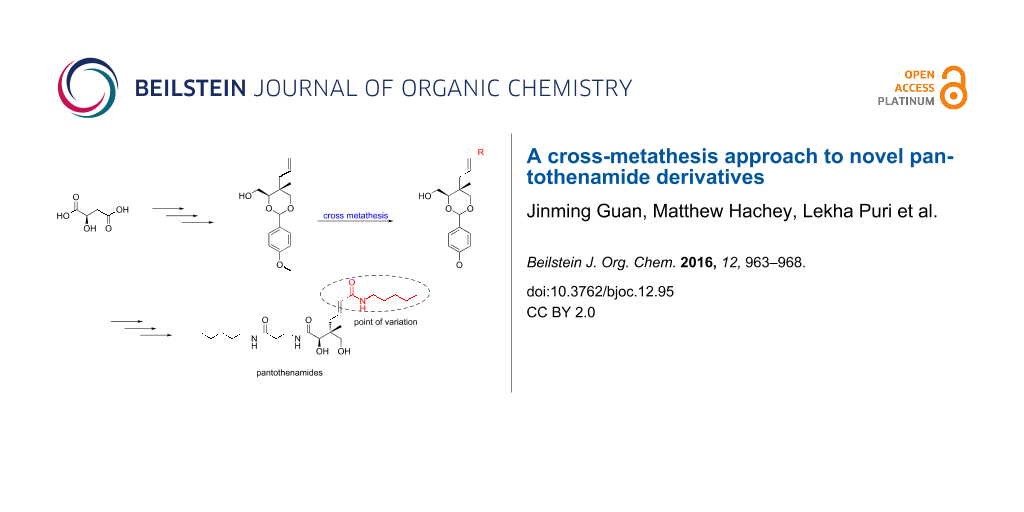
Abstract
Pantothenamides are known for their in vitro antimicrobial activity. Our group has previously reported a new stereoselective route to access derivatives modified at the geminal dimethyl moiety. This route however fails in the addition of large substituents. Here we report a new synthetic route that exploits the known allyl derivative, allowing for the installation of larger groups via cross-metathesis. The method was applied in the synthesis of a new pantothenamide with improved stability in human blood.

Graphical Abstract
Introduction
Bacteria, fungi, and parasites are all rapidly acquiring resistance to currently applied antimicrobials and as a result, our ability to treat infections effectively is diminishing. Efforts to control infections in this resistance era have taken a variety of paths from a renewed push for novel antimicrobial agents to a fresh understanding of antibiotic resistance mechanisms [1]. One successful research direction has been to revisit “older” unexploited structural classes of antimicrobials [2-5].
As first demonstrated in the 1970s [6], many pantothenamides show antimicrobial activity [7-12], including antibacterial, antifungal and/or antiplasmodial activity. For example, what has now become the benchmark pantothenamide, N-pentylpantothenamide (1, Figure 1), is active against Escherichia coli [6] and Staphylococcus aureus [13], at minimum inhibitory concentrations (MICs) in the low micromolar range. Pantothenamides are however rapidly degraded by enzymes, the pantetheinases, that normally hydrolyze pantetheine to pantothenate and cysteamine in human blood [10], which precludes their clinical application.
![[1860-5397-12-95-1]](/bjoc/content/figures/1860-5397-12-95-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of N-pentylpantothenamide (1) and of its methyl allyl derivative 2.
Figure 1: Chemical structures of N-pentylpantothenamide (1) and of its methyl allyl derivative 2.
The antimicrobial activity of pantothenamides is believed to arise from at least two mechanisms depending on the microbial strain. In E. coli pantothenamides may mimic pantetheine and are extended into a thiol-lacking coenzyme A (CoA) derivative by some of the natural CoA biosynthetic enzymes (pantothenate kinase or PanK, phosphopantetheine adenylyltransferase and dephosphocoenzyme A kinase). The resulting inactive CoA analog affects downstream pathways such as the acyl carrier protein activation required for fatty acid biosynthesis [13-16]. In S. aureus, however, recent studies suggest that the antimicrobial activity correlates better with an inhibition of PanK, the regulatory enzyme in CoA biosynthesis in several organisms [17].
Additional studies on pantothenamides are thus easily justified based on their different structural scaffold and new mechanism of action compared to the antimicrobials currently in use.
