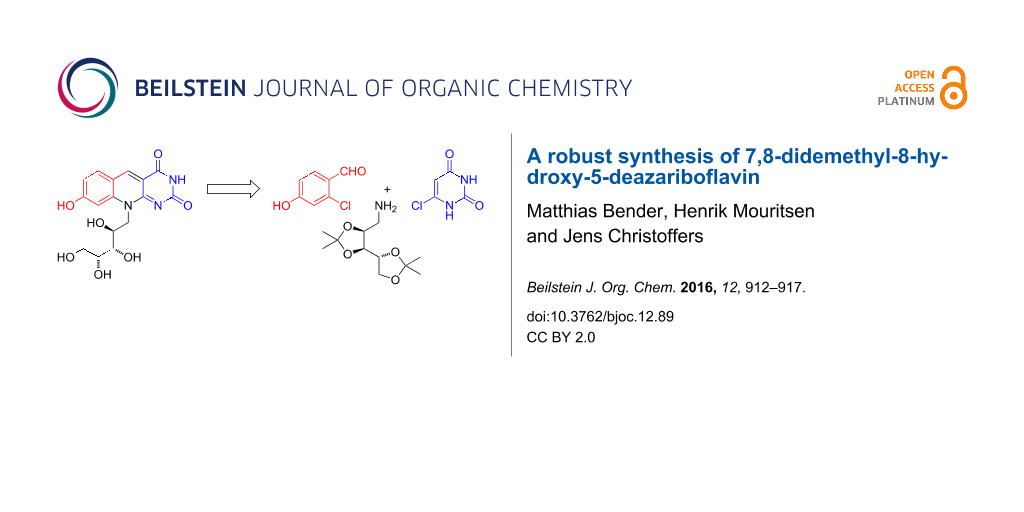
Abstract
The biosynthetic precursor of redox cofactor F420, 7,8-didemethyl-8-hydroxy-5-deazariboflavin, was prepared in four steps from 6-chlorouracil, 2-chloro-4-hydroxybenzaldehyde and bis-isopropylidene protected D-ribose. The latter aldehyde was transformed to the corresponding protected ribitylamine via the oxime, which was submitted to reduction with LiAlH4. Key advantage compared to previous syntheses is the utilization of a polyol-protective group which allowed the chromatographic purification of a key-intermediate product providing the target compound with high purity.

Graphical Abstract
Introduction
The deazariboflavin cofactor F420 plays an important role in bacterial methanogenesis [1,2] and was – in contrast to other, ubiquitous biological redox coenzymes like NAD+ or FAD – discovered relatively late (in 1972) [3].
Its biosynthetic precursor, 7,8-didemethyl-8-hydroxy-5-deazariboflavin (1, Scheme 1) without a phosphodiester extension is called FO and came into our focus of interest because it was for instance suggested to function as an antenna pigment in several cryptochrome proteins [4]. One or more avian cryptochromes in turn [4-7] are very likely to be the primary magnetoreceptor molecules which enable migratory birds to perceive the compass direction of the Earth’s magnetic field through a light-dependent, spin-based, radical-pair mechanism in the birds’ eyes [8-11]. A major challenge slowing down progress towards understanding the exact magnetoreceptive properties of avian cryptochromes is that, so far, avian cryptochromes have been very difficult to express recombinantly with their co-factors (most likely a FAD and a FO (1) or a FAD and a 5,10-methenyltetrahydrofolate (MTHF) still attached [4,12,13]). Consequently, no crystal structures of a functional avian cryptochrome are known at present. While FAD and MTHF are commercially available, FO (1) is not. However, this compound is potentially needed in order to reconstitute expressed apo-cryptochrome proteins to their functional state. Therefore, finding an efficient and reliable way to synthesize compound FO (1) is important.
![[1860-5397-12-89-i1]](/bjoc/content/inline/1860-5397-12-89-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Target structure of this synthetic study and two previous approaches.
Scheme 1: Target structure of this synthetic study and two previous approaches.
