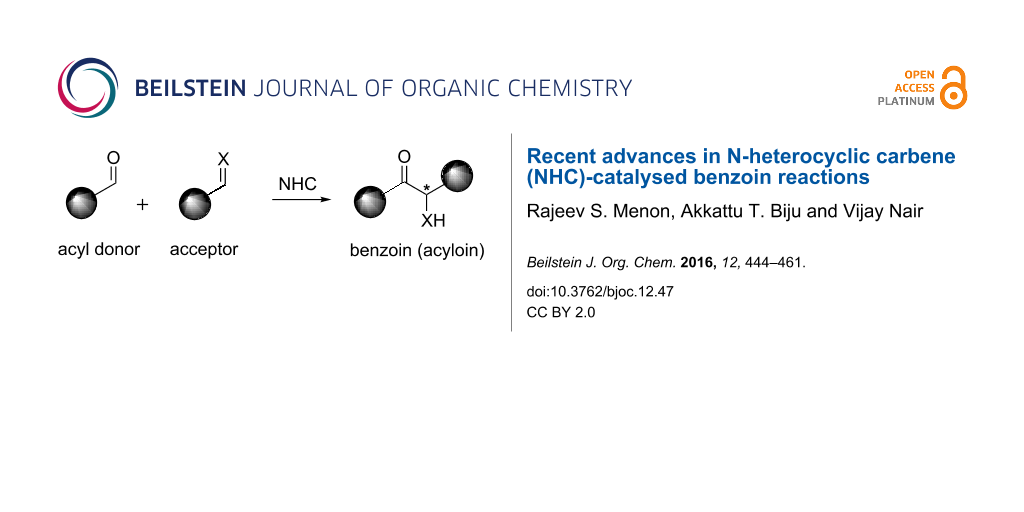
Abstract
N-Heterocyclic carbenes (NHCs) have emerged as a powerful class of organocatalysts that mediate a variety of organic transformations. The Benzoin reaction constitutes one of the earliest known carbon–carbon bond-forming reactions catalysed by NHCs. The rapid growth of NHC catalysis in general has resulted in the development of a variety of benzoin and benzoin-type reactions. An overview of such NHC-catalysed benzoin reactions is presented.

Graphical Abstract
Introduction
The benzoin reaction (or condensation) is named after the product it furnishes via a catalytic assembly of two molecules of aromatic aldehydes. One molecule of the aldehyde functions as an acyl anion and the other as a carbonyl electrophile to afford α-hydroxy ketones (benzoins). It is a 100% atom-economic process wherein a new stereocentre is produced. The reaction is sometimes referred to as acyloin condensation to encompass reactions of aliphatic aldehydes. The assembly of two molecules of the same aldehyde is known as homo-benzoin reaction and that of two different aldehydes is known as crossed benzoin reaction. Mechanistically the reaction involves polarity reversal (umpolung) of one aldehyde to generate an acyl anion equivalent and this event is mediated by the catalyst. Alkali metal cyanides and N-heterocyclic carbenes (NHCs) are the two main classes of catalysts that are known to mediate benzoin reactions. This review focuses on the recent advancements made in the area of NHC-catalysed benzoin reactions.
Historically, the first benzoin reaction was reported by Wöhler and Liebig in 1832. They discovered that the cyanide anion can catalyze the union of two molecules of aromatic aldehydes to afford α-hydroxy ketones [1]. More than a century later, a thiazolium salt-catalysed benzoin reaction was reported by Ukai [2]. This may be regarded as an early example of organocatalysis using an azolium salt. Breslow postulated in 1958 a mechanistic rationale for the thiazolium salt-catalysed benzoin reaction [3]. He depicted the catalytically active species as a thiazolium zwitterion (the resonance structure of an NHC) and proposed that the reaction proceeds via an enaminol intermediate. The latter is now popularly known as ‘Breslow intermediate’. This seminal discovery by Breslow paved the way for further developments in the area of carbene catalysis. Almost three decades later Bertrand and co-workers proved the existence of carbenes as catalytically active species in the benzoin reaction, with the synthesis of a stable phosphinocarbene [4]. Arduengo and co-workers isolated and characterised a stable NHC in 1991 [5]. These two reports on the isolation of NHCs implied that they are more stable and robust than previously considered. Subsequent years witnessed a renewal of interest in NHCs and a flurry of reports, mainly focusing on their catalytic activity, appeared in the literature [6,7].
The original mechanistic proposal by Breslow for the thiazolium salt-catalysed benzoin reaction can be delineated as follows (Scheme 1) [3]. Lapworth had suggested how the cyanide anion functions first as a nucleophile and then as a leaving group in cyanide-catalysed benzoin reactions [8]. Analogously, Breslow invoked the generation of a nucleophilic thiazolylidene species 1 via deprotonation of the thiazolium salt by base. The ylide 1 may also be represented as its resonance structure 1’ (carbene). Nucleophilic addition of 1 to aromatic aldehyde generates the tetrahedral intermediate 2. The latter then undergoes a proton shift to furnish an enaminol derivative 3. The aldehyde carbonyl carbon has now transformed into a nucleophilic entity by virtue of conjugation to the nitrogen and sulfur lone pairs. This acyl anion equivalent 3 is known as the "Breslow intermediate". Its reaction with another molecule of aldehyde leads to the formation of an alkoxide intermediate 4. Proton transfer and subsequent release of thiazolylidene 1 affords the final product, the α-hydroxy ketone 5. Breslow demonstrated that imidazolium-derived ylides also catalysed benzoin reactions. In most of the cases, the NHC-catalysed formation of benzoin from aldehydes is reversible in nature.
![[1860-5397-12-47-i1]](/bjoc/content/inline/1860-5397-12-47-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Breslow’s proposal on the mechanism of the benzoin condensation.
Scheme 1: Breslow’s proposal on the mechanism of the benzoin condensation.
In the following sections, detailed discussions on various types of benzoin reactions catalysed by NHCs are presented. In general, thiazolium salt-derived NHCs have found widespread application as catalysts for benzoin reactions, whereas triazolium-derived NHCs have emerged as popular catalysts for enantioselective benzoin transformations.
Review
Homo-benzoin reactions
The homo-benzoin condensation constitutes an overall catalytic dimerization of an aldehyde wherein the acyl anion derived from one molecule adds to another molecule of the aldehyde. (It may be noted that the term ‘homo’ implies the reaction between two molecules of the same aldehyde. It should not be misconstrued as a ‘homologous’ benzoin reaction). Benzoin reactions are reversible in basic medium and homo-benzoin products are often isolated as byproducts in other NHC-mediated reactions of aldehydes. The absence of chemoselectivity issues makes homo-benzoin reactions less challenging when compared to the cross-benzoin variant. NHC-mediated aerial oxidation of aldehydes to the corresponding carboxylic acids could compete with homo-benzoin reactions, but can be limited by careful exclusion of oxygen from the reaction mixture. A few recent reports of homo-benzoin reactions are discussed in the following passages.
Stetter’s report in 1976 of thiazolium salt-catalysed benzoin reactions may be regarded as the first report of an NHC-catalysed benzoin reaction on a synthetically useful scale [9]. Much later, in 2005, Xu and Xia used N-alkyl-substituted imidazolium carbene 6 to efficiently promote benzoin reactions. Although a high catalyst loading (50 mol %) was required, the reactions could be run at mild conditions. It was observed that neutral and electron rich aromatic aldehydes afford good yields of benzoin products whereas electron deficient aromatic aldehydes and aliphatic aldehydes reacted sluggishly (Scheme 2) [10].
![[1860-5397-12-47-i2]](/bjoc/content/inline/1860-5397-12-47-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Imidazolium carbene-catalysed homo-benzoin condensation.
Scheme 2: Imidazolium carbene-catalysed homo-benzoin condensation.
The easily accessible NHC precatalyst 7 endowed with long aliphatic side chains was used by Iwamoto and co-workers for promoting benzoin reactions in aqueous medium. The improved reactivity was attributed to the formation of micelles from the hydrophobic alkyl chains of the catalyst in aqueous medium. The reaction proceeded well with various aromatic and heteroaromatic aldehydes (Scheme 3) [11].
