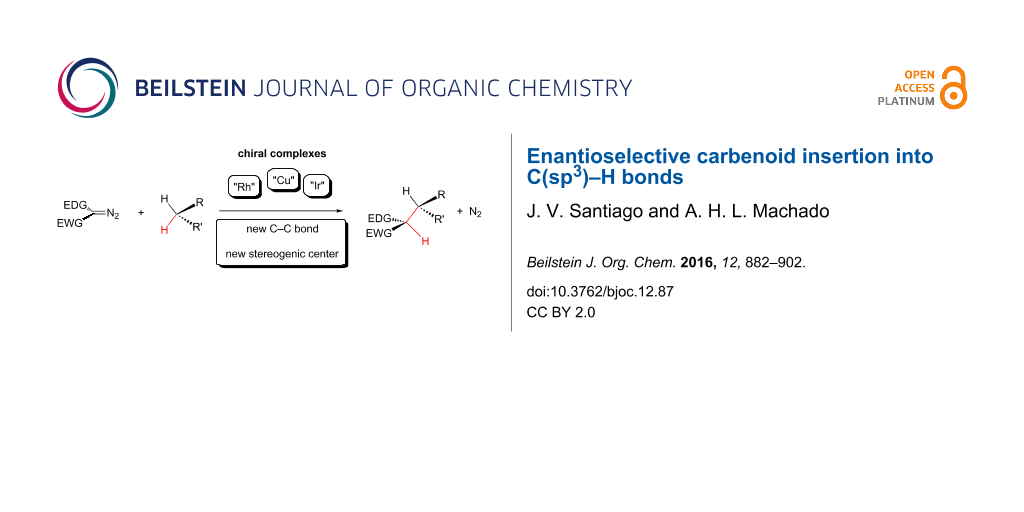
Abstract
The enantioselective carbenoid insertion into C(sp3)–H bonds is an important tool for the synthesis of complex molecules due to the high control of enantioselectivity in the formation of stereogenic centers. This paper presents a brief review of the early issues, related mechanistic studies and recent applications on this chemistry area.

Graphical Abstract
Introduction
One of the major challenges met in organic synthesis is the formation of carbon–carbon bonds, in particular in a stereoselective way. Nucleophilic substitution reactions, radical reactions, cross-coupling reactions and the Heck reaction are well-known approaches available to this goal. These reactions are based on the polar characteristic of the carbon–halogen or carbon–pseudohalogen bonds, as a result of the electronegativity difference between these atoms. Despite the proven success of these transformations, they are limited to pre-functionalization of the chemical structure of interest with halogen atoms or pseudohalogen functional groups.
One approach that has been gaining increasing attention, by not requiring the presence of a strongly polarized chemical bond, is the C(sp3)–H bonds activation by carbenoids [1]. The enantioselective insertion of these organometallic species into these non-polarized bonds is a recent topic in the chemical literature, when compared to the first reports of carbenoid chemistry around the 1950s.
Carbene is a molecule bearing a functional group with a divalent neutral carbon. This structural framework results in the presence of a nonbonding electron pair that may adopt two electronic configurations: singlet and triplet (Figure 1). A carbenoid is an organometallic complex where the carbene acts as a neutral ligand to a metal center. This ensures a greater stability of the carbene, allows the modulation of its reactivity, and controls the chemo-, regio- and stereoselectivity in reactions.
![[1860-5397-12-87-1]](/bjoc/content/figures/1860-5397-12-87-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Singlet carbene, triplet carbene and carbenoids.
Figure 1: Singlet carbene, triplet carbene and carbenoids.
The activation of the C(sp3)–H bond needs an appropriate interaction between the carbenoid intermediate and the carbon atom of the C(sp3)–H. Depending on the electronic demand of the substituent attached to the carbene carbon atom, the insertion reaction can be more or less selective. Very electrophilic carbenoid intermediates, for example, display little regio- and stereoselectivity, favoring the occurrence of side reactions. A less electrophilic carbenoid intermediate, on the other hand, has a lower reactivity, but its regio- and stereoselectivity are better [2].
The electrophilicity of the carbenoid intermediate is related to the substituents present in its structure. Electron-withdrawing groups (EWGs) increase the electrophilicity of the carbon atom in the carbenoid and electron-donating groups (EDGs) act with the opposite effect. Due to these observations, a carbenoid intermediate can be divided in three different classes based on the electronic nature of the chemical groups attached to its structure: acceptor, donor/acceptor and acceptor/acceptor (Figure 2) [2]. The modification of the substituents on the carbenoid intermediate can change its reactivity and hence the selectivity of the carbenoid reaction.
![[1860-5397-12-87-2]](/bjoc/content/figures/1860-5397-12-87-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Classification of the carbenoid intermediates by the electronic nature of the groups attached to the divalent carbene carbon.
Figure 2: Classification of the carbenoid intermediates by the electronic nature of the groups attached to th...
The most commonly used diazo compounds rely on the formation of a donor/acceptor carbenoid intermediate type. The EWG increases the electrophilicity and reactivity of the donor/acceptor carbenoid, while an EDG increases its stability and selectivity [2].
Despite the importance of the electronic factors to the reactivity and selectivity of carbenoid intermediates, steric and conformational effects are also determining factors for carbenoid chemistry. Steric as well as electronic factors and the chemical properties of the ligands around the metal center also determine significantly the type of insertion performed by the carbenoid intermediate. The complexes used for the formation of carbenoids in enantioselective insertion reactions must present a balance between steric and electronic factors, to promote the formation of a specific enantiomer.
The search for the best balance of these properties of the carbenoid intermediates was also sought through the use of different metals such as copper [3], rhodium [4], iron [5], ruthenium [6], iridium [7], osmium [8], and others. From these, copper and rhodium have been the most frequently used ones in carbenoid insertion reactions.
Copper carbenoids having a higher electrophilic character display a great reactivity, but little selectivity in insertion reactions. Despite these features, only recently the insertion of chiral copper carbenoids into a C(sp3)–H bond has gained special attention, as in the works of Muler and Boléa [9], Flynn [10], Stattery [11] and their respective co-workers. The most selective copper carbenoids are those generated from chiral bis(oxazoline) ligands in the presence of copper(I) triflate (CuOTf) (Figure 3).
![[1860-5397-12-87-3]](/bjoc/content/figures/1860-5397-12-87-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Chiral bis(oxazoline) ligands used in enantioselective copper carbenoid insertion.
Figure 3: Chiral bis(oxazoline) ligands used in enantioselective copper carbenoid insertion.
The rhodium carbenoid intermediates are prferably used in enantioselective insertion reactions. They are more often found as dirhodium(II) complexes. Only one of the two metal atoms present in the chemical structure effectively participates in the insertion reaction. The other rhodium atom withdraws electron density from the rhodium atom involved in the insertion mechanism increasing therefore its electrophilic character [12]. Four specific types of chiral rhodium(II) complexes can be found as catalyst in enantioselective insertion reactions of carbenoids in C(sp3)–H bonds: carboxylates [13-19], carboxamides [20-23], phosphates [24,25], and ortho-arylphosphines [26-30].
This work aims to review the chemical literature, since 2009 [31,32] until the end of 2015, concerning the development of catalytic systems able to promote enantioselective carbenoid insertion into C(sp3)–H bonds, the mechanistic aspects recently discovered to the known catalytic systems and the application of these synthetic tools to the organic synthesis of natural products.
Review
Historic perspective on the carbenoid reaction insertion into X–H bonds
One of the former works to address the chemistry of carbenoid was reported in 1952 by Peter Yates, although the author does not specifically use the term carbenoid, but – carbene–copper complex (Scheme 1) [3]. In his opinion, the copper catalyst promotes the decomposition of diazoketones to afford "free carbenes", the chemical intermediates responsible for the insertion reaction in X–H bonds (X = O, S, N, or C).
![[1860-5397-12-87-i1]](/bjoc/content/inline/1860-5397-12-87-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pioneering work of Peter Yates on the carbenoid insertion reaction into X–H bonds (where X = O, S, N, or C).
Scheme 1: Pioneering work of Peter Yates on the carbenoid insertion reaction into X–H bonds (where X = O, S, ...
Ledon et al, in 1973, showed a very important feature of the carbenoid insertions into C(sp3)–H bonds. The intramolecular reaction of the chiral diazomalonate (S)-3 led to the insertion of the carbenoid intermediate into the C(sp3)–H of the stereogenic center with full retention of the asymmetric carbon configuration (Scheme 2) [33].
![[1860-5397-12-87-i2]](/bjoc/content/inline/1860-5397-12-87-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Copper carbenoid insertion into C(sp3)–H bond of a stereogenic center with full retention of the asymmetric carbon configuration.
Scheme 2: Copper carbenoid insertion into C(sp3)–H bond of a stereogenic center with full retention of the as...
The authors demonstrated that an insertion reaction in C(sp3)–H bonds only occurs with considerable yield when small amounts of copper powder or copper salts, such as CuSO4 and CuCN, were employed. Even using the term "carbenoid", the work does not present the formation of a copper carbenoid intermediate. It only suggests an insertion reaction occurring through free carbenes with copper working only as a catalyst to promote the carbene formation.
In 1985, Taber and coworkers reported the synthesis of (+)-α-cuparenone (8) through the construction of a five-membered ring prepared by an enantioselective carbenoid insertion into a C(sp3)–H bond (Scheme 3) [34]. To carry out the cyclization, the carbenoid was formed by the action of Rh2(OAc)4 on the diazo compound 6. That intermediate intramolecularly inserted into the C(sp3)–H bond of the asymmetric carbon to yield ketoester 7 in 67% yield. This latter compound was converted to (+)-α-cuparenone (8) in 26% yield and 96% enantiomeric excess.
![[1860-5397-12-87-i3]](/bjoc/content/inline/1860-5397-12-87-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Carbenoid insertion into a C(sp3)–H bond as the key step of the Taber’s (+)-α-cuparenone (8) synthesis.
Scheme 3: Carbenoid insertion into a C(sp3)–H bond as the key step of the Taber’s (+)-α-cuparenone (8) synthe...