Up to the year 2015 the preparation of compound FO (1) was reported in the literature twice, both utilized commercially available chlorouracil 3 as C-ring synthon. The first report by Ashton et al. [14,15] used N-(D-ribityl)-3-hydroxyaniline (2) as A-ring fragment. The second report by Yoneda et al. [16-18] employed 4-benzyloxy-2-chlorobenzaldehyde (4) as building block for the A-ring. Both syntheses suffer from several drawbacks; these are the limited stability of intermediate products (e.g., oxidation of compound 2) and low yields together with low chemoselectivities. Major disadvantage in our hands turned out to be actually the high polarity and low solubility of intermediate products, which makes purification rather tedious. For example, the preparation of amine 5 by reduction of the oxime and its conversion with chlorouracil 3 turned out to be impossible when following a literature protocol [19]. We therefore decided to develop a robust synthesis of a protected derivative of amine 5 which could follow standard carbohydrate chemistry and might then allow purification and characterization of intermediate products. While we were following the synthetic approach by Yoneda et al. with a protected ribose derivative, Foss and co-workers were obviously facing the same preparative problem. To our surprise and delight (that we were not the only group struggling with the reports of Ashton and Yoneda), they have published very recently a synthesis of compound 1 by a modified route [20] and achieve 34% yield over six steps starting from D-ribose, 3-aminophenol and barbituric acid. Nevertheless, we would like to communicate herein our synthetic approach to compound FO (1), which finally turned out to reliably yield the final product with high purity.
Results and Discussion
Synthesis of aldehyde 9
We envisioned the reduction of an oxime to be the most convenient access to the doubly isopropylidene protected amine 10 [21,22]. Therefore, we prepared diisopropylidene ribose 9 according to a literature sequence of dithioacetalization, acetonide formation and dithioacetal deprotection [23-25]. In particular, we have chosen the conversion of D-ribose (6) with n-propylthiol (Scheme 2), since the product 7 (57%) was crystalline [26]. Diacetonide formation (product 8, 75%) proceeded as reported for the di(ethylthio) congener [24]. Finally, the dithioacetyl was cleaved while the isopropylidene moieties were retained, but not with mercury salts, as in original publications for the preparation of aldehyde 9 [27], but with the use of iodine (95% yield of product 9), as recommended by Ohlsson et al. [28,29].
![[1860-5397-12-89-i2]](/bjoc/content/inline/1860-5397-12-89-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of isopropylidene protected ribose 9 along a new, modified route. Reagents and conditions: (a) 2 equiv n-PrSH, conc. HCl/H2O, 0 °C, 1.5 h; (b) 20 equiv 2,2-dimethoxypropane, 0.1 equiv p-TosOH·H2O, acetone, 2 h, 23 °C; (c) 3 equiv I2, 5.5 equiv NaHCO3, acetone/H2O (10:1), 20 h, 23 °C.
Scheme 2: Preparation of isopropylidene protected ribose 9 along a new, modified route. Reagents and conditio...
Heterocyclic synthesis
From the aldehyde 9 the corresponding oxime was formed under buffered conditions [30] and the crude material was directly submitted to reduction with LiAlH4 [31] to furnish amine 10 as a practically pure compound (72%) (Scheme 3). The conversion of amine 10 with chlorouracil 3 proceeded with NEt3 as base in EtOH at elevated temperature. Now, the key advantage of using amine 10 with isopropylidene-protected hydroxy functions came into the game, because product 11 could be purified by column chromatography. We first investigated the conversion of this material with aldehyde 13 to furnish the isopropylidene-protected annulation product. However, subsequent deprotection gave compound 1 containing significant impurities. If compound 11 is first deprotected (product 12 was obtained in 99% yield without impurities) and then converted with aldehyde 13, the final target compound 1 could be crystallized from EtOH and washed with EtOH to furnish a pure, yellow crystalline material in 36% yield.
![[1860-5397-12-89-i3]](/bjoc/content/inline/1860-5397-12-89-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Final four steps of the synthesis. Reagents and conditions: (a) 1. 4.5 equiv NH2OH·HCl, 4 equiv NaHCO3, EtOH/H2O (10:1), 23 °C, 16 h; 2. 5 equiv LiAlH4, THF, 67 °C, 4 h; (b) 1.2 equiv amine 10, 1.0 equiv chlorouracil 3, 2.0 equiv NEt3, EtOH, sealed reaction tube, 150 °C, 1.5 h (c) TFA/H2O, 23 °C, 4 h; (d) 1 equiv aldehyde 13, 1.2 equiv NEt3, EtOH, 150 °C, 3 h.
Scheme 3: Final four steps of the synthesis. Reagents and conditions: (a) 1. 4.5 equiv NH2OH·HCl, 4 equiv NaH...