![[1860-5397-12-47-i3]](/bjoc/content/inline/1860-5397-12-47-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Homo-benzoin condensation in aqueous medium.
Scheme 3: Homo-benzoin condensation in aqueous medium.
Subsequently, the same group disclosed the application of bis(benzimidazolium) precursor 8 as a more efficient catalyst for the benzoin condensation in aqueous medium. Here, the NHC precatalyst incorporated a long aliphatic bridge between the two imidazolium entities. The aggregation of these units creates a hydrophobic environment in which the two aromatic aldehydes are subjected to catalysis (Scheme 4) [12].
![[1860-5397-12-47-i4]](/bjoc/content/inline/1860-5397-12-47-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Homobenzoin condensation catalysed by bis(benzimidazolium) salt 8.
Scheme 4: Homobenzoin condensation catalysed by bis(benzimidazolium) salt 8.
Asymmetric homo-benzoin reactions
Much of the progress in the area of NHC-catalysed asymmetric benzoin reactions has been covered in two excellent reviews [6,7]. Some additional recent examples are discussed below.
A selected list of chiral NHC catalysts that have been explored for mediating asymmetric benzoin reactions is presented in Scheme 5. The bis-triazolium catalyst 9 developed by You promoted asymmetric benzoin reactions in 95% ee [13]. Enders developed the pyroglutamic acid-derived triazolium salt 10 which mediated benzoin reactions in similar enantioselectivities [14].
![[1860-5397-12-47-i5]](/bjoc/content/inline/1860-5397-12-47-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: List of assorted chiral NHC-catalysts used for asymmetric homobenzoin condensation.
Scheme 5: List of assorted chiral NHC-catalysts used for asymmetric homobenzoin condensation.
The chiral triazolium catalyst 11 transfers chiral information to the benzoin products by engaging in hydrogen-bonding interactions [15]. Waser’s chiral bifunctional (thio)urea NHC 12 also relies on hydrogen bonding to mediate asymmetric benzoin reactions [16]. A 2-pyrdiyl appendage distinguishes Ukaji’s chiral triazolium catalyst 13 from similar salts [17]. Spirocyclic (1R)-camphor-derived triazolium salt 14developed by Rafiński also successfully catalysed asymmetric benzoin condensations [18].
The pentafluorophenyltriazolium catalyst 15 featured in the most efficient asymmetric benzoin reaction reported so far. Inoue and co-workers found that it promotes homocoupling of benzaldehyde at a low loading (4 mol %) to afford benzoin in 90% yield and >99% ee (Scheme 6) [19].
![[1860-5397-12-47-i6]](/bjoc/content/inline/1860-5397-12-47-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: A rigid bicyclic triazole precatalyst 15 in an efficient enantioselective benzoin reaction.
Scheme 6: A rigid bicyclic triazole precatalyst 15 in an efficient enantioselective benzoin reaction.
Cross-benzoin reactions
A cross-benzoin reaction unites two different aldehydes wherein one of them functions as the acyl anion equivalent. A total of four products are possible; a pair of homo-benzoin and cross-benzoin adducts each. A substrate-driven selectivity may be observed when one of the aldehydes is significantly less reactive due to electronic or steric reasons. The latter effect may be amplified by employing bulky NHCs. In general, NHC-mediated selective cross-benzoin reactions of electronically and sterically similar aldehydes remain as a highly challenging transformation.
In 1985, Inoue and co-workers reported the NHC-catalysed selective cross-benzoin reactions of aromatic and aliphatic aldehydes with formaldehyde leading to the formation of α-hydroxy ketones. Although an excellent selectivity was observed for the cross-benzoin product, the yields were low (Scheme 7) [20].
![[1860-5397-12-47-i7]](/bjoc/content/inline/1860-5397-12-47-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Inoue’s report of cross-benzoin reactions.
Scheme 7: Inoue’s report of cross-benzoin reactions.
Later Kuhl and Glorius employed an NHC generated from the thiazolium salt 17 to synthesise α-hydroxyketones 18 in good yields. This highly selective cross-benzoin reaction has a very broad substrate scope (Scheme 8) [21].
![[1860-5397-12-47-i8]](/bjoc/content/inline/1860-5397-12-47-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Cross-benzoin reactions catalysed by thiazolium salt 17.
Scheme 8: Cross-benzoin reactions catalysed by thiazolium salt 17.
Yang and co-workers developed an intermolecular cross coupling of aromatic aldehydes with acetaldehyde. The reaction showed an interesting divergence in reactivity controlled by the catalysts, viz., the thiazolium salt 19 and triazolium salt 20. The thiazolium-derived carbene preferentially mediated the formation of the Breslow intermediate from the aromatic aldehyde followed by coupling with acetaldehyde. In contrast, the triazolium-derived carbene prefered to activate acetaldehyde to generate the corresponding acyl anion equivalent followed by coupling with aromatic aldehydes (Scheme 9) [22]. It may be mentioned that Connon, Zeitler and co-workers have also reported the use of thiazolium and triazolium precatalysts for selective cross-benzoin reactions [23].
![[1860-5397-12-47-i9]](/bjoc/content/inline/1860-5397-12-47-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Catalyst-controlled divergence in cross-benzoin reactions.
Scheme 9: Catalyst-controlled divergence in cross-benzoin reactions.
Glorius introduced a number of thiazolium NHC precatalysts endowed with sterically bulky aryl groups on the nitrogen with varying backbone substitution. These NHCs exhibited high levels of reactivity and selectivity in intermolecular cross-benzoin reactions to afford a library of unsymmetrically substituted benzoins [24]. The presence of an ortho-substituent on the electrophilic aromatic aldehyde (which presumably hinders the direct addition of NHC to these aldehydes) was necessary for the high levels of selectivity (Scheme 10).
![[1860-5397-12-47-i10]](/bjoc/content/inline/1860-5397-12-47-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Chemoselective cross-benzoin reactions catalysed by a bulky NHC.
Scheme 10: Chemoselective cross-benzoin reactions catalysed by a bulky NHC.
The NHC-catalysed chemoselective intermolecular cross-benzoin condensation reaction of aromatic and aliphatic aldehydes was reported by Yang and co-workers. The chemoselectivity was achieved by using a large excess of the aliphatic aldehyde (molar ratio of 1:15) [25]. Thus, directing groups on the aromatic aldehydes were not a prerequisite for high levels of selectivity in contrast to the earlier example. Consecutive catalytic reactions were utilized in order to reuse the excess of aliphatic aldehydes employed for achieving selectivity. Interestingly, the reaction could be repeated up to five times without affecting the yield of product and chemoselectivity (Scheme 11).
![[1860-5397-12-47-i11]](/bjoc/content/inline/1860-5397-12-47-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Selective intermolecular cross-benzoin condensation reactions of aromatic and aliphatic aldehydes.
Scheme 11: Selective intermolecular cross-benzoin condensation reactions of aromatic and aliphatic aldehydes.
Morpholinone and piperidinone-derived triazolium precatalysts can catalyze highly chemoselectively the cross-benzoin reaction of aliphatic and aromatic aldehydes [26]. Smooth and selective benzoin reactions were observed with a wide variety of linear and branched aliphatic aldehydes as well as aromatic aldehydes (Scheme 12). Notably, the aliphatic aldehydes functioned as acyl anion equivalents leading to the formation of alkyl ketone (benzoin) products.
![[1860-5397-12-47-i12]](/bjoc/content/inline/1860-5397-12-47-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Chemoselective cross-benzoin reaction of aliphatic and aromatic aldehydes.
Scheme 12: Chemoselective cross-benzoin reaction of aliphatic and aromatic aldehydes.
Asymmetric cross-benzoin reactions
The development of enantioselective cross-benzoin reactions is an arduous task as both chemoselectivity and stereoselectivity must be controlled by a single catalyst. Unsurprisingly, most of the NHC-catalysed, enantioselective cross-benzoin reactions employ a combination of two distinct carbonyl components to minimize chemoselectivity issues. A selected group of asymmetric cross-benzoin reactions are described in the following section.
