Abstract
A straightforward strategy for the synthesis of dihydroquinazolines is presented, which allows for the preparation of 3,4- and 1,4-dihydroquinazolines with different substitution patterns from 2-aminobenzylamine (2-ABA) as common precursor. The required functionalization of both amino groups present in 2-ABA was achieved by different routes involving selective N-acylation and cesium carbonate-mediated N-alkylation reactions, avoiding protection/deprotection steps. The heterocycles were efficiently synthesized in short reaction times by microwave-assisted ring closure of the corresponding aminoamides promoted by ethyl polyphosphate (PPE).

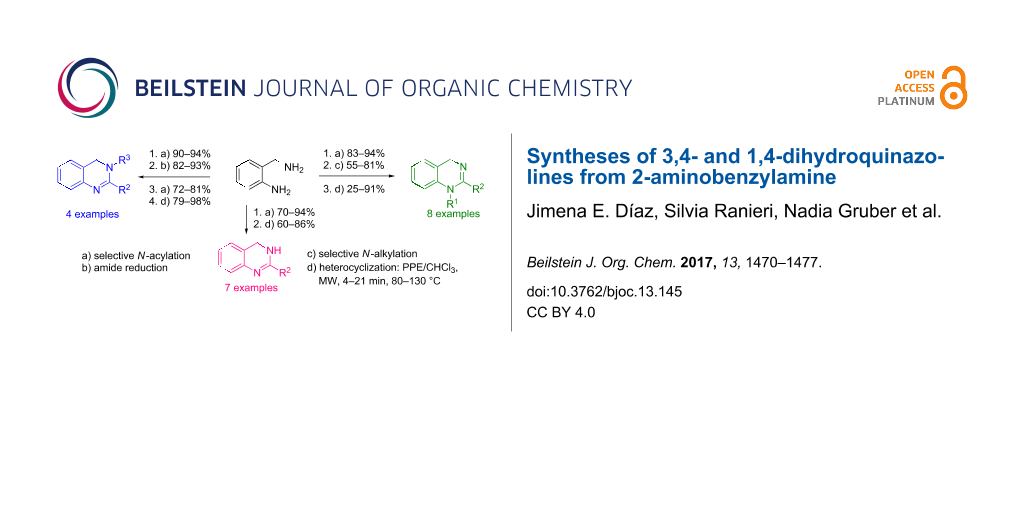
Graphical Abstract
Introduction
Nitrogen heterocycles are part of many drugs and represent structures with wide therapeutic potential. Therefore, much effort has been devoted to the development of efficient, general and expeditious methods for their synthesis [1,2]. Cyclic amidines are compounds of interest due to their multiple pharmacological properties, some of which derive from their interaction with a specific receptor [3,4]. Six-membered heterocylic amidines (1,4,5,6-tetrahydropyrimidines) display biological activity as anthelmintics used in medical practice (pyrantel, morantel and oxantel) [5,6], nicotinic agonists [7], antidepressants [8] and selective inhibitors of M1 acetylcholine receptors [9,10], among others. Their benzannulated analogues, dihydroquinazolines, also represent heterocyclic cores of pharmacological interest. Some derivatives containing this motif have shown antimicrobial [11] and antifungal properties [12]. Their activity as selective T-type calcium channel blockers [13-15] and as inhibitors of β-secretase (an important target for Alzheimer’s disease) has been reported [16]. They have also been studied as inhibitors of trypanothione reductase [17], an essential enzyme of the parasite Trypanosoma brucei, and as neuroprotective agents [18].
Different strategies have been used for the preparation of dihydroquinazolines. Some of them involve cyclic precursors such as quinazolinones, quinazolines or tetrahydroquinazolines, which can be reduced or oxidized to yield dihydroquinazolines [17,19-25]. Heterocyclization of acyclic precursors has also been applied for the synthesis of these heterocycles. Many methods use 2-aminobenzylamine (2-ABA) that reacts with amidines, carboxylic acids, orthoformates, or aldehydes in oxidative conditions to yield N-unsubstituted dihydroquinazolines [11,16,26-35]. A different approach is based on the heterocyclization of functionalized 2-aminobenzylamines [36,37] such as the cyclization of N-(2-aminobenzyl)amides, which in general requires drastic conditions or long reaction times [17,18,21,22,29,38-40]. Miscellaneous methods include the nucleophilic addition of secondary amines to carbodiimides followed by an intramolecular conjugate addition to an α,β-unsaturated ester [12-15] or copper-catalyzed annulation of N-arylamidines [41], among others [42]. Most of the already mentioned methods have been applied to the synthesis of 3,4-dihydroquinazolines. Among them, only a few were used for the preparation of 1,4-dihydroquinazolines, a heterocyclic framework quite unexplored.