Conclusion
We report on the new synthetic route to 7,8-didemethyl-8-hydroxy-5-deazariboflavin (1), which is the biosynthetic precursor compound to cofactor F420. In comparison with older routes to this target compound, the key innovation of our procedures is the introduction of two isopropylidene protective groups into the ribose side chain, which allowed the chromatographic purification of key intermediate product 11. The sequence started with the preparation of literature-known bis-isopropylidene protected D-ribose 9, which required a transitional protection of the aldehyde function as dithioacetal. With two deviations from the literature protocols (use of propyl- instead of ethylthiol and dithioacetal deprotection with iodine instead of mercury compounds) we have improved the synthesis of aldehyde 9. The latter compound was then transformed to the protected ribitylamine 10 via the oxime, which was reduced with LiAlH4. Primary amine 10 was then converted with chlorouracil 3, which was actually the key innovation of our new route, because the product 11 could be purified by column chromatography prior to deprotection to compound 12. The insufficient purity of compound 12 was the major drawback of one of the previously known syntheses. Conversion of aminouracil 12 to the target compound 1 proceeded straightforwardly.
Experimental
1-Amino-1-deoxy-2,3:4,5-bis-O-(isopropylidene)-D-ribitol (10)
Formation of the oxime: A solution of NaHCO3 (1.55 g, 18.4 mmol) and NH2OH·HCl (1.44 g, 20.7 mmol) in EtOH/H2O (10:1) was stirred for 30 min at ambient temperature. A solution of aldehyde 9 (1.06 g, 4.61 mmol) in EtOH (9 mL) was then added, and the resulting mixture stirred for further 16 h at ambient temperature. About half of the solvent was evaporated, the residue extracted with Et2O (3 × 20 mL) and the combined organic layers were dried (MgSO4) and evaporated after filtration to yield the crude oxime (985 mg, 4.00 mmol, 87%; mixture of E/Z isomers, ratio ca. 3:1), which was submitted to reduction without further purification. HRMS (ESI+): calcd for C11H19NNaO5 268.1161, found 268.1157 [M + Na]+.
Reduction to the amine 10: A solution of the above reported oxime (985 mg, 4.00 mmol) in abs. THF (anhydrous, 20 mL) was dropwise added to a cooled (ice/water bath) suspension of LiAlH4 (760 mg, 20.0 mmol) in abs. THF (20 mL). The reaction mixture was heated for 4 h under reflux, then cooled with an ice/water bath and treated with MgSO4·7 H2O (20 g). The suspension was stirred for 1 h at ambient temperature, then filtered through a short pad of MgSO4 and the residue washed with MTBE (100 mL). The solvent was removed in vacuum to give amine 10 (771 mg, 3.33 mmol, 83%) as colorless oil; [α]20D = −40 (CH2Cl2, 1 g/L); 1H NMR (500 MHz, CDCl3) δ 1.32 (s, 6H), 1.38 (s, 3H), 1.39 (s, 3H), 2.74 (brs, 2H), 2.90 (dd, J = 13.2 Hz, J = 7.7 Hz, 1H), 3.03 (dd, J = 13.2 Hz, J = 5.4 Hz, 1H), 3.89 (dd, J = 8.1 Hz, J = 5.1 Hz, 1H), 3.98 (dd, J = 9.1 Hz, J = 5.7 Hz, 1H), 4.03–4.11 (m, 2H), 4.19 (dt, J = 7.6 Hz, J = 5.5 Hz, 1H) ppm; 13C{1H} NMR (125 MHz, CDCl3) δ 25.42 (CH3), 25.43 (CH3), 26.7 (CH3), 28.1 (CH3), 41.2 (CH2), 68.0 (CH2), 73.2 (CH), 78.1 (CH), 79.2 (CH), 108.5 (C), 109.8 (C) ppm; IR (ATR): 3385 (w), 2986 (m), 2961 (m), 2935 (m), 2875 (w), 1571 (w), 1558 (w), 1481 (w), 1456 (m), 1400 (w), 1246 (s), 1213 (s), 1156 (s), 1061 (s), 980 (m), 894 (w), 845 (s), 791 (w), 754 (m) cm−1; HRMS (ESI+): calcd for C11H22NO4, 232.1543; found, 232.1539 [M + H+].