An NHC-catalysed union of aryl aldehydes and aryl trifluoromethyl ketones was developed in the laboratory of Enders. This direct intermolecular cross-benzoin reaction proceeded with high yields and chemoselectivity [27]. The reaction furnished excellent yields of α-hydroxy-α-trifluoromethyl ketones 25 possessing a quaternary stereocentre. The homo-benzoin condensation between two aldehydes is reversible under the reaction conditions. This eventually leads to the selective formation of the observed cross-benzoin product. Later, it was found that trifluoromethyl ketimines 26 also function as electrophiles under similar reaction conditions [28]. Although initial attempts of asymmetric transformations were not successful, enantioselective cross-benzoin reactions of heteroaromatic aldehydes (acyl donors) and aryl trifluoromethyl ketones were later developed using the chiral catalyst 27 (Scheme 13) [29].
![[1860-5397-12-47-i13]](/bjoc/content/inline/1860-5397-12-47-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Cross-benzoin reactions of trifluoromethyl ketones developed by Enders.
Scheme 13: Cross-benzoin reactions of trifluoromethyl ketones developed by Enders.
The electron-deficient triazolium-derived NHC 23 mediated efficient and chemoselective cross-benzoin reactions of aldehydes and α-ketoesters to produce acyloin products endowed with a quaternary stereocentre [30]. Remarkably, the competing hydroacylation reaction was not observed under these reaction conditions. A variety of aliphatic and aromatic aldehydes functioned as acyl donors, whereas several α-ketoesters could be employed as the electrophilic coupling partner to afford the desired products in moderate to good yields (Scheme 14). Interestingly, preliminary experiments to develop an enantioselective version of this reaction using a chiral NHC returned promising levels of enantioselectivity (76% ee).
![[1860-5397-12-47-i14]](/bjoc/content/inline/1860-5397-12-47-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Cross-benzoin reactions of aldehydes and α-ketoesters.
Scheme 14: Cross-benzoin reactions of aldehydes and α-ketoesters.
Subsequently, Gravel and co-workers reported a high yielding chemoselective and enantioselective intermolecular cross-benzoin reaction of aliphatic aldehydes and α-ketoesters. Notably, the reaction affords enantiomerically enriched tertiary alcohols. Excellent levels of enantioselection were obtained by using an electron-deficient valine-derived triazolium salt precatalyst 28 (Scheme 15) [31]. Moreover, diastereoselective reduction of the cross-benzoin products with NaBH4 afforded valuable syn-diol products.
![[1860-5397-12-47-i15]](/bjoc/content/inline/1860-5397-12-47-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Enantioselective cross-benzoin reactions of aliphatic aldehydes and α-ketoesters.
Scheme 15: Enantioselective cross-benzoin reactions of aliphatic aldehydes and α-ketoesters.
Goodman and Johnson disclosed a dynamic kinetic resolution of β-halo-α-ketoesters via NHC-catalysed asymmetric cross-benzoin reaction. Here, the cross-benzoin reaction of aromatic aldehydes with β-stereogenic-α-keto esters afforded fully substituted β-halo-α-glycolic acid derivatives in high diastereoselectivity and enantioselectivity [32]. The NHC generated from the amino indanol-derived chiral triazolium salt 29 provided the best results (Scheme 16). A variety of aromatic aldehydes and a series of β-halo α-ketoesters partake in the reaction to furnish the chiral glycolic acid derivatives.
![[1860-5397-12-47-i16]](/bjoc/content/inline/1860-5397-12-47-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Dynamic kinetic resolution of β-halo-α-ketoesters via cross-benzoin reaction.
Scheme 16: Dynamic kinetic resolution of β-halo-α-ketoesters via cross-benzoin reaction.
The enantioselective benzoin reaction between a variety of aldehydes and alkynones is catalysed by the NHC generated from chiral aminoindanol-triazolium salt 29. The reactions afforded substituted propargylic alcohols in high yields and enantioselectivity (Scheme 17). It is noteworthy that the catalytically generated Breslow intermediates undergo selective 1,2-addition with ynones and the competing Stetter-type reactivity was not observed [33].
![[1860-5397-12-47-i17]](/bjoc/content/inline/1860-5397-12-47-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Enantioselective benzoin reaction of aldehydes and alkynones.
Scheme 17: Enantioselective benzoin reaction of aldehydes and alkynones.
Aza-benzoin reactions
In aza-benzoin reactions, the acyl anions generated from aldehydes react with an aza electrophile. Imines possessing an electron-withdrawing N-substituent constitute the most commonly used aza electrophile and the reaction affords an α-aminocarbonyl compound as the product. The NHC-mediated addition of aldehyde-derived acyl anions to nitroso compounds leading to the formation of hydroxamic acid derivatives are also discussed in this section for convenience.
Acylimines function as electrophiles in NHC-catalysed aza-benzoin reaction with aldehydes. The reactive acylimine is generated in situ by the action of base on the sulfonylamide derivative 30 [34]. Meanwhile, the Breslow intermediate is produced from the aldehyde by the thiazolium 31-derived NHC. The union of these two reactive intermediates furnished α-amidoketones 32 in excellent yields (Scheme 18).
![[1860-5397-12-47-i18]](/bjoc/content/inline/1860-5397-12-47-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Aza-benzoin reaction of aldehydes and acylimines.
Scheme 18: Aza-benzoin reaction of aldehydes and acylimines.
A diastereoselective [4 + 1] annulation of phthalaldehyde with imines leading to the formation of cis-2-amino3-hydroxyindanones is catalysed by NHC 31. The imine electrophile is generated in situ from α-sulfonyl-N-Boc amine 33 (Scheme 19). Initial cross-aza-benzoin reaction of one of the aldehyde functionalities with the imine is followed by an intramolecular aldol reaction to furnish the indanone framework [35].
![[1860-5397-12-47-i19]](/bjoc/content/inline/1860-5397-12-47-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: NHC-catalysed diastereoselective synthesis of cis-2-amino 3-hydroxyindanones.
Scheme 19: NHC-catalysed diastereoselective synthesis of cis-2-amino 3-hydroxyindanones.
The thiazolium precatalyst 31 can also efficiently mediate cross-aza-benzoin reactions of aromatic and heteroaromatic aldehydes with unactivated aromatic imines 34 (Scheme 20) [36]. A control reaction of the corresponding benzoin (instead of the aldehyde) and imine 34 also afforded the α-amino ketone product 35 in 71% yield. This indicates that the reaction involves reversible formation of aldehyde-homobenzoin adducts.
![[1860-5397-12-47-i20]](/bjoc/content/inline/1860-5397-12-47-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Cross-aza-benzoin reactions of aldehydes with aromatic imines.
Scheme 20: Cross-aza-benzoin reactions of aldehydes with aromatic imines.
Enantioselective cross aza-benzoin reaction of aliphatic aldehydes with N-Boc-protected imines are promoted efficiently by NHC generated from the chiral triazolium salt 36. The aldehydes function as the acyl donor and the imines behave as the receptors (Scheme 21). Addition of NHC to the highly electrophilic N-Boc imines leads to the formation of corresponding aza-Breslow intermediates; however, it is reversible under the reaction conditions. Importantly, the chirally pure α-amino ketones formed in this reaction are valuable building blocks in organic synthesis [37].
![[1860-5397-12-47-i21]](/bjoc/content/inline/1860-5397-12-47-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Enantioselective cross aza-benzoin reaction of aliphatic aldehydes with N-Boc-imines.
Scheme 21: Enantioselective cross aza-benzoin reaction of aliphatic aldehydes with N-Boc-imines.
The NHC generated from the bicyclic pentafluoro triazolium salt promoted the chemoselective cross aza-benzoin reaction of aldehydes with N-PMP-imino esters to afford α-amino-β-keto esters in good yield (Scheme 22) [38]. A range of functional groups are tolerated under the optimised reaction conditions.
![[1860-5397-12-47-i22]](/bjoc/content/inline/1860-5397-12-47-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Chemoselective cross aza-benzoin reaction of aldehydes with N-PMP-imino esters.