The vast majority of reported pantothenamide derivatives are modified at the amine moiety [7,9,10]. Among the rare other variations, derivatives with different carbon-chain lengths in the β-alanine moiety have recently been reported by de Villiers et al. [12]. Interestingly, some of these show improved stability in the presence of blood pantetheinases. Modifications at the geminal dimethyl moiety of pantothenamides have also proven quite successful [11,18]. One of the promising derivatives reported is a methyl allyl (2, Figure 1) with a MIC of 3.2 μM against S. aureus and MRSA, compared to MICs of 7 μM for 1 against the same strains [18]. This result encouraged us to synthesize more derivatives with different substituents replacing the geminal dimethyl moiety. Although quite versatile and stereoselective, the reported synthetic methodology to access such analogs decreases in efficiency with increasing size of the group used to replace the methyl residue [11] . We report here a synthetic method to access larger substituents at the geminal dimethyl group. This route takes advantage of the reported path to 2 combined with cross-metathesis.
Results and Discussion
Establishing the synthetic route
Based on the success of the synthetic route published by Akinnusi et al. [18] to generate derivatives modified at the geminal dimethyl moiety of pantothenamides, it was envisaged that allyl derivatives, such as 2 or its precursors, could be good starting points to add larger moieties via cross-metathesis [19]. Two cross-metathesis catalysts were used here (Figure 2): Grubbs’ 2nd generation catalyst (3) and the more versatile Hoveyda–Grubbs’ 2nd generation catalyst (4). Compounds 8 and 9 were chosen for initial tests with this reaction (Scheme 1).
![[1860-5397-12-95-2]](/bjoc/content/figures/1860-5397-12-95-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of Grubbs’ catalysts used in this study.
Figure 2: Structure of Grubbs’ catalysts used in this study.
![[1860-5397-12-95-i1]](/bjoc/content/inline/1860-5397-12-95-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: From (R)-malic acid to allyl derivatives 8 and 9 tested in cross-metathesis. Details are provided in the main text. μ-wave: microwave.
Scheme 1: From (R)-malic acid to allyl derivatives 8 and 9 tested in cross-metathesis. Details are provided i...
The synthesis of compounds 8 and 9 was carried out as previously described (Scheme 1) except for some improvements in the esterification step [18]. Akinnusi et al. reported using reflux conditions (100 °C) with ethanol and thionyl chloride added in two portions separated by three hours [18]. Alternatively, it was found here that a microwave reactor reduced the overall reaction time down to 30 minutes, with an isolated yield of 90%, comparable to that of the thermal reaction.
Metathesis reaction optimization
In 2003, Grubbs published a classification system for cross-metathesis catalysts and substrates, defining the substrates by type ranging from I to IV depending on their level of homodimerization observed in a metathesis reaction [20]. Type I substrates are defined as resulting in fast homodimerization under the conditions of the reaction. Such substrates include terminal alkenes of low steric bulk. Type II and type III substrates, with the latter being bulkier, show slow or no detectable homodimerization, respectively. Type IV is a separate class, including substrates that are spectators to metathesis yet do not inactivate the catalyst. It has been shown that for optimal results (minimal homodimerization) the two olefins to undergo cross-metathesis should be of different types [20].
The two intermediates selected as possible reactants, compounds 8 and 9, were expected to present type II and III behavior, respectively. Acrylic acid (10a) is known to react well in metathesis with catalyst 3 and therefore was used to test the suitability of compounds 8 and 9 in metathesis at 40 °C overnight [21]. As expected based on its large number of accessible conformations, no metathesis product was observed with compound 8 when reacted with acrylic acid. Compound 9 however was more promising and demonstrated type III substrate characteristics (no homodimerization observed) when used with the Grubbs’ 2nd generation catalyst (3) [20]. The presence of a p-methoxybenzaldehyde acetal protecting group in 9 is believed to tie back the alcohols and prevent them from coordinating and deactivating the ruthenium catalyst. When testing the scope of the metathesis reaction with 9, a variety of partners were chosen, including not only acrylic acid (10a), but also 2-vinylpyridine (10b), propylacrylamide (10c), and pentylacrylamide (10d, Table 1). Based on the classification reported by Grubbs [20], these substrates are type II when used in the presence of catalyst 3. Propyl- and pentylacrylamide (10c and 10d) were synthesized from acrylic acid and the respective primary amines as shown in Scheme 2.