In the late 1980s, many studies have been published by Taber [35], Sonawane [36], Doyle [37] and their respective coworkers regarding the regiochemistry of carbenoid insertion into C(sp3)–H bonds, and also the steric and electronic factors related to this insertion.
The first example of an enantioselective carbenoid insertion reaction in chemical bonds catalyzed by chiral metal complexes was introduced in 1966 by Noyori and coworkers (Scheme 4) [38]. In addition to the novelty of the use of the chiral copper complex 11 for controlling the enantioselectivity, the authors proposed the participation of the copper carbenoid 13, formed from the reaction between the copper complex 11 and methyl diazoacetate 9 as active intermediate in the catalytic cycle of this transformation.
![[1860-5397-12-87-i4]](/bjoc/content/inline/1860-5397-12-87-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: First enantioselective carbenoid insertion into C–O bonds catalyzed by chiral metallic complexes.
Scheme 4: First enantioselective carbenoid insertion into C–O bonds catalyzed by chiral metallic complexes.
From the 1990s, the enantioselective carbenoid insertion into C(sp3)–H bonds starts to be better discussed in the literature. Ikegami and coworkers reported the enantioselective insertion of α-diazo-β-ketoesters into C(sp3)–H bonds catalyzed by rhodium carboxylate complexes in their homochiral form (Table 1) [39]. Modest enantiomeric excesses were provided by the three tested catalysts. The reactions carried out with complex 17a and 17b show very similar stereoselectivity, forming the R-enantiomer of compound 16 as the main product after decarboxylation reaction. The catalyst 17c showed opposite enantioselectivity when compared to the catalysts 17a and 17b, with the S-enantiomer formed as the major product.
Table 1: Enantioselective insertion of α-diazo-β-ketoesters into C(sp3)–H bonds catalyzed by chiral rhodium(II) complexes 17a, 17b and 17c.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-87-i25.svg?max-width=637&scale=1.0)
|
||||
| R | catalyst | yield (%) | ee (%) | configuration of 16 |
|---|---|---|---|---|
| CH3 | 17a | 76 | 24 | R |
| CH3 | 17b | 73 | 24 | R |
| CH3 | 17c | 75 | 10 | S |
| C5H11 | 17a | 43 | 29 | R |
| CH=CH2 | 17a | 44 | 38 | R |
| CH=CH2 | 17b | 39 | 35 | R |
| CH=CH2 | 17c | 44 | 30 | S |
| Ph | 17a | 96 | 46 | R |
| Ph | 17b | 87 | 43 | R |
| Ph | 17c | 73 | 13 | S |
In 1991, Doyle and coworkers published asymmetric synthesis of lactones from alkyl diazoacetates in high enantioselectivity by intramolecular rhodium carbenoid insertion into C(sp3)–H [40]. In this work, the authors introduced the enantiomeric rhodium(II) carboxamides complexes (R)-18 and (S)-18 (Figure 4).
![[1860-5397-12-87-4]](/bjoc/content/figures/1860-5397-12-87-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Chemical structures of complexes (R)-18 and (S)-18.
Figure 4: Chemical structures of complexes (R)-18 and (S)-18.
The authors could observe the enantioselective formation of the lactones 20 with high enantiomeric excess (Table 2). The carbenoid formed by (S)-18 favored the S configuration at the generated stereogenic center for most of the prepared lactones. The opposite preference, R configuration at the new stereogenic center of 20, was reported to the use of the enantiomeric rhodium complex (R)-18. When substrate 19f reacts under catalysis of rhodium(II) carboxamide complexes (R)-18 and (S)-18, the configuration of the new stereogenic center of 20f was reversed, probably due to the lack of the oxygen atom in the substituent R, as suggested by the authors.
Table 2: Enantioselective intramolecular insertion of carbenoids into C(sp3)–H bonds catalyzed by rhodium (II) carboxamides complexes (R)-18 and (S)-18.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-87-i26.svg?max-width=637&scale=1.0)
|
||||
| Reagent | catalyst | yield (%) | ee (%) | configuration of 20 |
|---|---|---|---|---|
| 19a | (S)-18 | 62 | 91 | S |
| 19a | (R)-18 | 73 | 91 | R |
| 19b | (S)-18 | 64 | 89 | S |
| 19b | (R)-18 | 63 | 89 | R |
| 19c | (S)-18 | 64 | 87 | S |
| 19c | (R)-18 | 69 | 87 | R |
| 19d | (S)-18 | 68 | 56 | S |
| 19d | (R)-18 | 70 | 57 | R |
| 19e | (S)-18 | 85 | 51 | S |
| 19f | (S)-18 | 42 | 46 | R |
| 19f | (R)-18 | 34 | 45 | S |
In 1997, Davies and Hansen reported the intermolecular carbenoid insertion into C(sp3)–H catalyzed by rhodium complex (S)-23 with good to excellent enantioselective control (Scheme 5) [41]. The best results were observed when the reaction was carried out at room temperature. A wide range of substituent groups were evaluated at para position of the aryldiazoacetate aromatic ring. The cyclic hydrocarbon reagents, also used as solvent, were cyclopentane, cyclohexane and cycloheptane.
![[1860-5397-12-87-i5]](/bjoc/content/inline/1860-5397-12-87-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Asymmetric carbenoid insertions into C(sp3)–H bonds of cycloalkanes catalyzed by chiral rhodium carboxylate complexes.
Scheme 5: Asymmetric carbenoid insertions into C(sp3)–H bonds of cycloalkanes catalyzed by chiral rhodium car...
Two factors are noteworthy in this work. Unlike the carboxamide complexes (R)-18 and (S)-18 previously reported by Doyle and coworkers (Table 2), where the complexation of the chiral ligand to rhodium atoms occurs through the carboxamide group, in the new chiral catalyst (S)-23 the rhodium atoms are complexed to the chiral ligands by the carboxylate group, similar to those chiral complexes presented by Ikegami and coworkers (Table 1). Another important feature of this work is, unlike to the work that preceded it, that the new stereogenic center is formed on the carbenoid carbon coordinated to the metal rhodium center and not on carbon-containing the C(sp3)–H bond activated by the carbenoid moiety.
The authors also reported in this work the insertion into the C(sp3)–H bond of tetrahydrofuran. This reaction showed good yield, regio-, diastereo- and enantioselectivity and represents the first example of the formation of a new stereogenic center out of the diazoacetate scaffold by an intermolecular carbenoid insertion into C(sp3)–H bond (Scheme 6).
![[1860-5397-12-87-i6]](/bjoc/content/inline/1860-5397-12-87-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: First diastereo and enantioselective intermolecular carbenoid insertion into tetrahydrofuran C(sp3)–H bond.
Scheme 6: First diastereo and enantioselective intermolecular carbenoid insertion into tetrahydrofuran C(sp3)...
Mechanism of the carbenoid insertion into the C(sp3)–H bond
Nakamura [12] and Doyle [37] were the first to do important contributions to the comprehension of the mechanism of this catalytic cycle. In a simplified form, the mechanism of the carbenoid insertion into a C(sp3)–H bond can be represented as outlined in Scheme 7.
![[1860-5397-12-87-i7]](/bjoc/content/inline/1860-5397-12-87-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Simplified mechanism of the carbenoid insertion into a C(sp3)–H bond.
Scheme 7: Simplified mechanism of the carbenoid insertion into a C(sp3)–H bond.
Nakamura et al. investigated the dirhodium tetracarboxylate-mediated carbenoid insertion reaction into C(sp3)–H bonds in more detail using the relationship between the transition-state structures and their corresponding free energies obtained by DFT investigation (Scheme 8) [12]. The insertion step primarily consists in the formation of the metal carbenoid 29 by the interaction of the diazo compound 28 and the dirhodium complex 27. In sequence, the reaction proceeds through the transition state TS-30 to release N2, and yields the carbenoid 31. The divalent carbon attached to the rhodium atom starts to interact with the hydrogen of the C(sp3)–H bond of the compound 32 to form the van der Waals complex 33 which undergoes through the transition state TS-34 to the product of the carbenoid insertion reaction 35, regenerating the dirhodium complex 27.
![[1860-5397-12-87-i8]](/bjoc/content/inline/1860-5397-12-87-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Nakamura’s carbenoid insertion into a C(sp3)–H bond catalytic cycle.
Scheme 8: Nakamura’s carbenoid insertion into a C(sp3)–H bond catalytic cycle.
In 2009, Davies and coworkers reported a DFT investigation of the relationship between the electronic characteristics of the substituent X attached to the carbenoid divalent carbon and the selectivity toward carbenoid insertion into σ C(sp3)–H bonds (Scheme 9) [42].
![[1860-5397-12-87-i9]](/bjoc/content/inline/1860-5397-12-87-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Investigation of the relationship between the electronic characteristics of the substituent X attached to the carbenoid divalent carbon and the selectivity of the carbenoid insertion into σ C(sp3)–H bonds.
Scheme 9: Investigation of the relationship between the electronic characteristics of the substituent X attac...
The authors found an exergonic carbenoid formation step and proposed two reasons for the selectivity toward insertion of this carbenoid into σ C(sp3)–H when X = Ph or H. The first reason concerns the relative stability of the carbenoids 38-Ph and 38-H. The first one, prepared from the donor/acceptor diazo compound 36-Ph, is 10.9 kcal more stable than the carbenoid 38-H obtained from the acceptor diazo compound 36-H. This observation was attributed to the stabilization of the partial positive charge on the divalent carbon of the transition state TS-40 provided by the phenyl donor group. The second reason concerns the large difference between the activation energy of these reactions which relies on the development of steric strain through the transitions state TS-40, less important when X = H. This study provided a simple empirical model able to predict the stereoselectivity of the intermolecular insertion of donor/acceptor dirhodium carbenoids into C(sp3)–H bonds properly validated by the results obtained by this research group during the development of the chiral catalyst (S)-23 (Scheme 10).
![[1860-5397-12-87-i10]](/bjoc/content/inline/1860-5397-12-87-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Empirical model to predict the stereoselectivity of the donor/acceptor dirhodium carbenoid insertion into C(sp3)–H catalyzed by (S)-23.
