Y. J. Org. Chem. 2001, 66, 7903–7906. doi:10.1021/jo010701u |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 53. | Wu, W.; Rao, W.; Er, Y. Q.; Loh, J. K.; Poh, C. Y.; Chan, P. W. H. Tetrahedron Lett. 2008, 49, 2620–2624. doi:10.1016/j.tetlet.2008.02.079 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 18. | Basavaiah, D.; Pandiaraju, S. Tetrahedron 1996, 52, 2261–2268. doi:10.1016/0040-4020(95)01055-6 |
| 19. | Deng, H.-P.; Shi, M. Eur. J. Org. Chem. 2012, 183–187. doi:10.1002/ejoc.201101365 |
| 20. | Mhasni, O.; Rezgui, F. Tetrahedron Lett. 2010, 51, 586–587. doi:10.1016/j.tetlet.2009.11.053 |
| 21. | Elleuch, H.; Ayadi, M.; Bouajila, J.; Rezgui, F. J. Org. Chem. 2016, 81, 1757–1761. doi:10.1021/acs.joc.5b02106 |
| 22. | Cohen, J. I.; Engel, R. Synthesis of Carbon-Phosphorus Bonds, 2nd ed.; CRC Press: Boca Raton, 2003. doi:10.1201/9780203998243 |
| 23. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. |
| 24. | Mucha, A.; Kafarski, P.; Berlicki, L. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 25. | Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328–2345. doi:10.1021/jm701260b |
| 26. | Brunel, J. M.; Buono, G. Top. Curr. Chem. 2002, 220, 79–105. doi:10.1007/3-540-45731-3_4 |
| 27. | Allen, M. C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J. M. J. Med. Chem. 1989, 32, 1652–1661. doi:10.1021/jm00127a041 |
| 28. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 29. | Allenberger, F.; Klare, I. J. Antimicrob. Chemother. 1999, 43, 211–217. doi:10.1093/jac/43.2.211 |
| 30. | Kafarski, P.; Lejczak, B. Phosphorus, Sulfur Silicon Relat. Elem. 1991, 63, 193–215. doi:10.1080/10426509108029443 |
| 31. | Smith, A. B., III; Yager, K. M.; Taylor, C. M. J. Am. Chem. Soc. 1995, 117, 10879–10888. doi:10.1021/ja00149a011 |
| 32. | Chung, S.-K.; Kang, D.-H. Tetrahedron: Asymmetry 1996, 7, 21–24. doi:10.1016/0957-4166(95)00411-4 |
| 33. | Bhattacharya, A. K.; Raut, D. S.; Ranna, K. C.; Polanki, I. K.; Khan, M. S.; Iram, S. Eur. J. Med. Chem. 2013, 66, 146–152. doi:10.1016/j.ejmech.2013.05.036 |
| 34. | Allen, J. G.; Atherton, F. R.; Hall, M. J.; Hassal, C. H.; Holmes, S. W.; Lambert, R. W.; Nisbet, L. J.; Ringrose, P. S. Nature 1978, 272, 56–58. doi:10.1038/272056a0 |
| 35. | Alonso, E.; Alonso, E.; Solís, A.; del Pozo, C. Synlett 2000, 698–700. doi:10.1055/s-2000-6615 |
| 36. | Hirschmann, R.; Smith, A. B.; Taylor, C. M.; Benkovic, P. A.; Taylor, S. D.; Yager, K. M.; Sprengler, P. A.; Benkovic, S. J. Science 1994, 265, 234–237. doi:10.1126/science.8023141 |
| 37. | Kamiya, T.; Hemmi, K.; Takeno, H.; Hashimoto, M. Tetrahedron Lett. 1980, 21, 95–98. doi:10.1016/S0040-4039(00)93633-5 |
| 38. | Hashimoto, M.; Hemmi, K.; Takeno, H.; Kamiya, T. Tetrahedron Lett. 1980, 21, 99–102. doi:10.1016/S0040-4039(00)93634-7 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 40. | Iguchi, E.; Okuhara, M.; Kohasaka, M.; Aoki, H.; Imanaka, H. J. Antibiot. 1980, 33, 18–23. doi:10.7164/antibiotics.33.18 |
| 41. | Wahbi, A.; Mhamdi, A.; Hassen, Z.; Touil, S. Green Chem. Lett. Rev. 2014, 7, 73–78. doi:10.1080/17518253.2014.895863 |
| 42. | Logusch, E. W.; Walker, D. M.; McDonald, J. F.; Leo, G. C.; Franz, J. E. J. Org. Chem. 1988, 53, 4069–4074. doi:10.1021/jo00252a034 |
| 43. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 44. | Foss, F. W., Jr.; Snyder, A. H.; Davis, M. D.; Rouse, M.; Oksua, M. D.; Lynch, K. R.; Mcdonald, T. L. Bioorg. Med. Chem. 2007, 15, 663–677. doi:10.1016/j.bmc.2006.10.060 |
| 45. | Logusch, E. W. Tetrahedron Lett. 1986, 27, 5935–5938. doi:10.1016/S0040-4039(00)85366-6 |
| 46. | Villiéras, J.; Rambaud, M. Org. Synth.. 1993; Vol. VIII, p 265. |
| 47. | Turki, T.; Villiéras, J.; Amri, H. Tetrahedron Lett. 2005, 46, 3071–3072. doi:10.1016/j.tetlet.2005.03.005 |
| 48. | Mrabet, H.; Zantour, H. Phosphorus, Sulfur Silicon Relat. Elem. 2004, 179, 25–33. doi:10.1080/10426500490257005 |
| 39. | Wiesner, J.; Henschker, D.; Hutchinson, D. B.; Beck, E.; Jomaa, H. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. doi:10.1128/AAC.46.9.2889-2894.2002 |
| 49. | Luche, J. L. J. Am. Chem. Soc. 1978, 100, 2226–2227. doi:10.1021/ja00475a040 |
| 50. | Gemal, A. L.; Luche, J. L. J. Am. Chem. Soc. 1981, 103, 5454–5459. doi:10.1021/ja00408a029 |
© 2016 Ayadi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)