1-Deoxy-1-[(1,2,3,6-tetrahydro-2,6-dioxopyrimidin-4 yl)amino]-2,3:4,5-bis-O-(isopropylidene)-D-ribitol (11)
A mixture of chlorouracil 3 (218 mg, 1.49 mmol), ribitylamine 10 (413 mg, 1.79 mmol), NEt3 (0.41 mL, 2.98 mmol) and EtOH (3 mL) was heated in a closed and sealed reaction vial at 150 °C for 1.5 h. After cooling to ambient temperature the mixture was concentrated in vacuum and the residue purified by column chromatography (SiO2, MeOH/CH2Cl2 1:20, Rf 0.06) to give title compound 11 (350 mg, 1.03 mmol, 69%) as a colorless solid; mp. 234–236 °C; [α]20D = –35 (MeOH, 1 g/L); 1H NMR (500 MHz, CDCl3) δ 1.33 (s, 6H), 1.39 (s, 3H), 1.41 (s, 3H), 3.32–3.36 (m, 1H), 3.53 (dd, J = 13.9 Hz, J = 3.5 Hz, 1H), 3.84–3.91 (m, 1H), 4.02–4.16 (m, 3H), 4.41 (ddd, J = 9.0 Hz, J = 5.9 Hz, J = 3.6 Hz, 1H), 4.78 (s, 1H) 5.94 (s, 1H), 8.99 (brs, 1H), 10.40 (brs, 1H) ppm; 13C{1H} NMR (125 MHz, CDCl3) δ 25.28 (CH3), 25.30 (CH3), 26.7 (CH3), 27.8 (CH3), 42.2 (CH2), 69.0 (CH2), 73.0 (CH), 74.0 (CH), 75.0 (CH), 77.9 (CH), 109.5 (C), 111.1 (C), 152.3 (C), 154.9 (C), 165.9 (C) ppm; IR (ATR): 3360 (w), 3138 (w), 2987 (w), 2934 (w), 2881 (w), 1714 (s), 1626 (s), 1455 (m), 1381 (m), 1281 (m), 1212 (m), 1155 (m), 1069 (s), 976 (w), 838 (s), 783 (s), 629 (m) cm−1; HRMS (ESI+): calcd for C15H23N3NaO6, 364.1479; found, 364.1480 [M + Na+].
1-Deoxy-1-[(2,6-dioxo-1,2,3,6-tetrahydro-4-pyrimidinyl)amino]-D-ribitol (12)
A solution of compound 11 (245 mg, 0.72 mmol) in TFA/H2O (2.2 mL, 10:1) was stirred for 4 h at 23 °C. Subsequently, all volatile materials were removed in vacuum, the residue was suspended in Et2O (3 mL) and this suspension heated to reflux for 30 min. After cooling to ambient temperature, the precipitate was collected on a glass frit, washed with EtOH (1 mL) and dried under vacuum to give the deprotected ribitol 12 (185 mg, 0.71 mmol, 99%) as a colorless solid; mp. 183–185 °C. [α]20D = –16.7 (DMSO, 1 g/L); 1H NMR (500 MHz, DMSO-d6) δ 3.01 (ddd, J = 12.5 Hz, J = 8.0 Hz, J = 4.6 Hz, 1H), 3.17 (ddd, J = 12.8 Hz, J = 5.6 Hz, J = 3.2 Hz, 1H), 3.37–3.41 (m, 2H), 3.48–3.51 (m, 1H), 3.57 (dd, J = 11.0 Hz, J = 3.3 Hz, 1H), 3.70–3.73 (m, 1H), 4.42 (s, 1H), 6.18 (s, 1H), 9.95 (brs, 1H), 10.15 (brs, 1H) ppm, signals for the four OH protons are not resolved (broad singlet from 4.5–5.0 ppm); 13C{1H} NMR (125 MHz, DMSO-d6) δ 43.9 (CH2), 63.2 (CH2), 69.5 (CH), 72.5 (CH), 72.8 (CH), 73.0 (CH), 150.8 (C), 154.4 (C), 164.5 (C) ppm; IR (ATR): 3296 (br m), 3144 (m), 3133 (m), 2980 (m), 2945 (m), 2889 (m), 1732 (m), 1709 (s), 1628 (s), 1603 (s), 1556 (m), 1483 (w), 1462 (w), 1345 (w), 1203 (w), 1048 (m), 1024 (m), 1000 (m) cm−1. HRMS (ESI+): calcd for C9H16N3O6, 262.1039; found, 262.1040 [M + H+].
