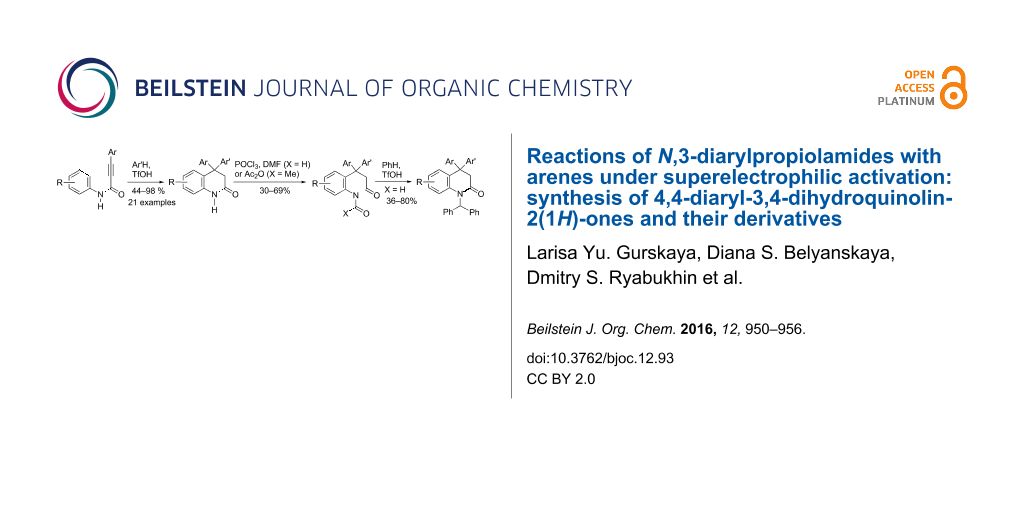
Abstract
The reaction of 3-aryl-N-(aryl)propiolamides with arenes in TfOH at room temperature for 0.5 h led to 4,4-diaryl-3,4-dihydroquinolin-2-(1H)-ones in yields of 44–98%. The obtained dihydroquinolinones were further transformed into the corresponding N-acyl or N-formyl derivatives. For the latter, the superelectrophilic activation of the N-formyl group by TfOH in the reaction with benzene resulted in the formation of N-(diphenylmethyl)-substituted dihydroquinolinones.

Graphical Abstract
Introduction
Quinoline derivatives are a very important class of heterocycles, which are used in chemistry, biology, medicine, and materials science. For instance, see a series of recent reviews on anti-malaria drugs containing a quinoline motif in the structure [1-3]. The synthesis of quinolines is an important task of organic chemistry [4,5]. Many of these synthetical protocols are based on alkynes, which are widely used for the preparation of various carbo- and heterocycles [6-10].
Based on our works on the synthesis of 4-arylquinolin-2(1H)-ones from acetylene compounds under superelectrophilic activation conditions [11-14], we continued to develop some methods for the syntheses of quinoline derivatives. Previously we showed, just in a few examples, that some 3-aryl-N-(aryl)propiolamides reacted with benzene under the action of Brønsted or Lewis superacids affording 4-aryl-4-phenyl-3,4-dihydroquinolin-2(1H)-ones [12,13].
The main goal of this work was a systematic study on reactions of 3-aryl-N-(aryl)propiolamides with arenes under the action of the Brønsted superacid TfOH (CF3SO3H, triflic acid), strong Lewis acids AlX3 (X = Cl, Br), or the conjugate Brønsted–Lewis superacid TfOH–SbF5.
Results and Discussion
The protonation of alkynamides 1 on both the oxygen atom of the amide group and a carbon atom of the acetylene bond in superacids or coordination of these basic centers with strong Lewis acids leads to the formation of dications A that are considered as superelectrophiles [15] (Scheme 1). These dications can either undergo an intramolecular cyclization to 4-arylquinolin-2(1H)-ones 3 or, alternatively, react with arenes as external π-nucleophiles. In this latter pathway, Friedel–Crafts alkenylation of arenes by species A leads to structures 4, which can then be diprotonated to the cations B and finally cyclized to 4,4-diaryl-3,4-dihydroquinolin-2(1H)-ones 2. It should be noted that quinolinones 3 do not react with arenes with formation of compounds 2 under superelectrophilic activation, and starting materials 3 remain unreacted [12,13].