Scheme 22: Chemoselective cross aza-benzoin reaction of aldehydes with N-PMP-imino esters.
Mattson and Scheidt developed a catalytic coupling reaction of acylsilanes with imines for the synthesis of aminoketones (Scheme 23). The reaction proceeds through the generation of the Breslow intermediate from the acylsilane followed by a cross-coupling with the imine [39].
![[1860-5397-12-47-i23]](/bjoc/content/inline/1860-5397-12-47-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: NHC-catalysed coupling reaction of acylsilanes with imines.
Scheme 23: NHC-catalysed coupling reaction of acylsilanes with imines.
In 2005, Miller and co-workers used the chiral thiazolium salt 38 to catalyse an enantioselective cross-aza-benzoin reaction. Racemisation of the products under the reaction conditions caused erosion of enantioselectivity. This problem was successfully addressed by using a hindered base, pentamethyl piperidine, which was inert towards the products (Scheme 24) [40].
![[1860-5397-12-47-i24]](/bjoc/content/inline/1860-5397-12-47-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Thiazolium salt-mediated enantioselective cross-aza-benzoin reaction.
Scheme 24: Thiazolium salt-mediated enantioselective cross-aza-benzoin reaction.
In 2013, Ye disclosed a remarkable NHC-catalysed enantioselective aza-benzoin reaction of enals and activated ketimines leading to the formation of functionalised α-aminoketones 39 in high enantioselectivity [41]. Notably, the homoenolate or enolate reactivity of the NHC-enal adduct was not observed in this case. The presence of a tertiary alcohol functionality and the steric bulk of the NHC-precatalyst 40 were essential for the selective formation of the aza-benzoin adduct. A variety of trifluoromethylated α-aminoketones could be synthesised in enantiomerically pure form using this method (Scheme 25).
![[1860-5397-12-47-i25]](/bjoc/content/inline/1860-5397-12-47-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Aza-benzoin reaction of enals with activated ketimines.
Scheme 25: Aza-benzoin reaction of enals with activated ketimines.
Isatin derived ketimines 41 were employed as electrophiles in the NHC-catalysed chemo- and stereoselective cross-aza-benzoin reaction with enals by Chi. The reaction afforded chiral quaternary aminooxindole derivatives. The NHC–enal adduct prefers to react via the acyl anion pathway and the competing homoenolate/enolate reactivity was not observed. The sterically non-congested, electron-deficient NHC-catalyst 42 presumably does not hinder bond formation at the catalyst-bound acyl carbon (Scheme 26) [42].
![[1860-5397-12-47-i26]](/bjoc/content/inline/1860-5397-12-47-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Isatin derived ketimines as electrophiles in cross aza-benzoin reaction with enals.
Scheme 26: Isatin derived ketimines as electrophiles in cross aza-benzoin reaction with enals.
The aza-benzoin reaction of aldehydes and phosphinoylimines catalysed by the bis(amino)cyclopropenylidene (BAC) carbene 43 was reported recently. The reaction showed excellent selectivity for the aza-benzoin products over the homo-benzoin adducts. A wide variety of aldehydes react with phosphinoylimines (generated from their sulfinic acid adducts 44) to afford N-phosphinoyl amnioketones (Scheme 27) [43]. The attempted enantioselective version of this reaction using a chiral BAC catalyst was, however, unsuccessful.
![[1860-5397-12-47-i27]](/bjoc/content/inline/1860-5397-12-47-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Aza-benzoin reaction of aldehydes and phosphinoylimines catalysed by the BAC-carbene.
Scheme 27: Aza-benzoin reaction of aldehydes and phosphinoylimines catalysed by the BAC-carbene.
As mentioned earlier, nitrosoarenes have been used as the electrophilic component in a few reactions of NHC-bound aldehydes. The addition of acyl anions occur at the nitrogen atom of the nitroso compound. A NHC-catalysed cascade reaction of o-vinylarylaldehydes with nitrosoarenes afforded functionalised 2,3-benzoxazin-4-ones 45 [44]. The initial intermolecular aza-benzoin reaction is followed by an intramolecular oxa-Michael reaction to form the observed product (Scheme 28).
![[1860-5397-12-47-i28]](/bjoc/content/inline/1860-5397-12-47-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Nitrosoarenes as the electrophilic component in benzoin-initiated cascade reaction.
Scheme 28: Nitrosoarenes as the electrophilic component in benzoin-initiated cascade reaction.
Enders reported a cascade reaction which is initiated by an NHC-catalysed aza-benzoin condensation between various aldehydes and nitrosobenzenes to generate the hydroxamic acids 47. This is followed by a redox esterification of the latter (47) with enals. The overall process constitutes a one-pot synthesis of hydroxamic esters 48 [45]. Notably, both steps can be performed using the single NHC catalyst 22 under same reaction conditions (Scheme 29). This two-step, one-pot synthesis of formahydroxamic esters constitutes a valuable addition to a thin list of NHC-mediated three-component reactions.
![[1860-5397-12-47-i29]](/bjoc/content/inline/1860-5397-12-47-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: One-pot synthesis of hydroxamic esters via aza-benzoin reaction.
Scheme 29: One-pot synthesis of hydroxamic esters via aza-benzoin reaction.
Intramolecular benzoin reactions
One of the earliest reports of an intramolecular benzoin condensation appeared in 1976. Cookson and Lane found that the treatment of anhydrous glutaraldehyde with thiazolium salt 49 and triethylamine resulted in the formation of 2-hydroxycyclopentanone. The latter underwent oxidation to afford 2-hydroxycyclopent-2-en-1-one 50 upon treatment with Cu(OAc)2 (Scheme 30) [46]. Hexanedial furnished the corresponding α-hydroxycyclohexanone under identical reaction conditions.
![[1860-5397-12-47-i30]](/bjoc/content/inline/1860-5397-12-47-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Cookson and Lane’s report of intramolecular benzoin condensation.
Scheme 30: Cookson and Lane’s report of intramolecular benzoin condensation.
An intramolecular cross-benzoin condensation between aldehyde and ketone moieties was developed by Suzuki in 2003. The isoxazole-fused cyclohexanone 51 endowed with an aryl aldehyde underwent a smooth cross-benzoin cyclisation in the presence of the thiazolium catalyst 19 and DBU. Although the presence of an isoxazole moiety is not a prerequisite for the success of this annulation, its rigid nature presumably renders the reaction highly stereoselective [47]. This simple and mild method allowed the construction of orthogonally protected polycyclic quinones from readily available starting materials. Later in 2006, they developed the enantioselective version of this reaction using an aminoindanol-derived triazolium salt 52 (Scheme 31) [48].
![[1860-5397-12-47-i31]](/bjoc/content/inline/1860-5397-12-47-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Intramolecular cross-benzoin condensation between aldehyde and ketone moieties.
Scheme 31: Intramolecular cross-benzoin condensation between aldehyde and ketone moieties.
Another intramolecular crossed aldehyde-ketone benzoin reaction of simple dicarbonyl systems was developed by Enders (Scheme 32). This method employs commercially available thiazolium salt 19 as precatalyst and affords five- and six-membered cyclic acyloins in good yields [49].
![[1860-5397-12-47-i32]](/bjoc/content/inline/1860-5397-12-47-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Intramolecular crossed aldehyde-ketone benzoin reactions.
Scheme 32: Intramolecular crossed aldehyde-ketone benzoin reactions.
The enantioselective NHC-catalysed crossed aldehyde-ketone benzoin reaction for the synthesis of five- and six-membered cyclic acyloins was also developed by Enders. NHC generated from the tetracyclic triazolium salt 53 gave the best results [50]. It is noteworthy that the absolute stereochemistry of the α-carbonyl quaternary center of benzo-fused carbocycles and chromanones is installed with excellent control (Scheme 33).
![[1860-5397-12-47-i33]](/bjoc/content/inline/1860-5397-12-47-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Enantioselective intramolecular crossed aldehyde-ketone benzoin reaction.
Scheme 33: Enantioselective intramolecular crossed aldehyde-ketone benzoin reaction.