Scheme 10: Empirical model to predict the stereoselectivity of the donor/acceptor dirhodium carbenoid insertio...
Recent studies concerning the enantioselective carbenoid insertion into C(sp3)–H bonds
From 2000, the study of carbenoid chemistry has become more comprehensive. The focus of most recently published works is the development of new catalysts for carbenoid insertion reactions into C(sp3)–H bonds and also the insertion into X–H bonds, where X = N, O, S, Si and others.
Copper-based chiral catalysts
In 2002, Müller and Boléa published a study evaluating the enantioselective insertion of copper carbenoids formed from phenyliodonium ylides and diazo compounds (Table 3) [9]. This work is particularly important because, at that time, the carbenoids derived from rhodium complexes were the most used for insertion reactions in C(sp3)–H bonds.
Table 3: Enantioselective intramolecular insertion of copper carbenoids derived from phenyliodonium ylides and diazo compounds.
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-87-i27.svg?max-width=637&scale=1.0)
|
||||
| La | yield (%) from 42aa | ee (%) | yield (%) from 42bb | ee (%) |
|---|---|---|---|---|
| 1a | 51 | (R)-42 | 38 | (R)-15 |
| 1b | 49 | (R)-38 | 32 | (R)-18 |
| 45a | 52 | (S)-72 | 14 | (S)-31 |
| 45b | 46 | (S)-17 | 55 | (S)-22 |
| 2 | 46 | (R)-59 | 35 | (R)-60 |
| 46c | 47 | (S)-67 | 17 | (S)-51 |
| 47 | 11 | (S)-57 | 32 | (S)-18 |
aCH2Cl2 at 0 °C; bClCH2CH2Cl at 65 °C; cLigand 46 was used in 70% de. When 42a was cyclized by 46 with de > 98%, the ee of the product 43 increased to 70% ee favoring the same stereoisomer (S).
Comparing the results of Table 3, the same enantiomer was obtained mainly for both carbenoid precursors, ylide 42a and the diazo compound 42b. The authors suggested the formation of the same chiral copper carbenoid intermediate by the reaction of the in situ prepared chiral copper complexes with both 42a and 42b to provide the observed insertion products.
The reactions with phenyliodonium ylides 42a showed better ee when compared to that done with diazo compounds 42b. The authors attributed this observation to the large difference between the reaction temperatures, 0 °C to ylides versus 65 °C to diazo compounds. Higher temperatures increase the carbenoid formation rate by the chiral copper complexes as well as the carbene formation rate by direct decomposition of the precursors 42a and 42b. The competition between carbene insertion and chiral carbenoid insertion into C(sp3)–H bonds decreases the enantioselectivity of this transformation.
To confirm the copper carbenoid formation and its participation on the insertion reactions, the authors prepared the carbenoid precursors (R)-48a and (R)-48b and submit them to Rh2(OAc)4 or Cu(hfa)2 catalysis (Table 4). All reactions maintained the configuration of the asymmetric carbon where de insertion happened, independent to the carbenoid precursor and the catalyst, a strong evidence of the carbenoid intermediates formation.
Table 4: Experimental evidences of the carbenoid formation from (R)-48a and (R)-48b and its intramolecular insertion into C(sp3)–H bonds.
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-87-i28.svg?max-width=637&scale=1.0)
|
||||
| precursor | catalyst | time | (R)-49 yield (%) | ee (%) |
|---|---|---|---|---|
| (R)-48a | [Rh2(OAc)4] | 30 min | 59 | >98 |
| (R)-48a | [Cu(hfa)2] | 3h | 54 | >98 |
| (R)-48b | [Rh2(OAc)4] | 3h | 57 | >98 |
| (R)-48b | [Cu(hfa)2] | 3h | 36 | >98 |
In 2010, Maguire et al. studied the enantioselective insertion of copper carbenoid derived from α-diazosulfones into C(sp3)–H bonds [10]. In this work, the authors produced cyclic sulfones (thiopyrans) 52 with high enantioselectivity by using a combination of 5 mol % of copper chloride salt, 6 mol % of ligand 1c and 6 mol % of sodium tetrakis[(3,5-tri-fluoromethyl)phenyl]borate (NaBARF). The cyclic sulfones 52 were obtained in good yields and excellent enantiomeric excesses (85–98%) favoring the cis-1,2-di-substituted stereoisomer (Table 5).
Table 5: Insertion of asymmetric copper carbenoid C(sp3)–H bonds to prepare thiopyrans 52.
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-87-i29.svg?max-width=637&scale=1.0)
|
|||||
| diazo compound | R | R1 | time (h) | 52 yield (%) | ee (%) |
|---|---|---|---|---|---|
| 51a | OCH3 | Ph | 5 | 47 | 98 |
| 51b | OCH3 | 4-tolyl | 5 | 64 | 96 |
| 51c | OCH3 | 4-anisyl | 22 | 56 | 91 |
| 51d | OCH3 | 4-nitrophenyl | 2,5 | – | – |
| 51e | OCH3 | benzyl | 7 | 42 | 96 |
| 51f | OCH3 | ethyl | 16 | 68 | 97 |
| 51g | OBn | octyl | 22 | 66 | 90 |
| 51h | CH3 | Ph | 22 | 30 | 85 |
| 51i | Ph | Ph | 6 | 49 | 97 |
The authors also performed the copper carbenoid insertion reaction to yield five-membered cyclic sulfones 54, under similar experimental conditions, in moderate yields and enantiomeric excesses of the trans stereoisomer (Table 6).
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-87-i30.svg?max-width=637&scale=1.0)
Independent to the size of the product, the authors emphasize the low dependence of the enantioselectivity with respect to the structural nature of the substrates where the lowest results are observed for the substrate 51h (R = CH3). In contrast, the reaction times showed to be more dependent on the chemical structure of the substrates.
This research group has focused a lot of efforts to better comprehend the scope of this catalytic system, especially on features concerning the BARF salt effect [43,44] and electronic effects on the aromatic rings of the chiral ligands [45].
In 2014, Maguire at al reported the syntheses of N-heterocycles by the enantioselective insertion of copper carbenoids to α-nitrogen C(sp3)–H bonds of amides (Scheme 11) [46]. A wide range of bis(oxazolines) were evaluated as chiral ligands in dry dichloromethane with NaBARF as additive. Three catalytic systems, based on ligands (−)-45a, (+)-45a and (4S)-1e, showed a better performance (>82% ee). The transformation was regio and stereoselective where the main product was trans-γ-lactam (56). The chiral rhodium complexes (S)-17, (S)-18 and (S)-23 were also evaluated and yielded similar regio- and diastereoselectivity, however, with lower enantioselectivity when compared to the bis(oxazoline)/CuCl2/NaBARF catalytic system.
![[1860-5397-12-87-i11]](/bjoc/content/inline/1860-5397-12-87-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Asymmetric insertion of copper carbenoids in C(sp3)–H bonds to prepare trans-γ-lactam.
Scheme 11: Asymmetric insertion of copper carbenoids in C(sp3)–H bonds to prepare trans-γ-lactam.
Attempts to heterogeneous catalysis using chiral copper complexes were also done. Fraile et al reported, in 2011, the copper catalyst 60 for enantioselective insertion of carbenoid into O-heterocycles C(sp3)–H bonds (Table 7) [47,48]. The reaction was performed under homogeneous and heterogeneous conditions, with laponite as support for the catalyst. The reaction afforded moderate yields, diastereomeric ratio and enantioselectivity under both conditions. The supported catalytic system was reused over three cycles with no performance decrease. The same heterogeneous catalyst was also used to perform an enantioselective insertion of the carbenoid into benzylic C(sp3)–H bonds and similar results were observed [49].
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-87-i31.svg?max-width=637&scale=1.0)
In 2011, the same research group developed a new heterogeneous copper catalyst for carbenoid insertion into C(sp3)–H bonds [50]. The solid support was based on SiO2/Al2O3 and, after addition of ligand 1c to the reaction media, the reaction afforded moderate yields, diastereomeric ratio and enantioselectivity. These catalysts were reused over three cycles with progressive yield and enantioselectivity decrease.
Iridium-based chiral catalysts
Most recently, chiral iridium complexes have been used as catalyst for insertion reactions in C(sp3)–H bonds. In 2009, Suematsu and Katsuki published the first study addressed to the use of iridium-based chiral complexes as catalyst for the formation of carbenoid intermediates (Figure 5) [51]. The authors conducted insertion reactions in C(sp3)–H bonds in a diastereo- and enantioselective manner. For enantioselective insertion reactions the authors tested two specific iridium complexes, 61a and 61b.
![[1860-5397-12-87-5]](/bjoc/content/figures/1860-5397-12-87-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Iridium catalysts used by Suematsu and Katsuki for carbenoid insertion into C(sp3)–H bonds.
Figure 5: Iridium catalysts used by Suematsu and Katsuki for carbenoid insertion into C(sp3)–H bonds.
The authors used a wide range of α-substituted α-diazoacetates for performing insertion into substrates such as tetrahydrofuran (24) and 1,4-cyclohexadiene (62). Using these two compounds, the authors evaluated the best conditions for carrying out the reaction. When the iridium-catalyzed insertion reaction of carbenoids into tetrahydrofurans C(sp3)–H bond was performed at room temperature, the authors reported the formation of dimers of α-substituted α-diazoacetates as the main products of this reaction. This issue was circumvented when low temperatures, −50 °C, were used and the insertion reaction occurred with considerable yields and good enantiomeric excess (Table 8). According to the authors, the low temperature could reduce some type of steric strain on the transition state of the insertion reaction and avoid the dimer formation.
Table 8: Intermolecular insertion of chiral iridium carbenoid into THF C(sp3)–H bond.