![[1860-5397-12-93-i1]](/bjoc/content/inline/1860-5397-12-93-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Transformations of 3-aryl-N-(aryl)propiolamides 1 into 4-arylquinolin-2(1H)-ones 3 or 4,4-diaryl-3,4-dihydroquinolin-2(1H)-ones 2 in the presence of arenes through the formation of intermediate cations A, B under the superelectrophilic activation.
Scheme 1: Transformations of 3-aryl-N-(aryl)propiolamides 1 into 4-arylquinolin-2(1H)-ones 3 or 4,4-diaryl-3,...
Such 4,4-diaryl-3,4-dihydroquinolin-2-(1H)-ones 2 are very rare objects, there is no general method for their synthesis [16,17]. Thus, developing of synthetic method to get these compounds is an actual goal of organic chemistry.
Reactions of the series of 3-aryl-N-(aryl)propiolamides 1a–u, bearing various donor–acceptor substituents on aryl rings, with benzene and other arenes in the presence of TfOH, TfOH–SbF5, AlCl3, or AlBr3 were studied. Initial amides 1a–u, reaction conditions, target 4,4-diaryl-3,4-dihydroquinolin-2-(1H)-ones 2a–x, and some byproducts 3a–c and intermediate compounds 4a,b are given in Table 1. Structures of compounds 2a–x, 3a–c, and 4a,b were determined by means of 1H, 13C, 19F NMR, HRMS methods (see Supporting Information File 1), and X-ray analysis in case of compound 2f (Figure 1).
Table 1: Reactions of amides 1a–u with benzene (and other arenes) under superelectrophilic activation, leading to dihydroquinolinones 2a–x.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-93-i4.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Starting amide 1 | Acid (reaction conditions) | Reaction product, dihydroquinolinone 2 | |||||
|---|---|---|---|---|---|---|---|---|
| no. | R | R′ | no. | R | R′ | Yield, % | ||
| 1 | 1a | H | H | TfOHa | 2a | H | H | 90 |
| 2 | 1a | TfOH-SbF5a | 2a | 85 | ||||
| 3 | 1a | AlCl3b | 2a | 91 | ||||
| 4 | 1a | AlBr3b | 2a | 52c | ||||
| 5 | 1b | 2-Me | H | TfOHa | 2b | 8-Me | H | 93 |
| 6 | 1b | TfOH-SbF5a | 2b | 38 | ||||
| 7 | 1c | 3-Me | H | TfOHa | 2c | 7-Me | H | 75 |
| 8 | 1c | TfOH-SbF5a | 2c | 83 | ||||
| 9 | 1d | 4-Me | H | TfOHa | 2d | 6-Me | H | 98 |
| 10 | 1d | TfOH-SbF5a | 2d | 53 | ||||
| 11 | 1e | 2-F | H | TfOHa | 2e | 8-F | H | 73 |
| 12 | 1e | TfOH-SbF5a | 2e | 95 | ||||
| 13 | 1e | AlBr3b | 2e | 55 | ||||
| 14 | 1f | 3-F | H | TfOHa | 2f | 7-F | H | 52 |
| 15 | 1f | AlBr3b | 2f | 58 | ||||
| 16 | 1g | 4-F | H | TfOHa | 2g | 6-F | H | 18d |
| 17 | 1g | TfOHe | 2g | 84 | ||||
| 18 | 1g | TfOH-SbF5a | 2g | 95 | ||||
| 19 | 1g | AlBr3b | 2g | 72 | ||||
| 20 | 1h | 4-Cl | H | TfOHa | 2h | 6-Cl | H | 23f |
| 21 | 1h | TfOHe | 2h | 88 | ||||
| 22 | 1h | AlBr3b | 2h | 52 | ||||
| 23 | 1i | 2,3-Me2 | H | TfOHa | 2i | 7,8-Me2 | H | 88 |
| 24 | 1j | 2,4-Me2 | H | TfOHa | 2j | 6,8-Me2 | H | 89 |
| 25 | 1k | 3,4-Me2 | H | TfOHa | 2k | 6,7-Me2 | H | 74 |
| 26 | 1l | 2-MeO | H | TfOHa | 2l | 8-MeO | H | 98 |
| 27 | 1m | 3-MeO | H | TfOHa | 2m | 7-MeO | H | 44 |