A combination of D-camphor-derived triazolium precatalyst 54 and DBU promoted enantioselective intramolecular cross-benzoin reaction of 55 to afford chromanone 56 in excellent yield and enantioselectivity (Scheme 34). The NHC-precatalyst is conveniently prepared from camphor in 5 steps [51].
![[1860-5397-12-47-i34]](/bjoc/content/inline/1860-5397-12-47-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Chromanone synthesis via enantioselective intramolecular cross-benzoin reaction.
Scheme 34: Chromanone synthesis via enantioselective intramolecular cross-benzoin reaction.
NHC generated from the N-tert-butyl-substituted imidazolium salt 57 catalysed the intramolecular cross-benzoin reaction of chalcones derived from o-phthalaldehydes. The reaction proceeded rapidly (20 min) at room temperature to afford good yields (75–94%) of naphthalenone-based tertiary alcohols 58 (Scheme 35) [52].
![[1860-5397-12-47-i35]](/bjoc/content/inline/1860-5397-12-47-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Intramolecular cross-benzoin reaction of chalcones.
Scheme 35: Intramolecular cross-benzoin reaction of chalcones.
The synthesis of bicyclic tertiary alcohols possessing two quaternary stereocentres at the bridgehead positions was achieved via an asymmetric intramolecular crossed benzoin reaction. A relatively high loading (30 mol %) of the NHC precatalyst 59 was necessary for efficient reactions (Scheme 36) [53].
![[1860-5397-12-47-i36]](/bjoc/content/inline/1860-5397-12-47-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Synthesis of bicyclic tertiary alcohols by intramolecular benzoin reaction.
Scheme 36: Synthesis of bicyclic tertiary alcohols by intramolecular benzoin reaction.
A multicatalytic Michael–benzoin cascade process for the asymmetric synthesis of functionalised cyclopentanones was disclosed in 2009 by Rovis. The chiral secondary amine 60 catalyzes the initial asymmetric Michael addition of an 1,3-diketone and an enal to afford a δ-ketoaldehyde 61. Subsequently, a cross-benzoin reaction of the latter promoted by the NHC precatalyst 22, installs the cyclopentenone system (Scheme 37). It may be noted that the absolute stereochemistry of the process is controlled by the prolinol catalyst 60 and the NHC precatalyst 22 is achiral. Control experiments revealed that the Michael addition is reversible but the NHC catalyst rapidly shuttles the intermediate δ-keto aldehyde 61 to the final product preventing the erosion of enantioselectivity [54]. This cascade reaction constitutes a fine example of symbiotic dual-catalysis wherein both catalysts perform better together in a one-pot reaction than they do independently over two steps.
![[1860-5397-12-47-i37]](/bjoc/content/inline/1860-5397-12-47-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: A multicatalytic Michael–benzoin cascade process for cyclopentanone synthesis.
Scheme 37: A multicatalytic Michael–benzoin cascade process for cyclopentanone synthesis.
A conceptually similar enamine-NHC dual-catalytic Michael–benzoin cascade was also developed by Rovis. The reaction proceeds via the generation of an enamine from the enolizable aldehyde 62 in presence of the prolinol catalyst 60 and its subsequent addition to the Michael acceptor 63. This is followed by NHC-mediated intramolecular cross-benzoin condensation to afford the cyclopentanone 64. Clear evidence for the co-operative relationship between the catalysts was obtained from control experiments. Chiral triazolium catalyst 65 preferentially converts only one of the diastereomeric Michael adducts into the benzoin product. The prolinol catalyst 60, on the other hand, mediates the epimerisation of the less reactive diastereomer. This synergy leads to the enrichment of the diastereomeric ratio of the final product 64 (Scheme 38) [55].
![[1860-5397-12-47-i38]](/bjoc/content/inline/1860-5397-12-47-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Enamine-NHC dual-catalytic, Michael–benzoin cascade reaction.
Scheme 38: Enamine-NHC dual-catalytic, Michael–benzoin cascade reaction.
Enders developed a closely related iminium-cross-benzoin cascade process involving enals and β-oxo sulfones to generate enantioenriched cyclopentanone derivatives with three contiguous stereocentres. A dual secondary amine/NHC catalytic system comprising of the prolinol 60 and NHC precatalyst 22 was found to give the best results (Scheme 39) [56]. The influence of these catalysts on the diastereoselectivity of the reaction was also studied using NMR techniques.
![[1860-5397-12-47-i39]](/bjoc/content/inline/1860-5397-12-47-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Iminium-cross-benzoin cascade reaction of enals and β-oxo sulfones.
Scheme 39: Iminium-cross-benzoin cascade reaction of enals and β-oxo sulfones.
An NHC-catalysed intramolecular benzoin condensation of carbohydrate-derived dialdehydes has been applied for the construction of carbocyclic sugars. Diastereoselective benzoin reactions of manno- and galacto-configured dialdehydes 66 were promoted by the triazolium carbene precatalyst 22 to produce single inosose stereoisomers 67 in high yields (Scheme 40) [57]. Stereospecific reduction and deprotection of the inosose derivatives furnished allo- and epi-inositol in good yields.
![[1860-5397-12-47-i40]](/bjoc/content/inline/1860-5397-12-47-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Intramolecular benzoin condensation of carbohydrate-derived dialdehydes.
Scheme 40: Intramolecular benzoin condensation of carbohydrate-derived dialdehydes.
The camphor-derived triazolium precatalyst 54 promoted enantioselective intramolecular benzoin reactions of N-tethered keto-aldehydes effectively. The substrates for the cyclisation are easily accessible and dihydroquinolinone systems possessing a quaternary stereocentre are produced in high yields and enantioselectivities (Scheme 41) [58].
![[1860-5397-12-47-i41]](/bjoc/content/inline/1860-5397-12-47-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Enantioselective intramolecular benzoin reactions of N-tethered keto-aldehydes.
Scheme 41: Enantioselective intramolecular benzoin reactions of N-tethered keto-aldehydes.
The chiral triazolium salt 68 derived from (1R)-camphor has been used in intramolecular cross-benzoin reactions of keto-aldehydes. The former efficiently catalysed stereoselective formation of chromanones 69 bearing quaternary stereocentres (Scheme 42) [59].
![[1860-5397-12-47-i42]](/bjoc/content/inline/1860-5397-12-47-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: Asymmetric cross-benzoin reactions promoted by camphor-derived catalysts.
Scheme 42: Asymmetric cross-benzoin reactions promoted by camphor-derived catalysts.
Cheng reported that the combination of NHC 24 and a Brønsted base (4-methoxyphenolate) promoted a formal dimerisation of 2-(aroylvinyl)arylaldehydes 70 to afford benzo[a]tetrahydrofluorenones 71 [60]. This stereoselective reaction proceeds via a benzoin–Michael–Michael cascade process (Scheme 43).
![[1860-5397-12-47-i43]](/bjoc/content/inline/1860-5397-12-47-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 43: NHC-Brønsted base co-catalysis in a benzoin–Michael–Michael cascade.
Scheme 43: NHC-Brønsted base co-catalysis in a benzoin–Michael–Michael cascade.
Further investigations in Cheng’s group revealed an intriguing divergent catalytic dimerisation of 2-formylcinnamates 72. Co-operative catalysis by NHC precatalyst 73 and a Lewis acid (titanium isopropoxide) afforded isochromenone derivatives 74 via a sequence of reactions initiated by a benzoin condensation. Treatment of 72 with the NHC precatalyst 73 alone, on the other hand, afforded isochromeno(4,3-c)isochromene derivatives 75 (Scheme 44) [61].
![[1860-5397-12-47-i44]](/bjoc/content/inline/1860-5397-12-47-i44.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 44: Divergent catalytic dimerization of 2-formylcinnamates.
Scheme 44: Divergent catalytic dimerization of 2-formylcinnamates.