Table 1: Optimization of the cross-metathesis reaction between 9 and various alkenes.
| Cross-metathesis partners | Conditions used | Isolated yield (%) using catalyst 3 | Isolated yield (%) using catalyst 4 |
|---|---|---|---|
| 10a | DCM, thermal 40 °C, 16 h, 5 mol % catalyst | 50 | N/Aa |
| 10b | DCM, thermal 40 °C, 16 h, 10 mol % catalyst | 14 | N/A |
| DCM, μ-waveb 60 °C, 60 min, 10 mol % catalyst | N/A | 67 | |
| 10c | DCM, thermal 40 °C, 16 h, 10 mol % catalyst | 25 | 30 |
| DCM, μ-wave 60 °C, 60 min, 10 mol % catalyst | N/A | 30 | |
| 10d | DCM, thermal 40 °C, 16 h, 10 mol % catalyst | 30 | 40 |
| DCM, μ-wave 60 °C, 60 min, 10 mol % catalyst | N/A | 60 | |
aN/A: not applicable; bμ-wave: microwave.
![[1860-5397-12-95-i2]](/bjoc/content/inline/1860-5397-12-95-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of compounds 10c and 10d.
Scheme 2: Synthesis of compounds 10c and 10d.
Although a reasonable yield (50%) was obtained at the first attempt of reacting 9 with acrylic acid (10a) in the presence of catalyst 3 (Table 1), the reaction with pentylacrylamide (10d) proved less successful (30%), yet superior to that of propylacrylamide (10c, 25%). These low yields might be explained by homodimerization of the acrylamide moiety. 2-Vinylpyridine (10b) reacted in even lower yields (14%), as expected based on the known deactivation of catalyst 3 in the presence of pyridinyl ligands [22].
A previous work by Grubbs and co-workers has demonstrated that after dissociation from the metal complex, phosphine ligands can attack the carbine [23]. Thus a phosphine-free catalyst was desirable, and the Hoveyda–Grubbs’ 2nd generation catalyst (4) was chosen. As predicted, the yields increased (Table 1) when catalyst 4 was used for the reaction of 9 with propylacrylamide (10c) and pentylacrylamide (10d). The trans-isomer was the only one detected by 1H NMR of the crude reaction mixture in all cases.
In the hope of further improving the yield of the reaction, a microwave reactor was employed. Indeed, a number of literature reports have demonstrated that microwave-assisted cross-metathesis reactions were complete in a fraction of the time required for the thermal process and provided a cleaner reaction [24-27]. In the cases of 2-vinylpyridine (10b) and pentylacrylamide (10d), not only did the reaction mixture appear cleaner by TLC, but a dramatic increase in yield (67% and 60%, respectively) was also observed when they were reacted with 9 in the presence of catalyst 4 at 60 °C for 60 minutes.
Synthesis of a novel pantothenamide derivative
With the successful optimization of the metathesis step, pentylacrylamide derivative 11d was chosen as the molecule to be extended to pantothenamide 16. As outlined in Scheme 3, 16 was assembled from 11d by first oxidation to the corresponding aldehyde 12 using Dess–Martin periodinane in wet DCM. Further oxidation to the carboxylic acid 13 was achieved using mild chlorite oxidation. The product was used in the next reaction without further purification due to its low stability.
![[1860-5397-12-95-i3]](/bjoc/content/inline/1860-5397-12-95-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The required amine 14 to be coupled to 13 was synthesized from Boc-protected β-alanine and pentylamine using a method reported by Hoegl et al. [11]. Deprotection of the Boc group with trifluoroacetic acid (TFA) provided the free amine 14, which was directly coupled to the carboxylic acid to yield 15. Finally, the desired compound 16 was obtained after acid deprotection of the p-methoxybenzaldehyde acetal in a 90% aqueous acetic acid solution (84% yield).