| 28 | 1n | 4-MeO | H | TfOHa | 2n | 6-MeO | H | 64 |
| 29 | 1o | 3-F, 4-MeO | H | TfOHa | 2o | 7-F, 6-MeO | H | 53 |
| 30 | 1p | H | 4-Me | TfOHa | 2p | H | 4-Me | 90 |
| 31 | 1q | 4-F | 4-Me | TfOHa | 2q | 6-F | 4-Me | 68 |
| 32 | 1q | AlBr3b | 2q | 53 | ||||
| 2g | 6-F | H | 13 | |||||
| 33 | 1r | H | 4-F | TfOHa | 2r | H | 4-F | 66 |
| 34 | 1s | 4-F | 4-Cl | TfOHa | 2s | 6-F | 4-Cl | 87 |
| 35 | 1s | AlBr3b | 2s | 70 | ||||
| 36 | 1t | 2,3-benzo | H | AlBr3b | 2t | 7,8-benzo | H | 22g |
| 37 | 1u | 3,4-benzo | H | AlBr3b | 2u | 5,6-benzo | H | 17h |
| 38 | 1a | H | H | TfOHa,i | 2v | H | 4-Cl | 50 |
| 39 | 1a | TfOHa,j | 2x | H | 3,4-Cl2 | 60 | ||
aRoom temperature, 0.5 h. b80 °C, 1 h. c4-Phenylquinolin-2(1H)-one 3a was also obtained in a yield of 41%. dN-(4-Fluorophenyl)amide of 3,3-diphenylpropenoic acid 4a [Ph2C=CHCONH(4-FC6H4)] was obtained as a major reaction product in a yield of 80%. eRoom temperature, 7 h. fN-(4-Chlorophenyl)amide of 3,3-diphenylpropenoic acid 4b [Ph2C=CHCONH(4-ClC6H4)] was obtained as a major reaction product in a yield of 75%. g4-Phenyl-7,8-benzoquinolin-2(1H)-one 3b was also obtained in a yield of 15%. h4-Phenyl-5,6-benzoquinolin-2(1H)-one 3c was also obtained in a yield of 17%. iReaction with chlorobenzene. jReaction with 1,2-dichlorobenzene.
![[1860-5397-12-93-1]](/bjoc/content/figures/1860-5397-12-93-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of 2f (ellipsoid contours of probability levels are 50%).
Figure 1: Molecular structure of 2f (ellipsoid contours of probability levels are 50%).
We found that the best conditions for the synthesis of dihydroquinolines 2 from amides 1 and arenes were as follows: TfOH, room temperature, 0.5 h. Some optimization of the reaction on minimization of the usage of arenes and TfOH were performed. The amount of arene may be decreased to 5 equivalents. But, concerning the use of a lower amount of TfOH, for instance, running the reaction in CH2Cl2 solution led to a dramatic decrease of the yields of the target products. Thus, it is better to conduct the reaction in neat TfOH, which is a good weak nucleophilic medium for the stabilization of the intermediate cationic species.
No concurrent formation of the corresponding 4-arylquinolinones 3 was observed, and in most cases the yields of the target products 2 are 44–98% (see Table 1). Also this reaction may be carried out under the action of other acids, e.g., TfOH–SbF5 or AlX3 (X = Cl, Br). But in the stronger acidic system TfOH–SbF5 (Hammet acidity function H0 −19, see [18]) the yields of compounds 2 were usually lower as compared to TfOH (H0 −14) (Table 1, entries 2, 6 and 10). For the Lewis acid AlBr3, additional formation of compounds 3a,b,c was detected (Table 1, entries 4, 36 and 37), or exchange of aryl groups in 2 took place (Table 1, entry 32, formation of 2g from 2q, as a result of the exchange of the p-tolyl moiety by a phenyl group).