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-87-i32.svg?max-width=637&scale=1.0)
|
|||||
| diazo compound | R1 | R2 | 25:26 | 25 yield (%) | 25 ee (%) |
|---|---|---|---|---|---|
| 21a | C6H5 | Me | 13:1 | 75 | 95 |
| 21b | p-MeOC6H4 | Me | >20:1 | 64 | 97 |
| 21c | p-ClC6H4 | Me | 19:1 | 82 | 94 |
| 21d | p-MeC6H4 | Me | 19:1 | 71 | 97 |
| 21e | p-BrC6H4 | Me | >20:1 | 76 | 93 |
| 21f | m-MeOC6H4 | Me | 9:1 | 75 | 97 |
| 21g | m-ClC6H4 | Me | >20:1 | 82 | 95 |
| 21h | 2-naphthyl | Me | >20:1 | 80 | 98 |
| 21i | o-MeOC6H4 | Me | >20:1 | 9 | 95 |
| 21j | Me | t-Bu | 13:1 | 70 | 90 |
For the insertion reaction of iridium carbenoid into the 1,4-cyclohexadiene (62) bis-allylic C(sp3)–H bond the authors observed the formation of two products, one resulting from iridium carbenoid insertion into the C(sp3)–H bond (63) and the other as a result of the cyclopropanation reaction (64, Table 9).
Table 9: Intermolecular insertion of chiral iridium carbenoids into the 1,4-cyclohexadiene (62) bis-allylic C(sp3)–H bond.
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-87-i33.svg?max-width=637&scale=1.0)
|
|||||
| diazo compound | R1 | R2 | 63:64 | 63 yield (%) | ee (%) o |
|---|---|---|---|---|---|
| 21a | C6H5 | Me | >20:1 | 91 | 94 |
| 21b | p-MeOC6H4 | Me | >20:1 | 39 | 90 |
| 21c | p-ClC6H4 | Me | >20:1 | 79 | 95 |
| 21f | m-MeOC6H4 | Me | >20:1 | 95 | 96 |
| 21g | m-ClC6H4 | Me | >20:1 | 80 | 99 |
| 21i | o-MeOC6H4 | Me | >20:1 | 54 | 97 |
| 21k | o-ClC6H4 | Me | >20:1 | 53 | 99 |
| 21l | 3,4-Cl2C6H3 | Me | >20:1 | 95 | 99 |
| 21m | 3-Thienyl | C2H4Cl | >20:1 | 67 | 97 |
| 21n | Me | Et | >20:1 | 68 | 83 |
| 21o | Me | t-Bu | >20:1 | 84 | >99 |
Both examples reported by Suematsu and Katsuki showed very good yields and excellent enantiomeric excesses of the products. This work is noteworthy because it is the first report in the literature of an enantioselective insertion of an iridium carbenoid into C(sp3)–H bonds.
Che and coworkers introduced the first porphyrin-based chiral iridium catalyst (−)-65 to insertion of carbenoids into C(sp3)–H bonds [52]. The reaction with 1,4-cyclohexadiene was promoted by 1 mol % of the catalyst at low temperatures to affords the product in high yields and enantioselectivity (Scheme 12).
![[1860-5397-12-87-i12]](/bjoc/content/inline/1860-5397-12-87-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Chiral porphyrin iridium complex catalyzes the carbenoid insertion into bis-allylic C(sp3)–H bonds.
Scheme 12: Chiral porphyrin iridium complex catalyzes the carbenoid insertion into bis-allylic C(sp3)–H bonds.
The same catalytic system was applied to carbenoid insertions into tetrahydrofuran C(sp3)–H bonds (Scheme 13). The reaction afforded the desired product in a regioselective way and high diastereoselectivity, ranging from 2.5:1 to >20:1, favoring the anti-product, in a complementary sense when compared to the results reported by Suematsu and Katsuki for iridium catalyst 61a (Table 8). Poor to excellent yields and high enantioselectivity were reported for the main product.
![[1860-5397-12-87-i13]](/bjoc/content/inline/1860-5397-12-87-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Chiral porphyrin iridium complex catalyzes the carbenoid insertion into tetrahydrofuran C(sp3)–H bonds.
Scheme 13: Chiral porphyrin iridium complex catalyzes the carbenoid insertion into tetrahydrofuran C(sp3)–H bo...
The chiral porphyrin-based iridium complex (−)-65 was also used by the same research group to catalyze the intramolecular carbenoid insertion into C(sp3)–H bonds and affords the synthesis of cis-β-lactones in a wide range of yields and enantioselectivities (Scheme 14) [53]. The reaction time was dependent on the chemical structure of the group Ar1 (p-CH3Ph = 24 h; p-FPh, m-ClPh, m-BrPh = 10 min) and the enantioselectivity drops from 70–80% ee to less than 50% ee when Ar1 is m-ClPh or m-BrPh.
![[1860-5397-12-87-i14]](/bjoc/content/inline/1860-5397-12-87-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Chiral porphyrin–iridium complex catalyzes the intramolecular carbenoid insertion into C(sp3)–H bonds to afford the synthesis of cis-β-lactones.
Scheme 14: Chiral porphyrin–iridium complex catalyzes the intramolecular carbenoid insertion into C(sp3)–H bon...
In 2013, Davies, Blakey and coworkers reported a new iridium catalyst to perform a carbenoid insertion into the C(sp3)–H bond (Scheme 15) [54]. The reactions were performed at room temperature and low catalyst loading (0.5 mol %) to afford the desired product in high yield and enantioselectivity. To reduce the amount of cyclohexadiene, the reaction was also performed in trifluorotoluene, resulting in a yield decrease (93% when 1,4-cyclohexadiene was solvent and reagent; 60% when 2,5 equivalents of cyclohexadiene and PhCF3 as solvent where used) but with almost the same enantioselectivity.
![[1860-5397-12-87-i15]](/bjoc/content/inline/1860-5397-12-87-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Chiral bis(oxazoline)–iridium complex catalyzes the carbenoid insertion into bis-allylic C(sp3)–H bonds.
Scheme 15: Chiral bis(oxazoline)–iridium complex catalyzes the carbenoid insertion into bis-allylic C(sp3)–H b...
Rhodium-based chiral catalysts
Since the pioneering reports by Ikegami [39], Doyle [40] and Davies [41] introducing their dirhodium chiral catalysts, these complexes have been the most frequently used and studied catalysts for enantioselective insertion of carbenoids into C(sp3)–H [55]. It is noteworthy the intensive contribution of the Davies research group which reported important works related to regioselectivity and stereoselectivity [56], and chemoselectivity [57] of this transformation.
In 2011, Davies et al reported a chiral rhodium complex based on a new cyclopropylcarboxylate ligand (Scheme 16) [58]. Among the various transformations promoted by this new catalyst we can find enantioselective carbenoid insertion into the endocyclic allylic C(sp3)–H bond of 71 followed by the Cope rearrangement and retro-Cope strategy previously described by the same research group [59]. The product was obtained in excellent yield, diastereo- and enantioselectivity.
![[1860-5397-12-87-i16]](/bjoc/content/inline/1860-5397-12-87-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: New cyclopropylcarboxylate-based chiral catalyst to enantioselective carbenoid insertion into the endocyclic allylic C(sp3)–H bond.
Scheme 16: New cyclopropylcarboxylate-based chiral catalyst to enantioselective carbenoid insertion into the e...
Later, the same authors showed a new chiral rhodium complex (R)-74 based on an analogue cyclopropylcarboxylate ligand (Scheme 17) [60]. This new ligand favors the regiochemistry of rhodium carbenoid insertion into primary C(sp3)–H activated bonds even in the presence of activated secondary C(sp3)–H bonds. This preference stems from the greater volume of the ligand and the consequent greater steric strain in the transition state that leads to the minor insertion product at activated secondary C(sp3)–H bonds. Substrates with benzylic bond, allylic and α-oxygen C(sp3)–H were submitted to the new catalyst, under dichloromethane reflux, and led to the preferential formation of the insertion products into primary carbon, (from 5:1 to >20:1), high yields and enantioselectivity, higher than 90% ee, 88% ee and 64% ee, respectively.
![[1860-5397-12-87-i17]](/bjoc/content/inline/1860-5397-12-87-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Regio- and enantioselective carbenoid insertion into the C(sp3)–H bond catalyzed by a new bulky cyclopropylcarboxylate-based chiral dirhodium complex (R)-74.
Scheme 17: Regio- and enantioselective carbenoid insertion into the C(sp3)–H bond catalyzed by a new bulky cyc...
The authors also employed this catalyst in the functionalization of the (−)-α-cedrene and a steroidal nucleus, both substrates containing primary, secondary and tertiary allylic C(sp3)–H bonds (Scheme 18). In both cases, was only observed the formation of the regioisomer derived from carbenoid insertion into the primary allylic position with excellent yield and high diastereoselectivity. For the steroidal substrate, the catalyst (R)-74 favored the formation of a new center with R configuration in a 6:1 diastereoisomeric ratio. The use of the enantiomeric catalyst, (S)-74, yielded the product with S configuration at the new stereogenic center with a higher diastereoisomeric ratio (>20:1).
![[1860-5397-12-87-i18]](/bjoc/content/inline/1860-5397-12-87-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Regio and diastereoselective carbenoid insertion into the C(sp3)–H bond catalyzed by a new bulky cyclopropylcarboxylate-based chiral dirhodium complex.
Scheme 18: Regio and diastereoselective carbenoid insertion into the C(sp3)–H bond catalyzed by a new bulky cy...
In 2014, Davies and coworkers expand the scope of catalyst (R)-74 by combining it with 2,2,2-trichloroethyl (TCE) aryldiazoacetates (Scheme 19) [61]. When compared with the use of traditional methylaryldiazoacetates (Scheme 17), an improved enantioselectivity of the insertion product 85 was observed combined with superior regiochemistry, favoring the rhodium carbenoid insertion into primary C(sp3)–H activated bonds even in the presence of activated secondary C(sp3)–H bonds.
![[1860-5397-12-87-i19]](/bjoc/content/inline/1860-5397-12-87-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: 2,2,2-Trichloroethyl (TCE) aryldiazoacetates to improve the scope, regio- and enantioselective of the carbenoid insertion into primary C(sp3)–Hs bond by (R)-74.
Scheme 19: 2,2,2-Trichloroethyl (TCE) aryldiazoacetates to improve the scope, regio- and enantioselective of t...