1-Deoxy-1-(8-hydroxy-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-10-yl)-D-ribitol (1)
A mixture of aldehyde 13 (90 mg, 0.57 mmol), aminouracil 12 (150 mg, 0.57 mmol), NEt3 (0.10 mL, 0.68 mmol) and EtOH (3 mL) was heated in a closed reaction vial at 150 °C for 3 h. After cooling to ambient temperature, the yellow precipitate was collected by filtration and washed with EtOH (3 mL) to give deazariboflavin 1 (75 mg, 0.21 mmol, 36%) as yellow solid, mp 240–250 °C (decomposition). [α]20D = –6.7 (DMSO, 1 g/L); 1H NMR (500 MHz, CDCl3) δ 3.44–3.66 (m, 4H), 4.20–4.25 (m, 1H), 4.48 (brs, 1H), 4.64–4.83 (m, 3H), 4.90–4.98 (m, 1H), 5.09–5.15 (m, 1H), 7.02 (dd, J = 8.7 Hz, J = 1.6 Hz, 1H), 7.39 (s, 1H), 8.01 (d, J = 8.8 Hz, 1H), 8.87 (s, 1H), 10.96 (s, 1H), 11.24 (brs, 1H) ppm; 13C{1H} NMR (125 MHz, CDCl3) δ 47.9 (CH2), 63.4 (CH2), 69.6 (CH), 72.8 (CH), 73.9 (CH), 102.2 (CH), 110.8 (C), 115.3 (C), 115.4 (CH), 133.8 (CH), 141.5 (CH), 144.1 (C), 156.6 (C), 158.2 (C), 162.5 (C), 164.3 (C) ppm; IR (ATR): 3124 (br m), 3015 (br w), 2803 (br m), 1699 (m), 1645 (m), 1590 (s), 1570 (s), 1513 (s), 1473 (s), 1392 (m), 1376 (m), 1345 (m), 1255 (m), 1233 (m), 1218 (m), 1149 (m), 1063 (m), 1036 (m), 909 (w), 854 (w), 818 (w), 793 (w), 767 (w), 722 (w), 691 (w) cm−1; HRMS (ESI+): calcd for C16H18N3O7, 364.1139; found, 364.1152 [M + H+]; UV–vis (DMSO, c = 0.11 g/L): λmax (lg ε) 411 (4.06), 455 nm (4.19); fluorescence (DMSO): λem = 482 (λirr = 412 or 456 nm), 441 nm (λirr = 412 nm).
Supporting Information
| Supporting Information File 1: General experimental methods, procedures and analytical data for the preparation of aldehyde 9 and copies of NMR spectra of all reported compounds. | ||
| Format: PDF | Size: 982.6 KB | Download |
References
-
Graham, D. E.; White, R. H. Nat. Prod. Rep. 2002, 19, 133–147. doi:10.1039/b103714p
Return to citation in text: [1] -
DiMarco, A. A.; Bobik, T. A.; Wolfe, R. S. Annu. Rev. Biochem. 1990, 59, 355–394. doi:10.1146/annurev.bi.59.070190.002035
Return to citation in text: [1] -
Cheeseman, P.; Toms-Wood, A.; Wolfe, R. S. J. Bacteriol. 1972, 112, 527–531.