Amides 1, bearing various donor (Me, MeO, benzo-) and acceptor (F, Cl) substituents on both aryl rings at the acetylene bond and at the nitrogen atom may be involved in this reaction. Substrates 1g, 1h, having electron withdrawing 4-F and 4-Cl substituents on the N-aryl ring, were hardly cyclized under the formation of dihydroquinolinones 2g, 2h. Thus, at room temperature for 0.5 h, apart from target compounds 2g, 2h, the corresponding amides of 3,3-diphenylpropenoic acid 4a, 4b were isolated (Table 1, entries 16 and 20). Increasing the reaction time to 7 h resulted in the solely formation of 2g and 2h (Table 1, entries 17 and 21). That clearly proved the participation of compounds 4 in the reaction pathway leading to dihydroquinoliones 2 (Scheme 1).
It should be noted, that substrates 1 with more than one possible reaction site, namely 3-substituted amides 1c,f and 3,4-disubstituted amides 1k,o gave regioselectively only 7-substituted quinolinones 2c,f (Table 1, entries 7, 8, 14 and 15) and 6,7-disubstituted quinolinones 2k,o (Table 1, entries 25 and 29), respectively.
Concerning the arene component of this reaction, apart from benzene, chlorobenzene and 1,2-dichlorobenzene it may take part in this Friedel–Crafts process (Table 1, entries 38 and 39). More donating aromatic substrates, such as toluene, isomeric xylenes, mesitylene, or pseudocumene, led to complex mixtures of oligomeric reaction products. Such activated aryl groups may undergo several electrophilic attack from intermediate cationic species A and B (Scheme 1), that complicated this reaction.
Thus, reaction of amides 1 with benzene and some other arenes in TfOH is an effective way to 4,4-diaryl-3,4-dihydroquinolin-2(1H)-ones 2.
To show the synthetic potential of dihydroquinolinones 2 their N-formylation and N-acetylation reactions were carried out. Compounds 2a,d,e and h were N-formylated in the system POCl3–DMF–CHCl3 under the Vilsmeier–Haack reaction conditions with formation of compounds 5a–d (Scheme 2). Apart from that, dihydroquinolinones 2a,e–h were acetylated into derivatives 6a–e (Scheme 2).
![[1860-5397-12-93-i2]](/bjoc/content/inline/1860-5397-12-93-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: N-Formylation and N-acylation of dihydroquinolinones 2.
Scheme 2: N-Formylation and N-acylation of dihydroquinolinones 2.
Then reactions of compounds 5 and 6 with arenes in TfOH were checked. Analogously to other heteroaromatic aldehydes of the series of pyridine [19], quinolone [20], pyrazol [21], and imidazole [22], the superelectrophilic activation of the formyl group in 5 was expected. Indeed, compounds 5a,b and d reacted with benzene in a Friedel–Crafts process affording N-diphenylmethyl substituted derivatives 7a–c, respectively (Scheme 3). Most probably, the reaction proceeds through the formation of O,O’-diprotanated intermediate C (Scheme 3), which are considered as superlectrophiles [15]. The close structural proximity of two positive charges in C substantially increases the electrophilic properties of the carbon of the O-protonated N-formyl group and explains its reactivity. The reactions of 5 with other arenes, such as 1,2-dichlorobenzene, toluene, or isomeric xylenes, gave deformylation products 2. In this case, probably, target compounds 7 were initially formed, but then protolytic cleavage of the N–CH(Ar)2 bond occurred leading to relatively stable diarylmethyl cations (+CH(Ar)2). That took place to less extent also for the N-diphenylmethyl group in 7a–c. It should be mentioned, that this is the first example of such a superelectrophilic activation of a N-formyl group, and this reaction did not take place for N-acyl derivatives 6.
![[1860-5397-12-93-i3]](/bjoc/content/inline/1860-5397-12-93-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Superelectrophilic activation of the N-formyl group of compounds 5 and their reaction with benzene.
Scheme 3: Superelectrophilic activation of the N-formyl group of compounds 5 and their reaction with benzene.