Another important contribution addressed by this work was the C(sp3)–H bond functionalization of methyl ethers even in the presence of other activated C(sp3)–H bonds. A wide range of methyl ethers were regioselectively functionalized, also with improved enantioselectivity, by the use of TCE aryldiazoacetates in combination with (R)-47, here exemplified by the reaction between methyl ether 81 and the TCE aryldiazoacetate 84. The comparison of this result with that presented at Scheme 17 shows a significant increase of the enantioselectivity.
TCE heteroaryldiazoacetates were also successfully employed for the formation of the rhodium carbenoid insertion products in superior yields when compared to the reaction with methyl aryldiazoacetates. TCE aryldiazoacetates reduced significantly the carbene dimerization allowing the reduction of the TCE diazoacetate addition time from 1.5 hours to 5 seconds.
The deactivated aryl methyl ether 84 was also functionalized by the use of TCE aryldiazoacetates in combination with (R)-47 in good yield and excellent enantioselectivity. The reaction between 84 and the methyl aryldiazoacetate 21e afforded the insertion product in only 15% yield.
In 2013, Davies, Yo et al reported a new strategy to construct 2,3-dihydrobenzofurans based on a sequential enantioselective rhodium catalyzed carbenoid insertion into a C(sp3)–H bond followed by a palladium C(sp2)–H bond activation to build a new C–O bond (Scheme 20) [62]. A wide range of benzyl silyl ethers and diazo compounds were tested providing the desired 2,3-dihydrobenzofuran in good yields and excellent diastereo- and enantioselectivity. Later, this strategy was further used by Davies, Zakarian and coworkers to access the total synthesis of (−)-maoecrystal V [63].
![[1860-5397-12-87-i20]](/bjoc/content/inline/1860-5397-12-87-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Sequential C–H functionalization approach to 2,3-dihydrobenzofurans.
Scheme 20: Sequential C–H functionalization approach to 2,3-dihydrobenzofurans.
During this study, the authors observed an unexpected result when ortho–halosubstituted diazo compounds were used. Here the formation of a β-lactone by the carbenoid insertion into the C(sp3)–H bond of the alkyl substituent of the alkoxy moiety of the ester (Scheme 21). The authors decided to investigate this observation and reported a more detailed study concerning the synthesis of cis-disubstituted β-lactones in high yield, diastereo- and enantioselectivity [64].
![[1860-5397-12-87-i21]](/bjoc/content/inline/1860-5397-12-87-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Enantioselective intramolecular rhodium carbenoid insertion into C(sp3)–H bonds to afford cis-disubstituted β-lactones.
Scheme 21: Enantioselective intramolecular rhodium carbenoid insertion into C(sp3)–H bonds to afford cis-disub...
Total syntheses of 2,3-dihydrobenzofurans containing natural products have also been recently reported independently by Hashimoto [65,66] and Kan [67] based on an enantioselective intramolecular rhodium carbenoid insertion into C(sp3)–H bonds.
In 2012, Pavlyuk and coworkers performed the synthesis of azacycloalkenes by rhodium carbenoid insertion into C(sp3)–H bonds, and subsequent ring closing olefin metathesis (RCM) [68]. The insertion of the rhodium carbenoids derived from vinyl diazoacetate into the C(sp3)–H bonds of the alkenylcarbamates 97a–d yields two reaction products (Table 10). The major one (99a–d) was the result of the cyclopropanation reaction of the double bond present in 97a–d. The minor product (100a–d) was the desired one, resulting from the insertion reaction on the C(sp3)–H bond α to the nitrogen atom.
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-87-i34.svg?max-width=637&scale=1.0)
The carbenoid insertion reaction into C(sp3)–H bonds was regioselective for substrates 97a–d, even when there was an allylic and α-nitrogen C(sp3)–H bond in substrate 97a. The authors also point out that the 66:67 ratio was 2:1 regardless of the rhodium source (Rh2(OAc)4, Rh2(pfb)4, Rh2(TFA)4, Rh2(TPA)4) or solvents (hexane, benzene) used in this reaction.
The dienes 100a–d were submitted to 2nd-generation Grubbs–Hoveyda catalyst (101), under dichloroethane reflux, to afford the desired azacycloalkenes 102a–c in 95–98% yield and 92–95 % ee (Table 11). Only the diene 100d did not cyclize and did not afford the nine-membered heterocycle by this methodology.
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-87-i35.svg?max-width=637&scale=1.0)
In 2015, Hashimoto et al reported the synthesis of methyl 2-vinyltetrahydropyran-3-carboxylates (104) by an enantioselective rhodium carbenoid insertion into C(sp3)–H bond strategy [69]. The desired product was obtained in very good yield and excellent diastereo- and enantioselectivity favoring the cis isomer (Scheme 22).
![[1860-5397-12-87-i22]](/bjoc/content/inline/1860-5397-12-87-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Enantioselective intramolecular rhodium carbenoid insertion into C(sp3)–H bonds to afford cis-2-vinyltetrahydropyran-3-carboxylates.
Scheme 22: Enantioselective intramolecular rhodium carbenoid insertion into C(sp3)–H bonds to afford cis-2-vin...
An interesting work was reported by Che and coworkers concerning the first rhodium porphyrin-based catalyst for enantioselective carbenoid insertion into C(sp3)–H bonds [70]. The reaction with acyclic alkanes showed regioselectivity in favor of the formation of the insertion product into primary carbons in modest stereoselectivity (Scheme 23). The preference for the reaction in less hindered carbon was attributed to high steric demand required by the chiral ligand in the transition state of the carbenoid insertion step in the C(sp3)–H bond.
![[1860-5397-12-87-i23]](/bjoc/content/inline/1860-5397-12-87-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: First rhodium porphyrin-based catalyst for enantioselective carbenoid insertion into C(sp3)–H bond.
Scheme 23: First rhodium porphyrin-based catalyst for enantioselective carbenoid insertion into C(sp3)–H bond.
Cyclic alkanes were also tested with yields ranging from 64–80% and enantioselectivities between 88 and 92% ee. The reaction with cyclohexane was conducted on a gram scale and, after 10 hours of reaction, 2.88 g (73% yield) were obtained of the carbenoid insertion product in 91% ee. Cyclohexene showed high regioselectivity for the carbenoid insertion of the allylic C(sp3)–H bond, 43% yield, 71% ee and a 60:40 diastereoisomeric ratio. The formation of the cyclopropanation product was also observed in 14% yield. Ethylbenzene (110) was used and also showed high regioselectivity favoring the carbenoid insertion into benzylic C(sp3)–H bonds (Scheme 24). The diastereoisomers 111 and 112 were obtained in 45% yield and 16%, respectively, and moderate stereoselectivity was observed in both products.
![[1860-5397-12-87-i24]](/bjoc/content/inline/1860-5397-12-87-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Rhodium porphyrin-based catalyst for enantioselective carbenoid insertion into benzylic C(sp3)–H bond.
Scheme 24: Rhodium porphyrin-based catalyst for enantioselective carbenoid insertion into benzylic C(sp3)–H bo...
Few examples of attempts to develop heterogeneous catalytic systems based on chiral rhodium complexes were also done. In 2010, Hashimoto and coworkers reported the synthesis of a highly robust polymer-supported chiral dirhodium(II) complex [71]. The chiral part of the catalyst was based on the N-phthaloyl-(S)-amino rhodium carboxylate (S)-17d. Two substrates were submitted to the enantioselective carbenoid insertion into the C(sp3)–H bond in toluene as solvent and at temperatures ranging from −78 °C to −60 °C. The desired products were obtained in yields up to 80% and enantioselectivity up to 90%, even after 15 recycles of the catalyst.
Jones, Davies and coworkers also recently published a new heterogeneous catalytic cycle base on homogeneous catalyst (S)-23 [72]. The chiral scaffold was covalently supported on silica surface through an organic linker and was tested in a tandem enantioselective carbenoid insertion into C(sp3)–H bond/Cope rearrangement. The reactions afforded good yields and excellent enantioselectivity. The recycling of the catalyst was evaluated in a cyclopropanation reaction and no significant decrease on its performance could be observed after five runs.
Conclusion
The efforts focused on the development of the enantioselective insertion of carbenoids into C(sp3)–H bonds have provided a wide range of catalytic systems to the chemical community able to perform this transformation and to introduce new C–C bonds in a enantiocontrolled way. The chiral rhodium catalysts are the state of art of this synthetic tool. However, rhodium is an expensive metal and increases the cost of the chemical process despite the low catalyst loads found in literature.
Despite the good results presented until today, the use of the reported chiral iridium catalysts is even more expensive than the use of other metals such as rhodium, copper and ruthenium, for example. Efforts should be directed toward the development of simpler ligands specially those based on inexpensive chiral building blocks like amino acids and sugars.
The examples of works focused on copper-based catalysts are growing in number and quality. Among the well-known metals able to react with diazo compounds to afford carbenoid intermediates, copper is inexpensive and has a wide range of well-established chiral ligands able to be tested and to inspire the rational design of new ligands.
For all metals commonly used in this transformation, more efforts should be focused towards the development of new and robust heterogeneous catalytic systems. This strategy can reduce the costs related to metals like rhodium or iridium and can also contribute to reduce the disposal of these metals in the environment.
Acknowledgements
We thank the Brazilian National Research Council (CNPq, Gran Nos. 477418/2013-9), the Brazilian Coordination for the Improvement of Higher Education Level Personnel (CAPES), Distrito Federal Research Foundation (FAPDF, Gran Nos. 193.001.049/2015), FINEP-MCT and DPP-UnB for financial support and fellowships.