In addition to the heterocyclization itself, the preparation of the acyclic precursors for the synthesis of dihydroquinazolines remains challenging as in some cases it includes a selective N-functionalization of the amino groups of 2-ABA. For example, the selective acylation of the aliphatic amino group was achieved by the treatment of the diamine with benzoic acid in the presence of Zr(azobenzene-4,4´-dicarboxylate) [43], with tert-butylperoxybenzoate [44] and with isopropenyl acetate [45], or by a DCC-mediated coupling with a carboxylic acid [38]. To our knowledge, selective acylations of N1-alkyl-2-ABAs have not been reported yet.
Alkylations of the aliphatic amino group of 2-ABA using triflates, halogen or CCl3 derivatives have also been reported. However, these reactions are frequently associated with long reaction times, poor yields and low selectivity [46,47]. The selective monofunctionalization of 2-ABA was achieved by using protective groups such as 9-BBN [48] or Boc [39], which require subsequent removal. Other alternative synthetic strategies use substrates like 2-nitrobenzoyl chloride, 2-nitrobenzaldehyde, 2-aminobenzamides, 2-aminobenzophenone, 2-nitrobenzyl bromide, among others, which require an additional reduction step [17,18,29,49-51].
Ethyl polyphosphate (PPE) and trimethylsilyl polyphosphate (PPSE) are mild irreversible dehydrating agents of the Lewis acid type. They have found application for synthetically useful transformations like conversion of amides into nitriles [52,53] or the Beckmann rearrangement [52,54]. They have also been widely employed in heterocyclic synthesis, including microwave-assisted reactions. The recognized wide functional group tolerance, stability and low environmental impact are additional advantages of these reagents. Cyclocondensation is a fundamental tool in heterocycles synthesis, and microwave (MW) heating has found interesting applications in this area [55,56], overcoming limitations arising from low reactivity and side reactions typical of conventional heating [57].
We had previously reported the MW-assisted synthesis of 1,4,5,6-tetrahydropyrimidines and their homologues by cyclodehydration reactions promoted by polyphosphoric acid (PPA) esters PPE and PPSE [58-60]. This strategy was successfully applied to the synthesis of 1-aryl-2-imino-1-azacycloalkanes [61], 2-oxazolines, 5,6-dihydro-4H-1,3-oxazines and 4,5,6,7-tetrahydro-1,3-oxazepines [62].
In continuation of this work we herein present an efficient strategy for the synthesis of dihydroquinazolines starting from the easily available 2-ABA as common precursor. The route allows for the preparation of 3,4-dihydroquinazolines 1 and 1,4-dihydroquinazolines 2 with different substitution patterns (Figure 1).
![[1860-5397-13-145-1]](/bjoc/content/figures/1860-5397-13-145-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 3,4-Dihydroquinazolines 1 and 1,4-dihydroquinazolines 2.
Figure 1: 3,4-Dihydroquinazolines 1 and 1,4-dihydroquinazolines 2.
Results and Discussion
The general synthetic pathways leading to the desired dihydroquinazolines 1 and 2 are depicted in Scheme 1.
![[1860-5397-13-145-i1]](/bjoc/content/inline/1860-5397-13-145-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathways for the preparation of 3,4-dihydroquinazolines 1 and 1,4-dihydroquinazolines 2.
Scheme 1: Synthetic pathways for the preparation of 3,4-dihydroquinazolines 1 and 1,4-dihydroquinazolines 2.
N-Alkyl-substituted 2-aminobenzylamines 3a–c were readily synthesized by reduction of the amides 4a–c employing a THF solution of borane (yields 82–93%; see Supporting Information File 1 and Scheme 2).