Additionally a DFT calculation of dications C1 and D1 derived from 5a and 6a, respectively, was carried out to estimate the electrophilic properties of these species. Charge distribution, contribution of atomic orbital into LUMO and global electrophilicity indices ω [23,24] were calculated (Table 2, and Figure 2). The calculations showed that the Cα carbon of the protonated N-formyl and N-acyl groups in species C1 and D1, respectively, have a rather large positive charge. Despite the large charge on C2 of the protonated carbonyl groups and its substantial contribution into LUMO (Figure 2), this carbon is not reactive, probably, due to steric reasons. A comparison of the electrophilicity indices ω of C1 and D1 (Table 2) revealed that the former is stronger electrophile. Also, perhaps, dication D1 was not reactive due to the bigger spatial volume of the methyl substituent in the N-acyl group compared to hydrogen of the N-formyl one in C1, that hampered the reaction with arene molecule.
Table 2: Selected electronic characteristics (DFT calculations) of dications C1 and D1 derived from protonation of 5a and 6a, respectively.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-93-i5.svg?max-width=637&scale=1.0)
|
|||||||||
| cation | EHOMO, eV | ELUMO, eV | ω,a eV | q(C2),b | q(Ca),b | q(CN),b | k(C2)LUMO,с % | k(Ca)LUMO,с % | k(CN)LUMO,с % |
|---|---|---|---|---|---|---|---|---|---|
| C1 | −7.11 | −4.56 | 6.69 | 0.84 | 0.60 | −0.44 | 14.8 | 11.5 | 1.1 |
| D1 | −7.22 | −4.49 | 6.28 | 0.82 | 0.84 | −0.50 | 24.9 | 11.9 | 3.1 |
aGlobal electrophilicity index ω = (EHOMO + ELUMO) 2/8(ELUMO − EHOMO). bNatural charges. cContribution of atomic orbitals into the molecular orbital.
![[1860-5397-12-93-2]](/bjoc/content/figures/1860-5397-12-93-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
A simple and effective approach to 4,4-diaryl-3,4-dihydroquinolin-2-(1H)-ones based on the reaction of 3-aryl-N(aryl)propiolamides with arenes under superelectrophilic activation in triflic acid has been developed. The synthetic potential of the obtained dihydroquinolinones has been demonstrated by their transformations into N-acyl, N-formyl, and N-diphenylmethyl derivatives.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization of compounds, 1H, 13C, 19F NMR spectra, and data on DFT calculations. | ||
| Format: PDF | Size: 8.7 MB | Download |
Acknowledgements
This work was supported by the Russian Scientific Foundation (grant no 14-13-00448). Spectral studies were performed at the Center for Magnetic Resonance, Center for Chemical Analysis and Materials Research, and the Research Center for X-ray Diffraction Studies of the Saint Petersburg State University, Saint Petersburg, Russia.
References
-
Njoroge, M.; Njuguna, N. M.; Mutai, P.; Ongarora, D. S. B.; Smith, P. W.; Chibale, K. Chem. Rev. 2014, 114, 11138–11163. doi:10.1021/cr500098f
Return to citation in text: [1] -
Teixeira, C.; Vale, N.; Pérez, B.; Gomes, A.; Gomes, J. R. B.; Gomes, P. Chem. Rev. 2014, 114, 11164–11220. doi:10.1021/cr500123g
Return to citation in text: [1] -
Barnet, D. S.; Guy, R. K. Chem. Rev. 2014, 114, 11221–11241. doi:10.1021/cr500543f
Return to citation in text: [1] -