References
-
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n
Return to citation in text: [1] -
Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217
Return to citation in text: [1] [2] [3] -
Yates, P. J. Am. Chem. Soc. 1952, 74, 5376–5381. doi:10.1021/ja01141a047
Return to citation in text: [1] [2] -
Taber, D. F.; Petty, E. H. J. Org. Chem. 1982, 47, 4808–4809. doi:10.1021/jo00145a050
Return to citation in text: [1] -
Mbuvi, H. M.; Woo, L. K. Organometallics 2008, 27, 637–645. doi:10.1021/om7007502
Return to citation in text: [1] -
Maas, G.; Werle, T.; Alt, M.; Mayer, D. Tetrahedron 1993, 49, 881–888. doi:10.1016/S0040-4020(01)80330-6
Return to citation in text: [1] -
Kubo, T.; Sakaguchi, S.; Ishii, Y. Chem. Commun. 2000, 625–626. doi:10.1039/B000518P
Return to citation in text: [1] -
Woo, L. K.; Smith, D. A. Organometallics 1992, 11, 2344–2346. doi:10.1021/om00043a011
Return to citation in text: [1] -
Müller, P.; Boléa, C. Helv. Chim. Acta 2002, 85, 483–494. doi:10.1002/1522-2675(200202)85:2<483::AID-HLCA483>3.0.CO;2-Q
Return to citation in text: [1] [2] -
Flynn, C. J.; Elcoate, C. J.; Lawrence, S. E.; Maguire, A. R. J. Am. Chem. Soc. 2010, 132, 1184–1185. doi:10.1021/ja909713a
Return to citation in text: [1] [2] -
Slattery, C. N.; Maguire, A. R. Tetrahedron Lett. 2013, 54, 2799–2801. doi:10.1016/j.tetlet.2013.03.078
Return to citation in text: [1] -
Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o
Return to citation in text: [1] [2] [3] -
Ye, T.; García, F. C.; McKervey, M. A. J. Chem. Soc., Perkin Trans. 1 1995, 1373–1379. doi:10.1039/P19950001373
Return to citation in text: [1] -
Ye, T.; McKervey, M. A.; Brandes, B. D.; Doyle, M. P. Tetrahedron Lett. 1994, 35, 7269–7272. doi:10.1016/0040-4039(94)85379-7
Return to citation in text: [1] -
Kennedy, M.; McKervey, M. A.; Maguire, A. R.; Roos, G. H. P. J. Chem. Soc., Chem. Commun. 1990, 361–362. doi:10.1039/C39900000361
Return to citation in text: [1] -
McKervey, M. A.; Ye, T. J. Chem. Soc., Chem. Commun. 1992, 823–824. doi:10.1039/C39920000823
Return to citation in text: [1] -
Roos, G. H. P.; McKervey, M. A. Synth. Commun. 1992, 22, 1751–1756. doi:10.1080/00397919208020494
Return to citation in text: [1] -
Doyle, M. P. Chem. Rev. 1986, 86, 919–939. doi:10.1021/cr00075a013
Return to citation in text: [1] -
Kitagaki, S.; Anada, M.; Kataoka, O.; Matsuno, K.; Umeda, C.; Watanabe, N.; Hashimoto, S.-i. J. Am. Chem. Soc. 1999, 121, 1417–1418. doi:10.1021/ja983748e
Return to citation in text: [1] -
Doyle, M. P.; Winchester, W. R.; Hoorn, J. A. A.; Lynch, V.; Simonsen, S. H.; Ghosh, R. J. Am. Chem. Soc. 1993, 115, 9968–9978. doi:10.1021/ja00075a013
Return to citation in text: [1] -
Timmons, D. J.; Doyle, M. P. J. Organomet. Chem. 2001, 617–618, 98–104. doi:10.1016/S0022-328X(00)00637-9
Return to citation in text: [1] -
Doyle, M. P.; Ren, T. The Influence of Ligands on Dirhodium(II) on Reactivity and Selectivity in Metal Carbene Reactions. In Progress in Inorganic Chemistry; Karlin, K. D., Ed.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2001; Vol. 49, pp 113–168. doi:10.1002/9780470166512.ch2
Return to citation in text: [1] -
Doyle, M. P. Russ. Chem. Bull. 1999, 48, 16–20. doi:10.1007/BF02494393
Return to citation in text: [1] -
McCarthy, N.; McKervey, M. A.; Ye, T.; McCann, M.; Murphy, E.; Doyle, M. P. Tetrahedron Lett. 1992, 33, 5983–5986. doi:10.1016/S0040-4039(00)61106-1
Return to citation in text: [1] -
Pirrung, M. C.; Zhang, J. Tetrahedron Lett. 1992, 33, 5987–5990. doi:10.1016/S0040-4039(00)61107-3
Return to citation in text: [1] -
Estevan, F.; Herbst, K.; Lahuerta, P.; Barberis, M.; Pérez-Prieto, J. Organometallics 2001, 20, 950–957. doi:10.1021/om0010060
Return to citation in text: [1] -
Estevan, F.; Lahuerta, P.; Pérez-Prieto, J.; Sanaú, M.; Stiriba, S.-E.; Ubeda, M. A. Organometallics 1997, 16, 880–886. doi:10.1021/om9608162
Return to citation in text: [1] -
Taber, D. F.; Malcolm, S. C.; Bieger, K.; Lahuerta, P.; Sanaú, M.; Stiriba, S.-E.; Pérez-Prieto, J.; Monge, M. A. J. Am. Chem. Soc. 1999, 121, 860–861. doi:10.1021/ja9819559
Return to citation in text: [1] -
Estevan, F.; Lahuerta, P.; Pérez-Prieto, J.; Pereira, I.; Stiriba, S.-E. Organometallics 1998, 17, 3442–3447. doi:10.1021/om971070q
Return to citation in text: [1] -
Lahuerta, P.; Pereira, I.; Pérez-Prieto, J.; Sanaú, M.; Stiriba, S.-E.; Taber, D. F. J. Organomet. Chem. 2000, 612, 36–45. doi:10.1016/S0022-328X(00)00357-0
Return to citation in text: [1] -
Davies, H. M. L.; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061–3071. doi:10.1039/b901170f
Return to citation in text: [1] -
Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242–3272. doi:10.1039/b816707a
Return to citation in text: [1] -
Ledon, H.; Linstrumelle, G.; Julia, S. Tetrahedron Lett. 1973, 14, 25–28. doi:10.1016/S0040-4039(01)95567-4
Return to citation in text: [1] -
Taber, D. F.; Petty, E. H.; Raman, K. J. Am. Chem. Soc. 1985, 107, 196–199. doi:10.1021/ja00287a035
Return to citation in text: [1] -
Taber, D. F.; Ruckle, R. E. J. Am. Chem. Soc. 1986, 108, 7686–7693. doi:10.1021/ja00284a037
Return to citation in text: [1] -
Sonawane, H. R.; Bellur, N. S.; Ahuja, J. R.; Kulkarni, D. G. J. Org. Chem. 1991, 56, 1434–1439. doi:10.1021/jo00004a019
Return to citation in text: [1] -
Doyle, M. P.; Westrum, L. J.; Wolthuis, W. N. E.; See, M. M.; Boone, W. P.; Bagheri, V.; Pearson, M. M. J. Am. Chem. Soc. 1993, 115, 958–964. doi:10.1021/ja00056a021
Return to citation in text: [1] [2] -
Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1966, 43, 5239–5244. doi:10.1016/S0040-4039(01)89263-7
Return to citation in text: [1] -
Hashimoto, S.-i.; Watanabe, N.; Ikegami, S. Tetrahedron Lett. 1990, 31, 5173–5174. doi:10.1016/S0040-4039(00)97834-1
Return to citation in text: [1] [2] -
Doyle, M. P.; Van Oeveren, A.; Westrum, L. J.; Protopopova, M. N.; Clayton, T. W., Jr. J. Am. Chem. Soc. 1991, 113, 8982–8984. doi:10.1021/ja00023a077
Return to citation in text: [1] [2] -
Davies, H. W. L.; Hansen, T. J. Am. Chem. Soc. 1997, 119, 9075–9076. doi:10.1021/ja971915p
Return to citation in text: [1] [2] -
Hansen, J.; Autschbach, J.; Davies, H. M. L. J. Org. Chem. 2009, 74, 6555–6563. doi:10.1021/jo9009968
Return to citation in text: [1] -
Slattery, C. N.; Clarke, L.-A.; O’Neill, S.; Ring, A.; Ford, A.; Maguire, A. R. Synlett 2012, 765–767. doi:10.1055/s-0031-1290598
Return to citation in text: [1] -
Slattery, C. N.; Clarke, L.-A.; Ford, A.; Maguire, A. R. Tetrahedron 2013, 69, 1297–1301. doi:10.1016/j.tet.2012.12.003
Return to citation in text: [1] -
Slattery, C. N.; O’Keeffe, S.; Maguire, A. R. Tetrahedron: Asymmetry 2013, 24, 1265–1275. doi:10.1016/j.tetasy.2013.09.009
Return to citation in text: [1] -
Clarke, L. A.; Ring, A.; Ford, A.; Sinha, A. S.; Lawrence, S. E.; Maguire, A. R. Org. Biomol. Chem. 2014, 12, 7612–7628. doi:10.1039/c4ob01430h
Return to citation in text: [1] -
Fraile, J. M.; García, J. I.; Mayoral, J. A.; Roldán, M. Org. Lett. 2007, 9, 731–733. doi:10.1021/ol070092y
Return to citation in text: [1] -
Fraile, J. M.; López-Ram-de-Viu, P.; Mayoral, J. A.; Roldán, M.; Santafé-Valero, J. Org. Biomol. Chem. 2011, 9, 6075–6081. doi:10.1039/c1ob05499f