![[1860-5397-13-145-i2]](/bjoc/content/inline/1860-5397-13-145-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of N-(2-aminobenzyl)amides 4a–k was achieved by selective N-acylation of 2-ABA or its N-substituted derivatives 3a–c, whose preparation is depicted in Scheme 2. In spite of the different nucleophilicity of both amino groups, the predictable formation of a hydrogen bond between the aromatic amino group (hydrogen bond donor) and the aliphatic NHR (hydrogen bond acceptor) could increase the nucleophilicity of the former and decrease the reactivity of the latter, favoring the chance for N,N´-diacylation. To avoid this, the reaction was performed employing a diluted solution of the amine in a biphasic system (CHCl3/aqueous NaOH), low temperature (0 °C) and the use of anhydrides as acylating agents, if commercially available. The results are shown in Table 1.
Table 1: Selective N-acylation of 2-ABA and its derivatives.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-145-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 4 | R3 | R2 | Acylation agent | Yield (%)a |
|---|---|---|---|---|---|
| 1 | a | H | CH3 | anhydride | 90 |
| 2 | b | H | CH2CH3 | anhydride | 93 |
| 3 | c | H | CH(CH3)2 | anhydride | 94 |
| 4 | d | H | C(CH3)3 | anhydride | 93 |
| 5 | e | H | C6H5 | anhydride | 94 |
| 6 | f | H | 2-CH3C6H4 | acyl chloride | 83 |
| 7 | g | H | 2-FC6H4 | acyl chloride | 70 |
| 8 | h | CH2CH3 | CH3 | anhydride | 81 |
| 9 | i | CH2CH3 | CH2CH3 | anhydride | 80 |
| 10 | j | CH2CH2CH3 | CH2CH3 | anhydride | 72 |
| 11 | k | CH2CH(CH3)2 | CH2CH3 | anhydride | 79 |
aYields correspond to pure compounds.
For R3 = H, products 4a–e were obtained in excellent yields when 2-ABA was treated with acid anhydrides as the acylating agents (Table 1, entries 1–5). Replacing anhydrides by acyl chlorides in the reaction the yields were slightly lower, and in some cases small amounts of the N,N’-diacylated products were also obtained (Table 1, entries 6 and 7). Under the same reaction conditions, the N-substituted derivatives 3 (R3 ≠ H) were selectively acylated with very good yields (Table 1, entries 8–11). Thus, precursors 4 were efficiently prepared by a sequence of N-acylation–reduction–N-acylation.
The acyclic precursors 5, necessary for the synthesis of dihydroquinazolines 2, were prepared by N-alkylation of the arylamino group present in compounds 4 using alkyl halides. In a previous work [63], we developed a methodology for the selective N-alkylation of anilines with ω-halonitriles in the presence of cesium carbonate and potassium iodide. There the use of the cesium base prevented N-dialkylation and therefore it was not necessary to use an excess of the amine. This led us to test the same reaction conditions for the synthesis of compounds 5. In this case, the presence of the amide functionality was regarded as an additional problem, as Cs2CO3-mediated alkylation of cyclic amides had already been reported in the literature [64]. In spite of this, compounds 5 were obtained in good to high yields (Table 2) employing the same experimental conditions [63]. The reaction times and temperatures were individually adjusted taking into account the boiling points of the alkylating agents and the reactivity of each substrate.
Table 2: Synthesis of compounds 5.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-145-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | 5 | R2 | R1 | X | Time (h) | Temperature (°C) | Yield (%)a |
| 1 | a | CH3 | CH2CH2CH3 | I | 5 | 75 | 81 |
| 2 | b | CH3 | CH2CH=CH2 | Br | 4.5 | 65 | 68 |
| 3 | c | CH3 | CH2C6H5 | Cl | 5 | 90 | 64 |
| 4 | d | CH3 | CH(CH3)2 | I | 6 | 85 | 60 |
| 5 | e | CH(CH3)2 | CH2CH2CH3 | I | 6.5 | 85 | 67 |
| 6 | f | C(CH3)3 | CH2CH2CH3 | I | 6 | 85 | 69 |
| 7 | g | C(CH3)3 | CH2C6H5 | Cl | 3 | 90 | 60 |
| 8 | h | C(CH3)3 | CH(CH3)2 | I | 7 | 85 | 75 |
| 9 | i | C6H5 | CH2CH2CH3 | I | 7 | 85 | 55 |
| 10 | j | 2-CH3C6H4 | CH2CH2CH3 | I | 10 | 85 | 71 |
aYields correspond to pure compounds.