A one-pot multicatalytic reaction for the asymmetric synthesis of complex tetracyclic tetrahydrocarbazole derivatives from readily available precursors was described by Melchiorre. A Diels–Alder reaction of indole-2,3-quinodimethane 76 (generated from 77 and the prolinol catalyst 78 ) with the enone 79 affords a tetrahydrocarbazole derivative 80. The NHC precatalyst 22 then promotes an intramolecular cross-benzoin condensation of the keto-aldehyde to furnish the tetracyclic product 81 (Scheme 45). The yields are moderate; however excellent diastereo- and enantioselectivities were observed for the one-pot reaction [62].
![[1860-5397-12-47-i45]](/bjoc/content/inline/1860-5397-12-47-i45.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 45: One-pot, multicatalytic asymmetric synthesis of tetrahydrocarbazole derivatives.
Scheme 45: One-pot, multicatalytic asymmetric synthesis of tetrahydrocarbazole derivatives.
In a similar fashion, an asymmetric multicatalytic cascade reaction involving the dienal 82 and unsaturated cyclic sulfonylimine 83 afforded spiro-fused cycloadducts 84 in good yield and enantioselectivity [63]. Initially, the trienamine 85 is generated by the action of prolinol catalyst 86 on the dienal 82. The former (85) then undergoes a Diels–Alder reaction with the sulfonylimine 83 to generate the keto-aldehyde 87. Finally, the NHC precatalyst 22 mediates a cross-benzoin reaction of the latter to furnish the spirocyclic product 84 (Scheme 46).
![[1860-5397-12-47-i46]](/bjoc/content/inline/1860-5397-12-47-i46.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 46: NHC-chiral secondary amine co-catalysis for the synthesis of complex spirocyclic scaffolds.
Scheme 46: NHC-chiral secondary amine co-catalysis for the synthesis of complex spirocyclic scaffolds.
Conclusion
The first report of a benzoin reaction by Wöhler and Liebig appeared merely four years after the former disclosed the paradigm-changing urea synthesis. However, detailed investigations of this reaction remained elusive due to a variety of reasons, the toxicity of cyanide catalysts being one of them. Breslow’s discovery in 1958 of the thiazolylidene-catalysed benzoin condensation via polarity reversal of aldehydes formed the conceptual basis for the later development of NHC-organocatalysis. The rekindling of interest in NHC-catalysed benzoin reactions coincided with the emergence of N-heterocyclic carbenes in the late twentieth century as non-toxic, readily available and versatile catalysts for a variety of organic transformations. Since then, a number of reports on a variety of benzoin reactions have appeared in the literature. They include homo, crossed, intramolecular and various asymmetric benzoin reactions leading to products that are difficult to access by other means. Aza-benzoin reactions, intramolecular benzoin condensations, use of aldehyde surrogates and use of non-carbonyl electrophiles (nitroso compounds) are some of the developments that revamped the synthetically unattractive, monotonous image of benzoin condensations. The driving force behind this remarkable evolution of benzoin reaction is NHC-catalysis. Benzoin chemistry is well-set to benefit, in the near future, from new developments in the rapidly growing realm of NHC-catalysis.
References
-
Wöhler, F.; Liebig, J. Ann. Pharm. 1832, 3, 249. doi:10.1002/jlac.18320030302
Return to citation in text: [1] -
Ukai, T.; Tanaka, R.; Dokawa, T. J. Pharm. Soc. Jpn. 1943, 63, 296.
Return to citation in text: [1] -
Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719. doi:10.1021/ja01547a064
Return to citation in text: [1] [2] -
Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 1988, 110, 6463. doi:10.1021/ja00227a028
Return to citation in text: [1] -
Arduengo, A. J., III; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361. doi:10.1021/ja00001a054
Return to citation in text: [1] -
Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307. doi:10.1021/acs.chemrev.5b00060
Return to citation in text: [1] [2] -
Enders, D.; Niemeir, O.; Henseler, A. Chem. Rev. 2007, 107, 5606. doi:10.1021/cr068372z
Return to citation in text: [1] [2] -
Lapworth, A. J. Chem. Soc., Trans. 1903, 83, 995. doi:10.1039/CT9038300995
Return to citation in text: [1] -
Stetter, H.; Rämsch, R. Y.; Kuhlmann, H. Synthesis 1976, 733. doi:10.1055/s-1976-24177
Return to citation in text: [1] -
Xu, L.-W.; Gao, Y.; Yin, J.-J.; Li, L.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 5317. doi:10.1016/j.tetlet.2005.06.015
Return to citation in text: [1] -
Iwamoto, K.; Hamaya, M.; Hashimoto, N.; Kimura, H.; Suzuki, Y.; Sato, M. Tetrahedron Lett. 2006, 47, 7175. doi:10.1016/j.tetlet.2006.07.153
Return to citation in text: [1] -
Iwamoto, K.; Kimura, H.; Oike, M.; Sato, M. Org. Biomol. Chem. 2008, 6, 912. doi:10.1039/B719430G
Return to citation in text: [1] -
Ma, Y.; Wei, S.; Wu, J.; Yang, F.; Liu, B.; Lan, J.; Yang, S.; You, J. Adv. Synth. Catal. 2008, 350, 2645. doi:10.1002/adsc.200800371
Return to citation in text: [1] -
Enders, D.; Han, J. Tetrahedron: Asymmetry 2008, 19, 1367. doi:10.1016/j.tetasy.2008.05.017
Return to citation in text: [1] -
O’Toole, S. E.; Connon, S. J. Org. Biomol. Chem. 2009, 7, 3584. doi:10.1039/B908517C
Return to citation in text: [1] -
Brand, J. P.; Siles, J. I. O.; Waser, J. Synlett 2010, 881. doi:10.1055/s-0029-1219543
Return to citation in text: [1] -
Soeta, T.; Tabatake, Y.; Inomata, K.; Ukaji, Y. Tetrahedron 2012, 68, 894. doi:10.1016/j.tet.2011.11.028
Return to citation in text: [1] -
Rafiński, Z.; Kozakiewicz, A.; Rafińska, K. Tetrahedron 2014, 70, 5739. doi:10.1016/j.tet.2014.06.066
Return to citation in text: [1] -
Baragwanath, L.; Rose, C. A.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2009, 74, 9214. doi:10.1021/jo902018j
Return to citation in text: [1] -
Matsumoto, T.; Ohishi, M.; Inoue, S. J. Org. Chem. 1985, 50, 603. doi:10.1021/jo00205a010
Return to citation in text: [1] -