Khusnutdinov, R. I.; Bayguzina, A. R.; Dzhemilev, U. M. J. Organomet. Chem. 2014, 768, 75–114. doi:10.1016/j.jorganchem.2014.06.008
Return to citation in text: [1] -
Madapa, S.; Tusi, Z.; Batra, S. Curr. Org. Chem. 2008, 12, 1116–1183. doi:10.2174/138527208785740300
Return to citation in text: [1] -
Yamamoto, Y. Chem. Soc. Rev. 2014, 43, 1575–1600. doi:10.1039/c3cs60369e
Return to citation in text: [1] -
Salvio, R.; Moliterno, M.; Bella, M. Asian J. Org. Chem. 2014, 3, 340–351. doi:10.1002/ajoc.201400021
Return to citation in text: [1] -
Chinchilla, R.; Najera, C. Chem. Rev. 2014, 114, 1783–1826. doi:10.1021/cr400133p
Return to citation in text: [1] -
Trofimov, B. A.; Schmidt, E. Yu. Russ. Chem. Rev. 2014, 83, 600–619. doi:10.1070/RC2014v083n07ABEH004425
Return to citation in text: [1] -
Vasilyev, A. V. Russ. Chem. Rev. 2013, 82, 187–204. doi:10.1070/RC2013v082n03ABEH004345
Return to citation in text: [1] -
Ryabukhin, D. S.; Vasil'ev, A. V. Russ. J. Org. Chem. 2008, 44, 1849–1851. doi:10.1134/S1070428008120257
Return to citation in text: [1] -
Ryabukhin, D. S.; Vasilyev, A. V.; Vyazmin, S. Yu. Russ. Chem. Bull. 2012, 61, 843–846. doi:10.1007/s11172-012-0117-3
Return to citation in text: [1] [2] [3] -
Ryabukhin, D. S.; Gurskaya, L. Yu.; Fukin, G. K.; Vasilyev, A. V. Tetrahedron 2014, 70, 6428–6443. doi:10.1016/j.tet.2014.07.028
Return to citation in text: [1] [2] [3] -
Ryabukhin, D. S.; Vasilyev, A. V. Tetrahedron Lett. 2015, 56, 2200–2202. doi:10.1016/j.tetlet.2015.03.060
Return to citation in text: [1] -
Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; Wiley: New York, 2008.
Return to citation in text: [1] [2] -
Natarajan, M.; Ramakrishnan, V. T. Indian J. Chem., Sect. B 1984, 23, 720–727.
Return to citation in text: [1] -
Petunin, P. A.; Pesis, A. S. Zh. Obshch. Khim. 1952, 22, 1187–1191.
Return to citation in text: [1] -
Olah, G. A.; Prakash, G. K. S.; Molnar, A.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley: New Jersey, 2009.
Return to citation in text: [1] -
Prakash, G. K. S.; Paknia, F.; Chacko, S.; Mathew, T.; Olah, G. A. Heterocycles 2008, 76, 783–799. doi:10.3987/COM-08-S(N)90
Return to citation in text: [1] -
Klumpp, D. A.; Jones, A.; Lau, S.; de Leon, S.; Garza, M. Synthesis 2000, 1117–1120. doi:10.1055/s-2000-6322
Return to citation in text: [1] -
Klumpp, D. A.; Kindelin, P. J.; Li, A. Tetrahedron Lett. 2005, 46, 2931–2935. doi:10.1016/j.tetlet.2005.02.152
Return to citation in text: [1] -
Sheets, M. R.; Li, A.; Bower, E. A.; Weigel, A. R.; Abbot, M. P.; Gallo, R. M.; Mitton, A. A.; Klumpp, D. A. J. Org. Chem. 2009, 74, 2502–2507. doi:10.1021/jo802798x
Return to citation in text: [1] -
Parr, R. G.; v. Szentpaly, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x
Return to citation in text: [1] -
Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p
Return to citation in text: [1]
| 1. | Njoroge, M.; Njuguna, N. M.; Mutai, P.; Ongarora, D. S. B.; Smith, P. W.; Chibale, K. Chem. Rev. 2014, 114, 11138–11163. doi:10.1021/cr500098f |
| 2. | Teixeira, C.; Vale, N.; Pérez, B.; Gomes, A.; Gomes, J. R. B.; Gomes, P. Chem. Rev. 2014, 114, 11164–11220. doi:10.1021/cr500123g |