Return to citation in text: [1] -
Liedvogel, M.; Mouritsen, H. J. R. Soc., Interface 2010, 7, S147–S162. doi:10.1098/rsif.2009.0411.focus
Return to citation in text: [1] [2] [3] -
Mouritsen, H.; Janssen-Bienhold, U.; Liedvogel, M.; Feenders, G.; Stalleicken, J.; Dirks, P.; Weiler, R. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14294–14299. doi:10.1073/pnas.0405968101
Return to citation in text: [1] -
Nießner, C.; Denzau, S.; Gross, J. C.; Peichl, L.; Bischof, H.-J.; Fleissner, G.; Wiltschko, W.; Wiltschko, R. PLoS One 2011, 6, e20091. doi:10.1371/journal.pone.0020091
Return to citation in text: [1] -
Bolte, P.; Bleibaum, F.; Einwich, A.; Günther, A.; Liedvogel, M.; Heyers, D.; Depping, A.; Wöhlbrand, L.; Rabus, R.; Janssen-Bienhold, U.; Mouritsen, H. PLoS One 2016, 11, e0147819. doi:10.1371/journal.pone.0147819
Return to citation in text: [1] -
Ritz, T.; Adem, S.; Schulten, K. Biophys. J. 2000, 78, 707–718. doi:10.1016/S0006-3495(00)76629-X
Return to citation in text: [1] -
Hein, C. M.; Engels, S.; Kishkinev, D.; Mouritsen, H. Nature 2011, 471, E11–E12. doi:10.1038/nature09875
Return to citation in text: [1] -
Mouritsen, H.; Hore, P. J. Curr. Opin. Neurobiol. 2012, 22, 343–352. doi:10.1016/j.conb.2012.01.005
Return to citation in text: [1] -
Engels, S.; Schneider, N.-L.; Lefeldt, N.; Hein, C. M.; Zapka, M.; Michalik, A.; Elbers, D.; Kittel, A.; Hore, P. J.; Mouritsen, H. Nature 2014, 509, 353–356. doi:10.1038/nature13290
Return to citation in text: [1] -
Liedvogel, M.; Maeda, K.; Henbest, K.; Schleicher, E.; Simon, T.; Timmel, C. R.; Hore, P. J.; Mouritsen, H. PLoS One 2007, 2, e1106. doi:10.1371/journal.pone.0001106
Return to citation in text: [1] -
Qin, S.; Yin, H.; Yang, C.; Dou, Y.; Liu, Z.; Zhang, P.; Yu, H.; Huang, Y.; Feng, J.; Hao, J.; Hao, J.; Deng, L.; Yan, X.; Dong, X.; Zhao, Z.; Jiang, T.; Wang, H.-W.; Luo, S.-J.; Xie, C. Nat. Mater. 2016, 15, 217–226. doi:10.1038/nmat4484
Return to citation in text: [1] -
Ashton, W. T.; Brown, R. D.; Jacobson, F.; Walsh, C. J. Am. Chem. Soc. 1979, 101, 4419–4420. doi:10.1021/ja00509a083
Return to citation in text: [1] -
Ashton, W. T.; Brown, R. D. J. Heterocycl. Chem. 1980, 17, 1709–1712. doi:10.1002/jhet.5570170813
Return to citation in text: [1] -
Tanaka, K.; Kimachi, T.; Kawase, M.; Yoneda, F. J. Chem. Soc., Chem. Commun. 1988, 524–526. doi:10.1039/c39880000524
Return to citation in text: [1] -
Kimachi, T.; Kawase, M.; Matsuki, S.; Tanaka, K.; Yoneda, F. J. Chem. Soc., Perkin Trans. 1 1990, 253–256. doi:10.1039/p19900000253
Return to citation in text: [1] -
Nagamatsu, T.; Hashiguchi, Y.; Yoneda, F. J. Chem. Soc., Perkin Trans. 1 1984, 561–565. doi:10.1039/p19840000561
Return to citation in text: [1] -
Kjer-Nielsen, L.; Patel, O.; Corbett, A. J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; Williamson, N. A.; Purcell, A. W.; Dudek, N. L.; McConville, M. J.; O'Hair, R. A. J.; Khairallah, G. N.; Godfrey, D. I.; Fairlie, D. P.; Rossjohn, J.; McCluskey, J. Nature 2012, 461, 717–723. doi:10.1038/nature11605
Return to citation in text: [1] -
Hossain, M. S.; Le, C. Q.; Joseph, E.; Nguyen, T. Q.; Johnson-Winters, K.; Foss, F., Jr. Org. Biomol. Chem. 2015, 13, 5082–5085. doi:10.1039/C5OB00365B