Kuhl, N.; Glorius, F. Chem. Commun. 2011, 47, 573. doi:10.1039/C0CC02416C
Return to citation in text: [1] -
Jin, M. Y.; Kim, S. M.; Han, H.; Ryu, D. H.; Yang, J. W. Org. Lett. 2011, 13, 880. doi:10.1021/ol102937w
Return to citation in text: [1] -
Rose, C. A.; Gundala, S.; Connon, S. J.; Zeitler, K. Synthesis 2011, 190. doi:10.1055/s-0030-1258363
Return to citation in text: [1] -
Piel, I.; Pawelczyk, M. D.; Hirano, K.; Fröhlich, R.; Glorius, F. Eur. J. Org. Chem. 2011, 5475. doi:10.1002/ejoc.201100870
Return to citation in text: [1] -
Jin, M. Y.; Kim, S. M.; Mao, H.; Ryu, D. H.; Song, C. E.; Yang, J. W. Org. Biomol. Chem. 2014, 12, 1547. doi:10.1039/C3OB42486C
Return to citation in text: [1] -
Langdon, S. M.; Wilde, M. M. D.; Thai, K.; Gravel, M. J. Am. Chem. Soc. 2014, 136, 7539. doi:10.1021/ja501772m
Return to citation in text: [1] -
Enders, D.; Henseler, A. Adv. Synth. Catal. 2009, 351, 1749. doi:10.1002/adsc.200900247
Return to citation in text: [1] -
Enders, D.; Henseler, A.; Lowins, S. Synthesis 2009, 4125. doi:10.1055/s-0029-1217070
Return to citation in text: [1] -
Enders, D.; Grossmann, A.; Fronert, J.; Raabe, G. Chem. Commun. 2010, 46, 6282. doi:10.1039/C0CC02013C
Return to citation in text: [1] -
Rose, C. A.; Gundala, S.; Fagan, C.-L.; Franz, J. F.; Connon, S. J.; Zeitler, K. Chem. Sci. 2012, 3, 735. doi:10.1039/C2SC00622G
Return to citation in text: [1] -
Thai, K.; Langdon, S. M.; Bilodeau, F.; Gravel, M. Org. Lett. 2013, 15, 2214. doi:10.1021/ol400769t
Return to citation in text: [1] -
Goodman, C. G.; Johnson, J. S. J. Am. Chem. Soc. 2014, 136, 14698. doi:10.1021/ja508521a
Return to citation in text: [1] -
Sánchez-Díez, E.; Fernández, M.; Uria, U.; Reyes, E.; Carrillo, L.; Vicario, J. L. Chem. – Eur. J. 2015, 21, 8384. doi:10.1002/chem.201501044
Return to citation in text: [1] -
Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J. J.; Reider, P. J. J. Am. Chem. Soc. 2001, 123, 9696. doi:10.1021/ja0165943
Return to citation in text: [1] -
Sun, F.-g.; Ye, S. Org. Biomol. Chem. 2011, 9, 3632. doi:10.1039/C1OB05092C
Return to citation in text: [1] -
Li, G.-Q.; Dai, L.-X.; You, S.-L. Chem. Commun. 2007, 852. doi:10.1039/B611646A
Return to citation in text: [1] -
DiRocco, D. A.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 5904. doi:10.1002/anie.201202442
Return to citation in text: [1] -
Uno, T.; Kobayashi, Y.; Takemoto, Y. Beilstein J. Org. Chem. 2012, 8, 1499. doi:10.3762/bjoc.8.169
Return to citation in text: [1] -
Mattson, A. E.; Scheidt, K. A. Org. Lett. 2004, 6, 4363. doi:10.1021/ol0481129
Return to citation in text: [1] -
Mennen, S. M.; Gipson, J. D.; Kim, Y. R.; Miller, S. J. J. Am. Chem. Soc. 2005, 127, 1654. doi:10.1021/ja042650z
Return to citation in text: [1] -
Sun, L.-H.; Liang, Z.-Q.; Jia, W.-Q.; Ye, S. Angew. Chem., Int. Ed. 2013, 52, 5803. doi:10.1002/anie.201301304
Return to citation in text: [1] -
Xu, J.; Mou, C.; Zhu, T.; Song, B.-A.; Chi, Y. R. Org. Lett. 2014, 16, 3272. doi:10.1021/ol501286e
Return to citation in text: [1] -
Wilde, M. M. D.; Gravel, M. Org. Lett. 2014, 16, 5308. doi:10.1021/ol5024807
Return to citation in text: [1] -
Sun, Z.-X.; Cheng, Y. Org. Biomol. Chem. 2012, 10, 4088. doi:10.1039/C2OB25137J
Return to citation in text: [1] -
Song, X.; Ni, Q.; Grossmann, A.; Enders, D. Chem. – Asian J. 2013, 8, 2965. doi:10.1002/asia.201300938
Return to citation in text: [1] -
Cookson, R. C.; Lane, R. M. J. Chem. Soc., Chem. Commun. 1976, 804. doi:10.1039/C39760000804
Return to citation in text: [1] -
Hachisu, Y.; Bode, J. W.; Suzuki, K. J. Am. Chem. Soc. 2003, 125, 8432. doi:10.1021/ja035308f
Return to citation in text: [1] -
Takikawa, H.; Hachisu, Y.; Bode, J. W.; Suzuki, K. Angew. Chem., Int. Ed. 2006, 45, 3492. doi:10.1002/anie.200600268
Return to citation in text: [1] -
Enders, D.; Niemeir, O. Synlett 2004, 2111. doi:10.1055/s-2004-831306
Return to citation in text: [1] -
Enders, D.; Niemeir, O.; Balensiefer, T. Angew. Chem., Int. Ed. 2006, 45, 1463. doi:10.1002/anie.200503885
Return to citation in text: [1] -
Li, Y.; Feng, Z.; You, S.-L. Chem. Commun. 2008, 2263. doi:10.1039/B801004H
Return to citation in text: [1] -
Kankala, S.; Edulla, R.; Modem, S.; Vadde, R.; Vasam, C. S. Tetrahedron Lett. 2011, 52, 3828. doi:10.1016/j.tetlet.2011.05.070
Return to citation in text: [1] -
Ema, T.; Akihara, K.; Obayashi, R.; Sakai, T. Adv. Synth. Catal. 2012, 354, 3283. doi:10.1002/adsc.201200499
Return to citation in text: [1] -
Lathrop, S. P.; Rovis, T. J. Am. Chem. Soc. 2009, 131, 13628. doi:10.1021/ja905342e
Return to citation in text: [1] -
Ozboya, K. E.; Rovis, T. Chem. Sci. 2011, 2, 1835. doi:10.1039/C1SC00175B
Return to citation in text: [1] -
Enders, D.; Grossmann, A.; Huang, H.; Raabe, G. Eur. J. Org. Chem. 2011, 4298. doi:10.1002/ejoc.201100690
Return to citation in text: [1] -
Stockton, K. P.; Greatrex, B. W.; Taylor, D. K. J. Org. Chem. 2014, 79, 5088. doi:10.1021/jo500645z
Return to citation in text: [1] -
Jia, M.-Q.; You, S.-L. ACS Catal. 2013, 3, 622. doi:10.1021/cs4000014
Return to citation in text: [1] -
Rafiński, Z.; Kozakiewicz, A. J. Org. Chem. 2015, 80, 7468. doi:10.1021/acs.joc.5b01029
Return to citation in text: [1] -
Tong, Y.-f.; Mao, J.-h.; Wu, S.; Zhao, Y.; Cheng, Y. J. Org. Chem. 2014, 79, 2075. doi:10.1021/jo4027758
Return to citation in text: [1] -
Dang, H.-Y.; Wang, Z.-T.; Cheng, Y. Org. Lett. 2014, 16, 5520. doi:10.1021/ol502791s
Return to citation in text: [1] -
Liu, Y.; Nappi, M.; Escudero-Adán, E. C.; Melchiorre, P. Org. Lett. 2012, 14, 1310. doi:10.1021/ol300192p
Return to citation in text: [1] -
Ma, C.; Gu, J.; Teng, B.; Zhou, Q.-Q.; Li, R.; Chen, Y.-C. Org. Lett. 2013, 15, 6206. doi:10.1021/ol4030474
Return to citation in text: [1]
| 27. | Enders, D.; Henseler, A. Adv. Synth. Catal. 2009, 351, 1749. doi:10.1002/adsc.200900247 |
| 28. | Enders, D.; Henseler, A.; Lowins, S. Synthesis 2009, 4125. doi:10.1055/s-0029-1217070 |