Biological studies
The antimicrobial activity of 16 was investigated. No visible growth inhibition was observed for E. coli or S. aureus in the presence of compound 16 (up to 512 μM). To determine if the lack of activity was due to poor target affinity or cell-permeability issues, kinetic studies with purified E. coli PanK were performed as previously reported [28]. Compound 16 was neither a good substrate nor an inhibitor of this enzyme, hence explaining its lack of antibacterial activity towards E. coli. On the other hand, compound 16 was found to inhibit the growth of Plasmodium falciparum with an IC50 value of 60 ± 11 μM (n = 3) in the absence of pantetheinase (Figure 3). Interestingly, its IC50 was unaffected by the presence of pantetheinase (51 μM ± 7, n = 6), confirming that modification of pantothenamides at the geminal dimethyl moiety is a viable strategy to overcome the blood stability issues that plague this family of compounds [11]. In order to verify if 16 was targeting CoA biosynthesis as reported for other pantothenamides, the IC50 was also measured in the presence of excess pantothenate (100 µM), in the presence or absence of pantetheinase. The dramatic loss of activity observed confirms that compound 16 acts on the CoA biosynthesis/utilization pathway.
![[1860-5397-12-95-3]](/bjoc/content/figures/1860-5397-12-95-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: In vitro antiplasmodial activity of compound 16 in growth medium containing pantetheinase (open symbols) or in medium in which the pantetheinase had been inactivated (dark symbols). The antiplasmodial activity of this compound can be antagonized by increasing the extracellular concentration of pantothenate from 1 µM (triangles) to 100 μM (circles).
Figure 3: In vitro antiplasmodial activity of compound 16 in growth medium containing pantetheinase (open sym...
Conclusion
In summary, we have successfully developed a new synthetic route that exploits the known allyl derivative 2, allowing for the installation of larger groups via cross-metathesis. Considering the importance of pantothenamides as a potential new class of antimicrobial agents, and the higher stability observed for 16 in blood, we expect this stereoselective synthetic route to find utility in accessing other new pantothenamide derivatives.
Acknowledgements
This work was supported by research grants from the Canadian Institute of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada (NSERC) to K.A. Also, thanks to the China Scholarship Council for financial support to J.G. We would like to thank the Canberra branch of the Australian Red Cross for the provision of red blood cells.
References
-
Brown, E. D.; Wright, G. D. Nature 2016, 529, 336–343. doi:10.1038/nature17042
Return to citation in text: [1] -
Pogue, J. M.; Marchaim, D.; Kaye, D.; Kaye, K. S. Pharmacotherapy 2011, 31, 912–921. doi:10.1592/phco.31.9.912
Return to citation in text: [1] -
Chen, L. F.; Kaye, D. Infect. Dis. Clin. North Am. 2009, 23, 1053–1075. doi:10.1016/j.idc.2009.06.004
Return to citation in text: [1] -
Wright, G. D. Can. J. Microbiol. 2014, 60, 147–154. doi:10.1139/cjm-2014-0063
Return to citation in text: [1] -
Page, M. G.; Heim, J. IDrugs 2009, 12, 561–565.
Return to citation in text: [1] -