Return to citation in text: [1] -
Peterson, K. E.; Cinelli, M. A.; Morrell, A. E.; Mehta, A.; Dexheimer, T. S.; Agama, K.; Antony, S.; Pommier, Y.; Cushman, M. J. Med. Chem. 2011, 54, 4937–4953. doi:10.1021/jm101338z
Return to citation in text: [1] -
Synder, J. R. Carbohydr. Res. 1990, 198, 1–13. doi:10.1016/0008-6215(90)84271-U
Return to citation in text: [1] -
Pearson, M. S. M.; Plantier-Royon, R.; Szymoniak, J.; Bertus, P.; Behr, J.-B. Synthesis 2007, 3589–3594. doi:10.1055/s-2007-990858
Return to citation in text: [1] -
Aslani-Shotorbani, G.; Buchanan, J. G.; Edgar, A. R.; Shahidi, P. K. Carbohydr. Res. 1985, 136, 37–52. doi:10.1016/0008-6215(85)85184-3
Return to citation in text: [1] [2] -
Kumar, V.; Dev, S. Tetrahedron 1987, 43, 5933–5948. doi:10.1016/S0040-4020(01)87799-1
Return to citation in text: [1] -
Zinner, H. Chem. Ber. 1950, 83, 275–277. doi:10.1002/cber.19500830315
Return to citation in text: [1] -
Sambaiah, T.; Fanwick, P. E.; Cushman, M. J. Org. Chem. 2001, 66, 4405–4408. doi:10.1021/jo001782h
Return to citation in text: [1] -
Ohlsson, J.; Magnusson, G. Carbohydr. Res. 2001, 331, 91–94. doi:10.1016/S0008-6215(00)00323-2
Return to citation in text: [1] -
Driguez, P.-A.; Barrere, B.; Quash, G.; Doutheau, A. Carbohydr. Res. 1994, 262, 297–310. doi:10.1016/0008-6215(94)84186-1
Return to citation in text: [1] -
Würdemann, M.; Christoffers, J. Eur. J. Org. Chem. 2013, 7421–7431. doi:10.1002/ejoc.201301039
Return to citation in text: [1] -
Schramm, H.; Christoffers, J. Tetrahedron: Asymmetry 2009, 20, 2724–2727. doi:10.1016/j.tetasy.2009.10.019
Return to citation in text: [1]
| 30. | Würdemann, M.; Christoffers, J. Eur. J. Org. Chem. 2013, 7421–7431. doi:10.1002/ejoc.201301039 |
| 31. | Schramm, H.; Christoffers, J. Tetrahedron: Asymmetry 2009, 20, 2724–2727. doi:10.1016/j.tetasy.2009.10.019 |
| 1. | Graham, D. E.; White, R. H. Nat. Prod. Rep. 2002, 19, 133–147. doi:10.1039/b103714p |
| 2. | DiMarco, A. A.; Bobik, T. A.; Wolfe, R. S. Annu. Rev. Biochem. 1990, 59, 355–394. doi:10.1146/annurev.bi.59.070190.002035 |
| 8. | Ritz, T.; Adem, S.; Schulten, K. Biophys. J. 2000, 78, 707–718. doi:10.1016/S0006-3495(00)76629-X |
| 9. | Hein, C. M.; Engels, S.; Kishkinev, D.; Mouritsen, H. Nature 2011, 471, E11–E12. doi:10.1038/nature09875 |
| 10. | Mouritsen, H.; Hore, P. J. Curr. Opin. Neurobiol. 2012, 22, 343–352. doi:10.1016/j.conb.2012.01.005 |
| 11. | Engels, S.; Schneider, N.-L.; Lefeldt, N.; Hein, C. M.; Zapka, M.; Michalik, A.; Elbers, D.; Kittel, A.; Hore, P. J.; Mouritsen, H. Nature 2014, 509, 353–356. doi:10.1038/nature13290 |
| 27. | Sambaiah, T.; Fanwick, P. E.; Cushman, M. J. Org. Chem. 2001, 66, 4405–4408. doi:10.1021/jo001782h |
| 4. | Liedvogel, M.; Mouritsen, H. J. R. Soc., Interface 2010, 7, S147–S162. doi:10.1098/rsif.2009.0411.focus |
| 5. | Mouritsen, H.; Janssen-Bienhold, U.; Liedvogel, M.; Feenders, G.; Stalleicken, J.; Dirks, P.; Weiler, R. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 14294–14299. doi:10.1073/pnas.0405968101 |
| 6. | Nießner, C.; Denzau, S.; Gross, J. C.; Peichl, L.; Bischof, H.-J.; Fleissner, G.; Wiltschko, W.; Wiltschko, R. PLoS One 2011, 6, e20091. doi:10.1371/journal.pone.0020091 |