| 3. | Barnet, D. S.; Guy, R. K. Chem. Rev. 2014, 114, 11221–11241. doi:10.1021/cr500543f |
| 12. | Ryabukhin, D. S.; Vasilyev, A. V.; Vyazmin, S. Yu. Russ. Chem. Bull. 2012, 61, 843–846. doi:10.1007/s11172-012-0117-3 |
| 13. | Ryabukhin, D. S.; Gurskaya, L. Yu.; Fukin, G. K.; Vasilyev, A. V. Tetrahedron 2014, 70, 6428–6443. doi:10.1016/j.tet.2014.07.028 |
| 23. | Parr, R. G.; v. Szentpaly, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922–1924. doi:10.1021/ja983494x |
| 24. | Chattaraj, P. K.; Giri, S.; Duley, S. Chem. Rev. 2011, 111, PR43–PR75. doi:10.1021/cr100149p |
| 11. | Ryabukhin, D. S.; Vasil'ev, A. V. Russ. J. Org. Chem. 2008, 44, 1849–1851. doi:10.1134/S1070428008120257 |
| 12. | Ryabukhin, D. S.; Vasilyev, A. V.; Vyazmin, S. Yu. Russ. Chem. Bull. 2012, 61, 843–846. doi:10.1007/s11172-012-0117-3 |
| 13. | Ryabukhin, D. S.; Gurskaya, L. Yu.; Fukin, G. K.; Vasilyev, A. V. Tetrahedron 2014, 70, 6428–6443. doi:10.1016/j.tet.2014.07.028 |
| 14. | Ryabukhin, D. S.; Vasilyev, A. V. Tetrahedron Lett. 2015, 56, 2200–2202. doi:10.1016/j.tetlet.2015.03.060 |
| 6. | Yamamoto, Y. Chem. Soc. Rev. 2014, 43, 1575–1600. doi:10.1039/c3cs60369e |
| 7. | Salvio, R.; Moliterno, M.; Bella, M. Asian J. Org. Chem. 2014, 3, 340–351. doi:10.1002/ajoc.201400021 |
| 8. | Chinchilla, R.; Najera, C. Chem. Rev. 2014, 114, 1783–1826. doi:10.1021/cr400133p |
| 9. | Trofimov, B. A.; Schmidt, E. Yu. Russ. Chem. Rev. 2014, 83, 600–619. doi:10.1070/RC2014v083n07ABEH004425 |
| 10. | Vasilyev, A. V. Russ. Chem. Rev. 2013, 82, 187–204. doi:10.1070/RC2013v082n03ABEH004345 |
| 22. | Sheets, M. R.; Li, A.; Bower, E. A.; Weigel, A. R.; Abbot, M. P.; Gallo, R. M.; Mitton, A. A.; Klumpp, D. A. J. Org. Chem. 2009, 74, 2502–2507. doi:10.1021/jo802798x |
| 4. | Khusnutdinov, R. I.; Bayguzina, A. R.; Dzhemilev, U. M. J. Organomet. Chem. 2014, 768, 75–114. doi:10.1016/j.jorganchem.2014.06.008 |
| 5. | Madapa, S.; Tusi, Z.; Batra, S. Curr. Org. Chem. 2008, 12, 1116–1183. doi:10.2174/138527208785740300 |
| 15. | Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; Wiley: New York, 2008. |
| 18. | Olah, G. A.; Prakash, G. K. S.; Molnar, A.; Sommer, J. Superacid Chemistry, 2nd ed.; Wiley: New Jersey, 2009. |
| 20. | Klumpp, D. A.; Jones, A.; Lau, S.; de Leon, S.; Garza, M. Synthesis 2000, 1117–1120. doi:10.1055/s-2000-6322 |
| 16. | Natarajan, M.; Ramakrishnan, V. T. Indian J. Chem., Sect. B 1984, 23, 720–727. |
| 17. | Petunin, P. A.; Pesis, A. S. Zh. Obshch. Khim. 1952, 22, 1187–1191. |
| 21. | Klumpp, D. A.; Kindelin, P. J.; Li, A. Tetrahedron Lett. 2005, 46, 2931–2935. doi:10.1016/j.tetlet.2005.02.152 |
| 12. | Ryabukhin, D. S.; Vasilyev, A. V.; Vyazmin, S. Yu. Russ. Chem. Bull. 2012, 61, 843–846. doi:10.1007/s11172-012-0117-3 |
| 13. | Ryabukhin, D. S.; Gurskaya, L. Yu.; Fukin, G. K.; Vasilyev, A. V. Tetrahedron 2014, 70, 6428–6443. doi:10.1016/j.tet.2014.07.028 |
| 15. | Olah, G. A.; Klumpp, D. A. Superelectrophiles and Their Chemistry; Wiley: New York, 2008. |
| 19. | Prakash, G. K. S.; Paknia, F.; Chacko, S.; Mathew, T.; Olah, G. A. Heterocycles 2008, 76, 783–799. doi:10.3987/COM-08-S(N)90 |
© 2016 Gurskaya et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)