Clifton, G.; Bryant, S. R.; Skinner, C. G. Arch. Biochem. Biophys. 1970, 137, 523–528. doi:10.1016/0003-9861(70)90470-4
Return to citation in text: [1] [2] -
Spry, C.; Kirk, K.; Saliba, K. J. FEMS Microbiol. Rev. 2008, 32, 56–106. doi:10.1111/j.1574-6976.2007.00093.x
Return to citation in text: [1] [2] -
Saliba, K. J.; Ferru, I.; Kirk, K. Antimicrob. Agents Chemother. 2005, 49, 632–637. doi:10.1128/AAC.49.2.632-637.2005
Return to citation in text: [1] -
Spry, C.; van Schalkwyk, D. A.; Strauss, E.; Saliba, K. J. Infect. Disord.: Drug Targets 2010, 10, 200–216. doi:10.2174/187152610791163390
Return to citation in text: [1] [2] -
Spry, C.; Macuamule, C.; Lin, Z.; Virga, K. G.; Lee, R. E.; Strauss, E.; Saliba, K. J. PLoS One 2013, 8, e54974. doi:10.1371/journal.pone.0054974
Return to citation in text: [1] [2] [3] -
Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E. S. C.; Habib, E.; Saliba, K. J.; Auclair, K. Bioorg. Med. Chem. Lett. 2014, 24, 3274–3277. doi:10.1016/j.bmcl.2014.06.013
Return to citation in text: [1] [2] [3] [4] [5] -
de Villiers, M.; Macuamule, C.; Spry, C.; Hyun, Y.-M.; Strauss, E.; Saliba, K. J. ACS Med. Chem. Lett. 2013, 4, 784–789. doi:10.1021/ml400180d
Return to citation in text: [1] [2] -
Choudhry, A. E.; Mandichak, T. L.; Broskey, J. P.; Egolf, R. W.; Kinsland, C.; Begley, T. P.; Seefeld, M. A.; Ku, T. W.; Brown, J. R.; Zalacain, M.; Ratnam, K. Antimicrob. Agents Chemother. 2003, 47, 2051–2055. doi:10.1128/AAC.47.6.2051-2055.2003
Return to citation in text: [1] [2] -
Strauss, E.; Begley, T. P. J. Biol. Chem. 2002, 277, 48205–48209. doi:10.1074/jbc.M204560200
Return to citation in text: [1] -
Zhang, Y.-M.; Frank, M. W.; Virga, K. G.; Lee, R. E.; Rock, C. O.; Jackowski, S. J. Biol. Chem. 2004, 279, 50969–50975. doi:10.1074/jbc.M409607200
Return to citation in text: [1] -
Thomas, J.; Cronan, J. E. Antimicrob. Agents Chemother. 2010, 54, 1374–1377. doi:10.1128/AAC.01473-09
Return to citation in text: [1] -
de Villiers, M.; Barnard, L.; Koekemoer, L.; Snoep, J. L.; Strauss, E. FEBS J. 2014, 281, 4731–4753. doi:10.1111/febs.13013
Return to citation in text: [1] -
Akinnusi, T. O.; Vong, K.; Auclair, K. Bioorg. Med. Chem. 2011, 19, 2696–2706. doi:10.1016/j.bmc.2011.02.053
Return to citation in text: [1] [2] [3] [4] [5] -
Hachey, M. Modifying the pantoyl moiety of N-alkylpantothenamides through stereoselective alkylations and olefin cross-metathesis. M.Sc. Thesis, McGill University, Montreal, Canada, 2013.
Return to citation in text: [1] -
Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882
Return to citation in text: [1] [2] [3] [4] -
Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::AID-ANIE1277>3.0.CO;2-E
Return to citation in text: [1] -
Storvick, J. M.; Ankoudinova, E.; King, B. R.; Van Epps, H.; O'Neil, G. W. Tetrahedron Lett. 2011, 52, 5858–5861. doi:10.1016/j.tetlet.2011.08.153
Return to citation in text: [1] -
Hong, S. H.; Wenzel, A. G.; Salguero, T. T.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2007, 129, 7961–7968. doi:10.1021/ja0713577
Return to citation in text: [1] -
Bargiggia, F. C.; Murray, W. V. J. Org. Chem. 2005, 70, 9636–9639. doi:10.1021/jo0514624
Return to citation in text: [1] -
Graham, T. J. A.; Gray, E. E.; Burgess, J. M.; Goess, B. C. J. Org. Chem. 2010, 75, 226–228. doi:10.1021/jo9020375
Return to citation in text: [1] -