| 29. | Enders, D.; Grossmann, A.; Fronert, J.; Raabe, G. Chem. Commun. 2010, 46, 6282. doi:10.1039/C0CC02013C |
| 36. | Li, G.-Q.; Dai, L.-X.; You, S.-L. Chem. Commun. 2007, 852. doi:10.1039/B611646A |
| 37. | DiRocco, D. A.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 5904. doi:10.1002/anie.201202442 |
| 34. | Murry, J. A.; Frantz, D. E.; Soheili, A.; Tillyer, R.; Grabowski, E. J. J.; Reider, P. J. J. Am. Chem. Soc. 2001, 123, 9696. doi:10.1021/ja0165943 |
| 32. | Goodman, C. G.; Johnson, J. S. J. Am. Chem. Soc. 2014, 136, 14698. doi:10.1021/ja508521a |
| 33. | Sánchez-Díez, E.; Fernández, M.; Uria, U.; Reyes, E.; Carrillo, L.; Vicario, J. L. Chem. – Eur. J. 2015, 21, 8384. doi:10.1002/chem.201501044 |
| 30. | Rose, C. A.; Gundala, S.; Fagan, C.-L.; Franz, J. F.; Connon, S. J.; Zeitler, K. Chem. Sci. 2012, 3, 735. doi:10.1039/C2SC00622G |
| 31. | Thai, K.; Langdon, S. M.; Bilodeau, F.; Gravel, M. Org. Lett. 2013, 15, 2214. doi:10.1021/ol400769t |
| 38. | Uno, T.; Kobayashi, Y.; Takemoto, Y. Beilstein J. Org. Chem. 2012, 8, 1499. doi:10.3762/bjoc.8.169 |
| 39. | Mattson, A. E.; Scheidt, K. A. Org. Lett. 2004, 6, 4363. doi:10.1021/ol0481129 |
| 40. | Mennen, S. M.; Gipson, J. D.; Kim, Y. R.; Miller, S. J. J. Am. Chem. Soc. 2005, 127, 1654. doi:10.1021/ja042650z |
| 47. | Hachisu, Y.; Bode, J. W.; Suzuki, K. J. Am. Chem. Soc. 2003, 125, 8432. doi:10.1021/ja035308f |
| 48. | Takikawa, H.; Hachisu, Y.; Bode, J. W.; Suzuki, K. Angew. Chem., Int. Ed. 2006, 45, 3492. doi:10.1002/anie.200600268 |
| 45. | Song, X.; Ni, Q.; Grossmann, A.; Enders, D. Chem. – Asian J. 2013, 8, 2965. doi:10.1002/asia.201300938 |
| 46. | Cookson, R. C.; Lane, R. M. J. Chem. Soc., Chem. Commun. 1976, 804. doi:10.1039/C39760000804 |
| 44. | Sun, Z.-X.; Cheng, Y. Org. Biomol. Chem. 2012, 10, 4088. doi:10.1039/C2OB25137J |
| 41. | Sun, L.-H.; Liang, Z.-Q.; Jia, W.-Q.; Ye, S. Angew. Chem., Int. Ed. 2013, 52, 5803. doi:10.1002/anie.201301304 |
| 42. | Xu, J.; Mou, C.; Zhu, T.; Song, B.-A.; Chi, Y. R. Org. Lett. 2014, 16, 3272. doi:10.1021/ol501286e |
| 50. | Enders, D.; Niemeir, O.; Balensiefer, T. Angew. Chem., Int. Ed. 2006, 45, 1463. doi:10.1002/anie.200503885 |
| 5. | Arduengo, A. J., III; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361. doi:10.1021/ja00001a054 |
| 14. | Enders, D.; Han, J. Tetrahedron: Asymmetry 2008, 19, 1367. doi:10.1016/j.tetasy.2008.05.017 |
| 4. | Igau, A.; Grutzmacher, H.; Baceiredo, A.; Bertrand, G. J. Am. Chem. Soc. 1988, 110, 6463. doi:10.1021/ja00227a028 |
| 15. | O’Toole, S. E.; Connon, S. J. Org. Biomol. Chem. 2009, 7, 3584. doi:10.1039/B908517C |
| 6. | Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307. doi:10.1021/acs.chemrev.5b00060 |
| 7. | Enders, D.; Niemeir, O.; Henseler, A. Chem. Rev. 2007, 107, 5606. doi:10.1021/cr068372z |
| 56. | Enders, D.; Grossmann, A.; Huang, H.; Raabe, G. Eur. J. Org. Chem. 2011, 4298. doi:10.1002/ejoc.201100690 |
| 13. | Ma, Y.; Wei, S.; Wu, J.; Yang, F.; Liu, B.; Lan, J.; Yang, S.; You, J. Adv. Synth. Catal. 2008, 350, 2645. doi:10.1002/adsc.200800371 |
| 57. | Stockton, K. P.; Greatrex, B. W.; Taylor, D. K. J. Org. Chem. 2014, 79, 5088. doi:10.1021/jo500645z |
| 9. | Stetter, H.; Rämsch, R. Y.; Kuhlmann, H. Synthesis 1976, 733. doi:10.1055/s-1976-24177 |
| 11. | Iwamoto, K.; Hamaya, M.; Hashimoto, N.; Kimura, H.; Suzuki, Y.; Sato, M. Tetrahedron Lett. 2006, 47, 7175. doi:10.1016/j.tetlet.2006.07.153 |
| 54. | Lathrop, S. P.; Rovis, T. J. Am. Chem. Soc. 2009, 131, 13628. doi:10.1021/ja905342e |
| 12. | Iwamoto, K.; Kimura, H.; Oike, M.; Sato, M. Org. Biomol. Chem. 2008, 6, 912. doi:10.1039/B719430G |
| 52. | Kankala, S.; Edulla, R.; Modem, S.; Vadde, R.; Vasam, C. S. Tetrahedron Lett. 2011, 52, 3828. doi:10.1016/j.tetlet.2011.05.070 |
| 6. | Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307. doi:10.1021/acs.chemrev.5b00060 |
| 7. | Enders, D.; Niemeir, O.; Henseler, A. Chem. Rev. 2007, 107, 5606. doi:10.1021/cr068372z |
| 10. | Xu, L.-W.; Gao, Y.; Yin, J.-J.; Li, L.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 5317. doi:10.1016/j.tetlet.2005.06.015 |
| 53. | Ema, T.; Akihara, K.; Obayashi, R.; Sakai, T. Adv. Synth. Catal. 2012, 354, 3283. doi:10.1002/adsc.201200499 |
| 18. | Rafiński, Z.; Kozakiewicz, A.; Rafińska, K. Tetrahedron 2014, 70, 5739. doi:10.1016/j.tet.2014.06.066 |
| 16. | Brand, J. P.; Siles, J. I. O.; Waser, J. Synlett 2010, 881. doi:10.1055/s-0029-1219543 |
| 17. | Soeta, T.; Tabatake, Y.; Inomata, K.; Ukaji, Y. Tetrahedron 2012, 68, 894. doi:10.1016/j.tet.2011.11.028 |
| 61. | Dang, H.-Y.; Wang, Z.-T.; Cheng, Y. Org. Lett. 2014, 16, 5520. doi:10.1021/ol502791s |
| 62. | Liu, Y.; Nappi, M.; Escudero-Adán, E. C.; Melchiorre, P. Org. Lett. 2012, 14, 1310. doi:10.1021/ol300192p |
| 59. | Rafiński, Z.; Kozakiewicz, A. J. Org. Chem. 2015, 80, 7468. doi:10.1021/acs.joc.5b01029 |
| 60. | Tong, Y.-f.; Mao, J.-h.; Wu, S.; Zhao, Y.; Cheng, Y. J. Org. Chem. 2014, 79, 2075. doi:10.1021/jo4027758 |
| 25. | Jin, M. Y.; Kim, S. M.; Mao, H.; Ryu, D. H.; Song, C. E.; Yang, J. W. Org. Biomol. Chem. 2014, 12, 1547. doi:10.1039/C3OB42486C |
| 26. | Langdon, S. M.; Wilde, M. M. D.; Thai, K.; Gravel, M. J. Am. Chem. Soc. 2014, 136, 7539. doi:10.1021/ja501772m |
| 23. | Rose, C. A.; Gundala, S.; Connon, S. J.; Zeitler, K. Synthesis 2011, 190. doi:10.1055/s-0030-1258363 |
| 24. | Piel, I.; Pawelczyk, M. D.; Hirano, K.; Fröhlich, R.; Glorius, F. Eur. J. Org. Chem. 2011, 5475. doi:10.1002/ejoc.201100870 |
| 22. | Jin, M. Y.; Kim, S. M.; Han, H.; Ryu, D. H.; Yang, J. W. Org. Lett. 2011, 13, 880. doi:10.1021/ol102937w |
| 19. | Baragwanath, L.; Rose, C. A.; Zeitler, K.; Connon, S. J. J. Org. Chem. 2009, 74, 9214. doi:10.1021/jo902018j |
| 63. | Ma, C.; Gu, J.; Teng, B.; Zhou, Q.-Q.; Li, R.; Chen, Y.-C. Org. Lett. 2013, 15, 6206. doi:10.1021/ol4030474 |
| 20. | Matsumoto, T.; Ohishi, M.; Inoue, S. J. Org. Chem. 1985, 50, 603. doi:10.1021/jo00205a010 |
© 2016 Menon et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)