Return to citation in text: [1] -
Fraile, J. M.; Mayoral, J. A.; Muñoz, A. M.; Santafé-Valero, J. Tetrahedron 2013, 69, 7360–7364. doi:10.1016/j.tet.2013.06.088
Return to citation in text: [1] -
Fraile, J. M.; Mayoral, J. A.; Ravasio, N.; Roldán, M.; Sordelli, L.; Zaccheria, F. J. Catal. 2011, 281, 273–278. doi:10.1016/j.jcat.2011.05.008
Return to citation in text: [1] -
Suematsu, H.; Katsuki, T. J. Am. Chem. Soc. 2009, 131, 14218–14219. doi:10.1021/ja9065267
Return to citation in text: [1] -
Wang, J.-C.; Xu, Z.-J.; Guo, Z.; Deng, Q.-H.; Zhuo, C.-Y.; Wan, X.-L.; Che, C.-M. Chem. Commun. 2012, 48, 4299–4301. doi:10.1039/c2cc30441d
Return to citation in text: [1] -
Wang, J.-C.; Zhang, Y.; Xu, Z.-J.; Lo, V. K.-Y.; Che, C.-M. ACS Catal. 2013, 3, 1144–1148. doi:10.1021/cs4001656
Return to citation in text: [1] -
Owens, C. P.; Varela-Álvarez, A.; Boyarskikh, V.; Musaev, D. G.; Davies, H. M. L.; Blakey, S. B. Chem. Sci. 2013, 4, 2590–2596. doi:10.1039/c3sc50886b
Return to citation in text: [1] -
Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; McKervey, M. A. Chem. Rev. 2015, 115, 9981–10080. doi:10.1021/acs.chemrev.5b00121
Return to citation in text: [1] -
Davies, H. M. L.; Morton, D. Chem. Soc. Rev. 2011, 40, 1857–1869. doi:10.1039/C0CS00217H
Return to citation in text: [1] -
Nadeau, E.; Ventura, D. L.; Brekan, J. A.; Davies, H. M. L. J. Org. Chem. 2010, 75, 1927–1939. doi:10.1021/jo902644f
Return to citation in text: [1] -
Qin, C.; Boyarskikh, V.; Hansen, J. H.; Hardcastle, K. I.; Musaev, D. G.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 19198–19204. doi:10.1021/ja2074104
Return to citation in text: [1] -
Davies, H. M. L.; Lian, Y. Acc. Chem. Res. 2012, 45, 923–935. doi:10.1021/ar300013t
Return to citation in text: [1] -
Qin, C.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 9792–9796. doi:10.1021/ja504797x
Return to citation in text: [1] -
Guptill, D. M.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 17718–17721. doi:10.1021/ja5107404
Return to citation in text: [1] -
Wang, H.; Li, G.; Engle, K. M.; Yu, J.-Q.; Davies, H. M. L. J. Am. Chem. Soc. 2013, 135, 6774–6777. doi:10.1021/ja401731d
Return to citation in text: [1] -
Lu, P.; Mailyan, A.; Gu, Z.; Guptill, D. M.; Wang, H.; Davies, H. M. L.; Zakarian, A. J. Am. Chem. Soc. 2014, 136, 17738–17749. doi:10.1021/ja510573v
Return to citation in text: [1] -
Fu, L.; Wang, H.; Davies, H. M. L. Org. Lett. 2014, 16, 3036–3039. doi:10.1021/ol5011505
Return to citation in text: [1] -
Natori, Y.; Tsutsui, H.; Sato, N.; Nakamura, S.; Nambu, H.; Shiro, M.; Hashimoto, S. J. Org. Chem. 2009, 74, 4418–4421. doi:10.1021/jo900502d
Return to citation in text: [1] -
Ito, M.; Namie, R.; Krishnamurthi, J.; Miyamae, H.; Takeda, K.; Nambu, H.; Hashimoto, S. Synlett 2014, 288–292. doi:10.1055/s-0033-1340291
Return to citation in text: [1] -
Wakimoto, T.; Miyata, K.; Ohuchi, H.; Asakawa, T.; Nukaya, H.; Suwa, Y.; Kan, T. Org. Lett. 2011, 13, 2789–2791. doi:10.1021/ol200728w
Return to citation in text: [1] -
McMills, M. C.; Humes, R. J.; Pavlyuk, O. M. Tetrahedron Lett. 2012, 53, 849–851. doi:10.1016/j.tetlet.2011.12.019
Return to citation in text: [1] -
Ito, M.; Kondo, Y.; Nambu, H.; Anada, M.; Takeda, K.; Hashimoto, S. Tetrahedron Lett. 2015, 56, 1397–1400. doi:10.1016/j.tetlet.2015.01.125
Return to citation in text: [1] -
Thu, H.-Y.; Tong, G. S.-M.; Huang, J.-S.; Chan, S. L.-F.; Deng, Q.-H.; Che, C.-M. Angew. Chem., Int. Ed. 2008, 47, 9747–9751. doi:10.1002/anie.200803157
Return to citation in text: [1] -
Takeda, K.; Oohara, T.; Anada, M.; Nambu, H.; Hashimoto, S. Angew. Chem., Int. Ed. 2010, 49, 6979–6983. doi:10.1002/anie.201003730
Return to citation in text: [1] -
Chepiga, K. M.; Feng, Y.; Brunelli, N. A.; Jones, C. W.; Davies, H. M. L. Org. Lett. 2013, 15, 6136–6139. doi:10.1021/ol403006r
Return to citation in text: [1]
| 40. | Doyle, M. P.; Van Oeveren, A.; Westrum, L. J.; Protopopova, M. N.; Clayton, T. W., Jr. J. Am. Chem. Soc. 1991, 113, 8982–8984. doi:10.1021/ja00023a077 |
| 41. | Davies, H. W. L.; Hansen, T. J. Am. Chem. Soc. 1997, 119, 9075–9076. doi:10.1021/ja971915p |
| 12. | Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o |
| 45. | Slattery, C. N.; O’Keeffe, S.; Maguire, A. R. Tetrahedron: Asymmetry 2013, 24, 1265–1275. doi:10.1016/j.tetasy.2013.09.009 |
| 46. | Clarke, L. A.; Ring, A.; Ford, A.; Sinha, A. S.; Lawrence, S. E.; Maguire, A. R. Org. Biomol. Chem. 2014, 12, 7612–7628. doi:10.1039/c4ob01430h |
| 10. | Flynn, C. J.; Elcoate, C. J.; Lawrence, S. E.; Maguire, A. R. J. Am. Chem. Soc. 2010, 132, 1184–1185. doi:10.1021/ja909713a |
| 43. | Slattery, C. N.; Clarke, L.-A.; O’Neill, S.; Ring, A.; Ford, A.; Maguire, A. R. Synlett 2012, 765–767. doi:10.1055/s-0031-1290598 |
| 44. | Slattery, C. N.; Clarke, L.-A.; Ford, A.; Maguire, A. R. Tetrahedron 2013, 69, 1297–1301. doi:10.1016/j.tet.2012.12.003 |
| 42. | Hansen, J.; Autschbach, J.; Davies, H. M. L. J. Org. Chem. 2009, 74, 6555–6563. doi:10.1021/jo9009968 |
| 9. | Müller, P.; Boléa, C. Helv. Chim. Acta 2002, 85, 483–494. doi:10.1002/1522-2675(200202)85:2<483::AID-HLCA483>3.0.CO;2-Q |
| 37. | Doyle, M. P.; Westrum, L. J.; Wolthuis, W. N. E.; See, M. M.; Boone, W. P.; Bagheri, V.; Pearson, M. M. J. Am. Chem. Soc. 1993, 115, 958–964. doi:10.1021/ja00056a021 |
| 12. | Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o |
| 47. | Fraile, J. M.; García, J. I.; Mayoral, J. A.; Roldán, M. Org. Lett. 2007, 9, 731–733. doi:10.1021/ol070092y |
| 48. | Fraile, J. M.; López-Ram-de-Viu, P.; Mayoral, J. A.; Roldán, M.; Santafé-Valero, J. Org. Biomol. Chem. 2011, 9, 6075–6081. doi:10.1039/c1ob05499f |
| 49. | Fraile, J. M.; Mayoral, J. A.; Muñoz, A. M.; Santafé-Valero, J. Tetrahedron 2013, 69, 7360–7364. doi:10.1016/j.tet.2013.06.088 |
| 50. | Fraile, J. M.; Mayoral, J. A.; Ravasio, N.; Roldán, M.; Sordelli, L.; Zaccheria, F. J. Catal. 2011, 281, 273–278. doi:10.1016/j.jcat.2011.05.008 |
| 41. | Davies, H. W. L.; Hansen, T. J. Am. Chem. Soc. 1997, 119, 9075–9076. doi:10.1021/ja971915p |
| 55. | Ford, A.; Miel, H.; Ring, A.; Slattery, C. N.; Maguire, A. R.; McKervey, M. A. Chem. Rev. 2015, 115, 9981–10080. doi:10.1021/acs.chemrev.5b00121 |
| 39. | Hashimoto, S.-i.; Watanabe, N.; Ikegami, S. Tetrahedron Lett. 1990, 31, 5173–5174. doi:10.1016/S0040-4039(00)97834-1 |
| 40. | Doyle, M. P.; Van Oeveren, A.; Westrum, L. J.; Protopopova, M. N.; Clayton, T. W., Jr. J. Am. Chem. Soc. 1991, 113, 8982–8984. doi:10.1021/ja00023a077 |
| 53. | Wang, J.-C.; Zhang, Y.; Xu, Z.-J.; Lo, V. K.-Y.; Che, C.-M. ACS Catal. 2013, 3, 1144–1148. doi:10.1021/cs4001656 |
| 54. | Owens, C. P.; Varela-Álvarez, A.; Boyarskikh, V.; Musaev, D. G.; Davies, H. M. L.; Blakey, S. B. Chem. Sci. 2013, 4, 2590–2596. doi:10.1039/c3sc50886b |
| 51. | Suematsu, H.; Katsuki, T. J. Am. Chem. Soc. 2009, 131, 14218–14219. doi:10.1021/ja9065267 |
| 52. | Wang, J.-C.; Xu, Z.-J.; Guo, Z.; Deng, Q.-H.; Zhuo, C.-Y.; Wan, X.-L.; Che, C.-M. Chem. Commun. 2012, 48, 4299–4301. doi:10.1039/c2cc30441d |