Michaut, A.; Boddaert, T.; Coquerel, Y.; Rodriguez, J. Synthesis 2007, 18, 2867–2871. doi:10.1055/s-2007-983825
Return to citation in text: [1] -
de la Hoz, A.; Díaz-Ortiz, Á.; Moreno, A. Chem. Soc. Rev. 2005, 34, 164–178. doi:10.1039/B411438H
Return to citation in text: [1] -
Awuah, E.; Ma, E.; Hoegl, A.; Vong, K.; Habib, E.; Auclair, K. Bioorg. Med. Chem. 2014, 22, 3083–3090. doi:10.1016/j.bmc.2014.04.030
Return to citation in text: [1]
| 28. | Awuah, E.; Ma, E.; Hoegl, A.; Vong, K.; Habib, E.; Auclair, K. Bioorg. Med. Chem. 2014, 22, 3083–3090. doi:10.1016/j.bmc.2014.04.030 |
| 11. | Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E. S. C.; Habib, E.; Saliba, K. J.; Auclair, K. Bioorg. Med. Chem. Lett. 2014, 24, 3274–3277. doi:10.1016/j.bmcl.2014.06.013 |
| 6. | Clifton, G.; Bryant, S. R.; Skinner, C. G. Arch. Biochem. Biophys. 1970, 137, 523–528. doi:10.1016/0003-9861(70)90470-4 |
| 18. | Akinnusi, T. O.; Vong, K.; Auclair, K. Bioorg. Med. Chem. 2011, 19, 2696–2706. doi:10.1016/j.bmc.2011.02.053 |
| 7. | Spry, C.; Kirk, K.; Saliba, K. J. FEMS Microbiol. Rev. 2008, 32, 56–106. doi:10.1111/j.1574-6976.2007.00093.x |
| 8. | Saliba, K. J.; Ferru, I.; Kirk, K. Antimicrob. Agents Chemother. 2005, 49, 632–637. doi:10.1128/AAC.49.2.632-637.2005 |
| 9. | Spry, C.; van Schalkwyk, D. A.; Strauss, E.; Saliba, K. J. Infect. Disord.: Drug Targets 2010, 10, 200–216. doi:10.2174/187152610791163390 |
| 10. | Spry, C.; Macuamule, C.; Lin, Z.; Virga, K. G.; Lee, R. E.; Strauss, E.; Saliba, K. J. PLoS One 2013, 8, e54974. doi:10.1371/journal.pone.0054974 |
| 11. | Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E. S. C.; Habib, E.; Saliba, K. J.; Auclair, K. Bioorg. Med. Chem. Lett. 2014, 24, 3274–3277. doi:10.1016/j.bmcl.2014.06.013 |
| 12. | de Villiers, M.; Macuamule, C.; Spry, C.; Hyun, Y.-M.; Strauss, E.; Saliba, K. J. ACS Med. Chem. Lett. 2013, 4, 784–789. doi:10.1021/ml400180d |
| 19. | Hachey, M. Modifying the pantoyl moiety of N-alkylpantothenamides through stereoselective alkylations and olefin cross-metathesis. M.Sc. Thesis, McGill University, Montreal, Canada, 2013. |
| 6. | Clifton, G.; Bryant, S. R.; Skinner, C. G. Arch. Biochem. Biophys. 1970, 137, 523–528. doi:10.1016/0003-9861(70)90470-4 |
| 18. | Akinnusi, T. O.; Vong, K.; Auclair, K. Bioorg. Med. Chem. 2011, 19, 2696–2706. doi:10.1016/j.bmc.2011.02.053 |
| 2. | Pogue, J. M.; Marchaim, D.; Kaye, D.; Kaye, K. S. Pharmacotherapy 2011, 31, 912–921. doi:10.1592/phco.31.9.912 |
| 3. | Chen, L. F.; Kaye, D. Infect. Dis. Clin. North Am. 2009, 23, 1053–1075. doi:10.1016/j.idc.2009.06.004 |
| 4. | Wright, G. D. Can. J. Microbiol. 2014, 60, 147–154. doi:10.1139/cjm-2014-0063 |
| 5. | Page, M. G.; Heim, J. IDrugs 2009, 12, 561–565. |
| 11. | Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E. S. C.; Habib, E.; Saliba, K. J.; Auclair, K. Bioorg. Med. Chem. Lett. 2014, 24, 3274–3277. doi:10.1016/j.bmcl.2014.06.013 |
| 17. | de Villiers, M.; Barnard, L.; Koekemoer, L.; Snoep, J. L.; Strauss, E. FEBS J. 2014, 281, 4731–4753. doi:10.1111/febs.13013 |
| 12. | de Villiers, M.; Macuamule, C.; Spry, C.; Hyun, Y.-M.; Strauss, E.; Saliba, K. J. ACS Med. Chem. Lett. 2013, 4, 784–789. doi:10.1021/ml400180d |