| 7. | Bolte, P.; Bleibaum, F.; Einwich, A.; Günther, A.; Liedvogel, M.; Heyers, D.; Depping, A.; Wöhlbrand, L.; Rabus, R.; Janssen-Bienhold, U.; Mouritsen, H. PLoS One 2016, 11, e0147819. doi:10.1371/journal.pone.0147819 |
| 28. | Ohlsson, J.; Magnusson, G. Carbohydr. Res. 2001, 331, 91–94. doi:10.1016/S0008-6215(00)00323-2 |
| 29. | Driguez, P.-A.; Barrere, B.; Quash, G.; Doutheau, A. Carbohydr. Res. 1994, 262, 297–310. doi:10.1016/0008-6215(94)84186-1 |
| 4. | Liedvogel, M.; Mouritsen, H. J. R. Soc., Interface 2010, 7, S147–S162. doi:10.1098/rsif.2009.0411.focus |
| 24. | Aslani-Shotorbani, G.; Buchanan, J. G.; Edgar, A. R.; Shahidi, P. K. Carbohydr. Res. 1985, 136, 37–52. doi:10.1016/0008-6215(85)85184-3 |
| 19. | Kjer-Nielsen, L.; Patel, O.; Corbett, A. J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; Williamson, N. A.; Purcell, A. W.; Dudek, N. L.; McConville, M. J.; O'Hair, R. A. J.; Khairallah, G. N.; Godfrey, D. I.; Fairlie, D. P.; Rossjohn, J.; McCluskey, J. Nature 2012, 461, 717–723. doi:10.1038/nature11605 |
| 21. | Peterson, K. E.; Cinelli, M. A.; Morrell, A. E.; Mehta, A.; Dexheimer, T. S.; Agama, K.; Antony, S.; Pommier, Y.; Cushman, M. J. Med. Chem. 2011, 54, 4937–4953. doi:10.1021/jm101338z |
| 22. | Synder, J. R. Carbohydr. Res. 1990, 198, 1–13. doi:10.1016/0008-6215(90)84271-U |
| 16. | Tanaka, K.; Kimachi, T.; Kawase, M.; Yoneda, F. J. Chem. Soc., Chem. Commun. 1988, 524–526. doi:10.1039/c39880000524 |
| 17. | Kimachi, T.; Kawase, M.; Matsuki, S.; Tanaka, K.; Yoneda, F. J. Chem. Soc., Perkin Trans. 1 1990, 253–256. doi:10.1039/p19900000253 |
| 18. | Nagamatsu, T.; Hashiguchi, Y.; Yoneda, F. J. Chem. Soc., Perkin Trans. 1 1984, 561–565. doi:10.1039/p19840000561 |
| 23. | Pearson, M. S. M.; Plantier-Royon, R.; Szymoniak, J.; Bertus, P.; Behr, J.-B. Synthesis 2007, 3589–3594. doi:10.1055/s-2007-990858 |
| 24. | Aslani-Shotorbani, G.; Buchanan, J. G.; Edgar, A. R.; Shahidi, P. K. Carbohydr. Res. 1985, 136, 37–52. doi:10.1016/0008-6215(85)85184-3 |
| 25. | Kumar, V.; Dev, S. Tetrahedron 1987, 43, 5933–5948. doi:10.1016/S0040-4020(01)87799-1 |
| 14. | Ashton, W. T.; Brown, R. D.; Jacobson, F.; Walsh, C. J. Am. Chem. Soc. 1979, 101, 4419–4420. doi:10.1021/ja00509a083 |
| 15. | Ashton, W. T.; Brown, R. D. J. Heterocycl. Chem. 1980, 17, 1709–1712. doi:10.1002/jhet.5570170813 |
| 4. | Liedvogel, M.; Mouritsen, H. J. R. Soc., Interface 2010, 7, S147–S162. doi:10.1098/rsif.2009.0411.focus |
| 12. | Liedvogel, M.; Maeda, K.; Henbest, K.; Schleicher, E.; Simon, T.; Timmel, C. R.; Hore, P. J.; Mouritsen, H. PLoS One 2007, 2, e1106. doi:10.1371/journal.pone.0001106 |
| 13. | Qin, S.; Yin, H.; Yang, C.; Dou, Y.; Liu, Z.; Zhang, P.; Yu, H.; Huang, Y.; Feng, J.; Hao, J.; Hao, J.; Deng, L.; Yan, X.; Dong, X.; Zhao, Z.; Jiang, T.; Wang, H.-W.; Luo, S.-J.; Xie, C. Nat. Mater. 2016, 15, 217–226. doi:10.1038/nmat4484 |
| 20. | Hossain, M. S.; Le, C. Q.; Joseph, E.; Nguyen, T. Q.; Johnson-Winters, K.; Foss, F., Jr. Org. Biomol. Chem. 2015, 13, 5082–5085. doi:10.1039/C5OB00365B |
© 2016 Bender et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)