| 57. | Nadeau, E.; Ventura, D. L.; Brekan, J. A.; Davies, H. M. L. J. Org. Chem. 2010, 75, 1927–1939. doi:10.1021/jo902644f |
| 58. | Qin, C.; Boyarskikh, V.; Hansen, J. H.; Hardcastle, K. I.; Musaev, D. G.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 19198–19204. doi:10.1021/ja2074104 |
| 56. | Davies, H. M. L.; Morton, D. Chem. Soc. Rev. 2011, 40, 1857–1869. doi:10.1039/C0CS00217H |
| 1. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 13. | Ye, T.; García, F. C.; McKervey, M. A. J. Chem. Soc., Perkin Trans. 1 1995, 1373–1379. doi:10.1039/P19950001373 |
| 14. | Ye, T.; McKervey, M. A.; Brandes, B. D.; Doyle, M. P. Tetrahedron Lett. 1994, 35, 7269–7272. doi:10.1016/0040-4039(94)85379-7 |
| 15. | Kennedy, M.; McKervey, M. A.; Maguire, A. R.; Roos, G. H. P. J. Chem. Soc., Chem. Commun. 1990, 361–362. doi:10.1039/C39900000361 |
| 16. | McKervey, M. A.; Ye, T. J. Chem. Soc., Chem. Commun. 1992, 823–824. doi:10.1039/C39920000823 |
| 17. | Roos, G. H. P.; McKervey, M. A. Synth. Commun. 1992, 22, 1751–1756. doi:10.1080/00397919208020494 |
| 18. | Doyle, M. P. Chem. Rev. 1986, 86, 919–939. doi:10.1021/cr00075a013 |
| 19. | Kitagaki, S.; Anada, M.; Kataoka, O.; Matsuno, K.; Umeda, C.; Watanabe, N.; Hashimoto, S.-i. J. Am. Chem. Soc. 1999, 121, 1417–1418. doi:10.1021/ja983748e |
| 65. | Natori, Y.; Tsutsui, H.; Sato, N.; Nakamura, S.; Nambu, H.; Shiro, M.; Hashimoto, S. J. Org. Chem. 2009, 74, 4418–4421. doi:10.1021/jo900502d |
| 66. | Ito, M.; Namie, R.; Krishnamurthi, J.; Miyamae, H.; Takeda, K.; Nambu, H.; Hashimoto, S. Synlett 2014, 288–292. doi:10.1055/s-0033-1340291 |
| 2. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 20. | Doyle, M. P.; Winchester, W. R.; Hoorn, J. A. A.; Lynch, V.; Simonsen, S. H.; Ghosh, R. J. Am. Chem. Soc. 1993, 115, 9968–9978. doi:10.1021/ja00075a013 |
| 21. | Timmons, D. J.; Doyle, M. P. J. Organomet. Chem. 2001, 617–618, 98–104. doi:10.1016/S0022-328X(00)00637-9 |
| 22. | Doyle, M. P.; Ren, T. The Influence of Ligands on Dirhodium(II) on Reactivity and Selectivity in Metal Carbene Reactions. In Progress in Inorganic Chemistry; Karlin, K. D., Ed.; John Wiley & Sons: Hoboken, NJ, U.S.A., 2001; Vol. 49, pp 113–168. doi:10.1002/9780470166512.ch2 |
| 23. | Doyle, M. P. Russ. Chem. Bull. 1999, 48, 16–20. doi:10.1007/BF02494393 |
| 2. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 11. | Slattery, C. N.; Maguire, A. R. Tetrahedron Lett. 2013, 54, 2799–2801. doi:10.1016/j.tetlet.2013.03.078 |
| 63. | Lu, P.; Mailyan, A.; Gu, Z.; Guptill, D. M.; Wang, H.; Davies, H. M. L.; Zakarian, A. J. Am. Chem. Soc. 2014, 136, 17738–17749. doi:10.1021/ja510573v |
| 2. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 12. | Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o |
| 64. | Fu, L.; Wang, H.; Davies, H. M. L. Org. Lett. 2014, 16, 3036–3039. doi:10.1021/ol5011505 |
| 7. | Kubo, T.; Sakaguchi, S.; Ishii, Y. Chem. Commun. 2000, 625–626. doi:10.1039/B000518P |
| 9. | Müller, P.; Boléa, C. Helv. Chim. Acta 2002, 85, 483–494. doi:10.1002/1522-2675(200202)85:2<483::AID-HLCA483>3.0.CO;2-Q |
| 61. | Guptill, D. M.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 17718–17721. doi:10.1021/ja5107404 |
| 6. | Maas, G.; Werle, T.; Alt, M.; Mayer, D. Tetrahedron 1993, 49, 881–888. doi:10.1016/S0040-4020(01)80330-6 |
| 10. | Flynn, C. J.; Elcoate, C. J.; Lawrence, S. E.; Maguire, A. R. J. Am. Chem. Soc. 2010, 132, 1184–1185. doi:10.1021/ja909713a |
| 62. | Wang, H.; Li, G.; Engle, K. M.; Yu, J.-Q.; Davies, H. M. L. J. Am. Chem. Soc. 2013, 135, 6774–6777. doi:10.1021/ja401731d |
| 5. | Mbuvi, H. M.; Woo, L. K. Organometallics 2008, 27, 637–645. doi:10.1021/om7007502 |
| 59. | Davies, H. M. L.; Lian, Y. Acc. Chem. Res. 2012, 45, 923–935. doi:10.1021/ar300013t |
| 4. | Taber, D. F.; Petty, E. H. J. Org. Chem. 1982, 47, 4808–4809. doi:10.1021/jo00145a050 |
| 8. | Woo, L. K.; Smith, D. A. Organometallics 1992, 11, 2344–2346. doi:10.1021/om00043a011 |
| 60. | Qin, C.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 9792–9796. doi:10.1021/ja504797x |
| 31. | Davies, H. M. L.; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061–3071. doi:10.1039/b901170f |
| 32. | Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J.-Q. Chem. Soc. Rev. 2009, 38, 3242–3272. doi:10.1039/b816707a |
| 24. | McCarthy, N.; McKervey, M. A.; Ye, T.; McCann, M.; Murphy, E.; Doyle, M. P. Tetrahedron Lett. 1992, 33, 5983–5986. doi:10.1016/S0040-4039(00)61106-1 |
| 25. | Pirrung, M. C.; Zhang, J. Tetrahedron Lett. 1992, 33, 5987–5990. doi:10.1016/S0040-4039(00)61107-3 |
| 26. | Estevan, F.; Herbst, K.; Lahuerta, P.; Barberis, M.; Pérez-Prieto, J. Organometallics 2001, 20, 950–957. doi:10.1021/om0010060 |
| 27. | Estevan, F.; Lahuerta, P.; Pérez-Prieto, J.; Sanaú, M.; Stiriba, S.-E.; Ubeda, M. A. Organometallics 1997, 16, 880–886. doi:10.1021/om9608162 |
| 28. | Taber, D. F.; Malcolm, S. C.; Bieger, K.; Lahuerta, P.; Sanaú, M.; Stiriba, S.-E.; Pérez-Prieto, J.; Monge, M. A. J. Am. Chem. Soc. 1999, 121, 860–861. doi:10.1021/ja9819559 |
| 29. | Estevan, F.; Lahuerta, P.; Pérez-Prieto, J.; Pereira, I.; Stiriba, S.-E. Organometallics 1998, 17, 3442–3447. doi:10.1021/om971070q |
| 30. | Lahuerta, P.; Pereira, I.; Pérez-Prieto, J.; Sanaú, M.; Stiriba, S.-E.; Taber, D. F. J. Organomet. Chem. 2000, 612, 36–45. doi:10.1016/S0022-328X(00)00357-0 |
| 69. | Ito, M.; Kondo, Y.; Nambu, H.; Anada, M.; Takeda, K.; Hashimoto, S. Tetrahedron Lett. 2015, 56, 1397–1400. doi:10.1016/j.tetlet.2015.01.125 |
| 70. | Thu, H.-Y.; Tong, G. S.-M.; Huang, J.-S.; Chan, S. L.-F.; Deng, Q.-H.; Che, C.-M. Angew. Chem., Int. Ed. 2008, 47, 9747–9751. doi:10.1002/anie.200803157 |
| 67. | Wakimoto, T.; Miyata, K.; Ohuchi, H.; Asakawa, T.; Nukaya, H.; Suwa, Y.; Kan, T. Org. Lett. 2011, 13, 2789–2791. doi:10.1021/ol200728w |
| 68. | McMills, M. C.; Humes, R. J.; Pavlyuk, O. M. Tetrahedron Lett. 2012, 53, 849–851. doi:10.1016/j.tetlet.2011.12.019 |
| 38. | Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1966, 43, 5239–5244. doi:10.1016/S0040-4039(01)89263-7 |
| 39. | Hashimoto, S.-i.; Watanabe, N.; Ikegami, S. Tetrahedron Lett. 1990, 31, 5173–5174. doi:10.1016/S0040-4039(00)97834-1 |
| 36. | Sonawane, H. R.; Bellur, N. S.; Ahuja, J. R.; Kulkarni, D. G. J. Org. Chem. 1991, 56, 1434–1439. doi:10.1021/jo00004a019 |
| 37. | Doyle, M. P.; Westrum, L. J.; Wolthuis, W. N. E.; See, M. M.; Boone, W. P.; Bagheri, V.; Pearson, M. M. J. Am. Chem. Soc. 1993, 115, 958–964. doi:10.1021/ja00056a021 |
| 34. | Taber, D. F.; Petty, E. H.; Raman, K. J. Am. Chem. Soc. 1985, 107, 196–199. doi:10.1021/ja00287a035 |
| 35. | Taber, D. F.; Ruckle, R. E. J. Am. Chem. Soc. 1986, 108, 7686–7693. doi:10.1021/ja00284a037 |
| 71. | Takeda, K.; Oohara, T.; Anada, M.; Nambu, H.; Hashimoto, S. Angew. Chem., Int. Ed. 2010, 49, 6979–6983. doi:10.1002/anie.201003730 |
| 33. | Ledon, H.; Linstrumelle, G.; Julia, S. Tetrahedron Lett. 1973, 14, 25–28. doi:10.1016/S0040-4039(01)95567-4 |
| 72. | Chepiga, K. M.; Feng, Y.; Brunelli, N. A.; Jones, C. W.; Davies, H. M. L. Org. Lett. 2013, 15, 6136–6139. doi:10.1021/ol403006r |
© 2016 Santiago and Machado; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)