| 13. | Choudhry, A. E.; Mandichak, T. L.; Broskey, J. P.; Egolf, R. W.; Kinsland, C.; Begley, T. P.; Seefeld, M. A.; Ku, T. W.; Brown, J. R.; Zalacain, M.; Ratnam, K. Antimicrob. Agents Chemother. 2003, 47, 2051–2055. doi:10.1128/AAC.47.6.2051-2055.2003 |
| 14. | Strauss, E.; Begley, T. P. J. Biol. Chem. 2002, 277, 48205–48209. doi:10.1074/jbc.M204560200 |
| 15. | Zhang, Y.-M.; Frank, M. W.; Virga, K. G.; Lee, R. E.; Rock, C. O.; Jackowski, S. J. Biol. Chem. 2004, 279, 50969–50975. doi:10.1074/jbc.M409607200 |
| 16. | Thomas, J.; Cronan, J. E. Antimicrob. Agents Chemother. 2010, 54, 1374–1377. doi:10.1128/AAC.01473-09 |
| 11. | Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E. S. C.; Habib, E.; Saliba, K. J.; Auclair, K. Bioorg. Med. Chem. Lett. 2014, 24, 3274–3277. doi:10.1016/j.bmcl.2014.06.013 |
| 18. | Akinnusi, T. O.; Vong, K.; Auclair, K. Bioorg. Med. Chem. 2011, 19, 2696–2706. doi:10.1016/j.bmc.2011.02.053 |
| 10. | Spry, C.; Macuamule, C.; Lin, Z.; Virga, K. G.; Lee, R. E.; Strauss, E.; Saliba, K. J. PLoS One 2013, 8, e54974. doi:10.1371/journal.pone.0054974 |
| 13. | Choudhry, A. E.; Mandichak, T. L.; Broskey, J. P.; Egolf, R. W.; Kinsland, C.; Begley, T. P.; Seefeld, M. A.; Ku, T. W.; Brown, J. R.; Zalacain, M.; Ratnam, K. Antimicrob. Agents Chemother. 2003, 47, 2051–2055. doi:10.1128/AAC.47.6.2051-2055.2003 |
| 7. | Spry, C.; Kirk, K.; Saliba, K. J. FEMS Microbiol. Rev. 2008, 32, 56–106. doi:10.1111/j.1574-6976.2007.00093.x |
| 9. | Spry, C.; van Schalkwyk, D. A.; Strauss, E.; Saliba, K. J. Infect. Disord.: Drug Targets 2010, 10, 200–216. doi:10.2174/187152610791163390 |
| 10. | Spry, C.; Macuamule, C.; Lin, Z.; Virga, K. G.; Lee, R. E.; Strauss, E.; Saliba, K. J. PLoS One 2013, 8, e54974. doi:10.1371/journal.pone.0054974 |
| 20. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 18. | Akinnusi, T. O.; Vong, K.; Auclair, K. Bioorg. Med. Chem. 2011, 19, 2696–2706. doi:10.1016/j.bmc.2011.02.053 |
| 18. | Akinnusi, T. O.; Vong, K.; Auclair, K. Bioorg. Med. Chem. 2011, 19, 2696–2706. doi:10.1016/j.bmc.2011.02.053 |
| 24. | Bargiggia, F. C.; Murray, W. V. J. Org. Chem. 2005, 70, 9636–9639. doi:10.1021/jo0514624 |
| 25. | Graham, T. J. A.; Gray, E. E.; Burgess, J. M.; Goess, B. C. J. Org. Chem. 2010, 75, 226–228. doi:10.1021/jo9020375 |
| 26. | Michaut, A.; Boddaert, T.; Coquerel, Y.; Rodriguez, J. Synthesis 2007, 18, 2867–2871. doi:10.1055/s-2007-983825 |
| 27. | de la Hoz, A.; Díaz-Ortiz, Á.; Moreno, A. Chem. Soc. Rev. 2005, 34, 164–178. doi:10.1039/B411438H |
| 11. | Hoegl, A.; Darabi, H.; Tran, E.; Awuah, E.; Kerdo, E. S. C.; Habib, E.; Saliba, K. J.; Auclair, K. Bioorg. Med. Chem. Lett. 2014, 24, 3274–3277. doi:10.1016/j.bmcl.2014.06.013 |
| 22. | Storvick, J. M.; Ankoudinova, E.; King, B. R.; Van Epps, H.; O'Neil, G. W. Tetrahedron Lett. 2011, 52, 5858–5861. doi:10.1016/j.tetlet.2011.08.153 |
| 23. | Hong, S. H.; Wenzel, A. G.; Salguero, T. T.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2007, 129, 7961–7968. doi:10.1021/ja0713577 |
| 20. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 20. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 20. | Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. doi:10.1021/ja0214882 |
| 21. | Choi, T.-L.; Chatterjee, A. K.; Grubbs, R. H. Angew. Chem., Int. Ed. 2001, 40, 1277–1279. doi:10.1002/1521-3773(20010401)40:7<1277::AID-ANIE1277>3.0.CO;2-E |
© 2016 Guan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)