As expected, the reaction was selective toward the N-monoalkylation of the arylamino group in the presence of the amide. Satisfactory results were also obtained using the more hindered isopropyl iodide (Table 2, entries 4 and 8).
Next, the conversion of N-(2-aminobenzyl)acetamide (4a) into 2-methyl-3,4-dihydroquinazoline (1a) was chosen for the optimization of the reaction conditions for the synthesis of compounds 1. No background reaction was observed when heating a solution of compound 4a in chloroform for 10 minutes at 130 °C under MW irradiation in a closed vessel. Using a solution of PPSE in DCM as cyclodehydrating agent, after 30 minutes of reaction under reflux using conventional heating compound 1a was obtained in 86% yield. Analogous results were obtained using either PPE/CHCl3 or PPSE/CH2Cl2 under microwave irradiation at 110 ºC for 4 minutes. Due to the easier preparation and the inexpensive reagents required for its synthesis [65], PPE was chosen as cyclodehydrating agent for the preparation of compounds 1. Results and optimized experimental conditions of the MW-assisted ring-closure reaction are depicted in Table 3. Due to the different reactivity of each substrate, irradiation times and temperatures were individually adjusted.
Table 3: Synthesis of 3,4-dihydroquinazolines 1.
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-145-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Product 1 | R3 | R2 | Time (min) | Temperature (°C) | Yield (%)a |
|---|---|---|---|---|---|---|
| 1 | a | H | CH3 | 4 | 110 | 86 |
| 2 | b | H | CH2CH3 | 4 | 110 | 84 |
| 3 | c | H | CH(CH3)2 | 9 | 120 | 76 |
| 4 | d | H | C(CH3)3 | 12 | 130 | 73 |
| 5 | e | H | C6H5 | 12 | 130 | 71 |
| 6 | f | H | 2-CH3C6H4 | 15 | 130 | 79 |
| 7 | g | H | 2-FC6H4 | 15 | 130 | 60 |
| 8 | h | CH2CH3 | CH3 | 4.5 | 100 | 98 |
| 9 | i | CH2CH3 | CH2CH3 | 8 | 115 | 79 |
| 10 | j | CH2CH2CH3 | CH2CH3 | 8 | 115 | 86 |
| 11 | k | CH2CH(CH3)2 | CH2CH3 | 8 | 115 | 80 |
aYields correspond to pure compounds.
For the synthesis of compounds 1 the reaction conditions mainly depended on the steric and electronic features of the amido group present in the substrates. Acetamides and propionamides 4a,b,h–k reacted readily under mild reaction conditions (Table 3, entries 1, 2, and 8–11), while for isobutyramide 4c a higher temperature was necessary (Table 3, entry 3). Finally, the more sterically hindered pivalamide 4d and the less reactive benzamides 4e–g required harsher reaction conditions to achieve complete conversion of the substrates and the products were afforded in generally lower yields (Table 3, entries 4–7).
In order to widen the scope of the method, we examined next the applicability of the PPE/MW system to the synthesis of 1,4-dihydroquinazolines 2. Treatment of precursor 5a with PPE/CHCl3 under microwave irradiation led to 1,4-dihydroquinazoline 2a, accompanied by a low percentage of a collateral product. This compound was identified as 2-methyl-1-propylquinazolin-4(1H)-one (6a) (Scheme 3, (a), R1 = Pr, R2 = Me), arising from spontaneous benzylic oxidation of 2a. An analogous reaction had been previously reported for 3,4-dihydroquinazolines upon exposure to air (Scheme 3, (b)) [19,24,40,66].
![[1860-5397-13-145-i3]](/bjoc/content/inline/1860-5397-13-145-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Benzylic oxidation of 1,4-dihydroquinazolines (a) and 3,4-dihydroquinazolines (b).
Scheme 3: Benzylic oxidation of 1,4-dihydroquinazolines (a) and 3,4-dihydroquinazolines (b).
The fact that quinazolin-4(1H)-ones 6, which are fully conjugated push–pull molecules, are more resonance stabilized than their isomeric counterparts quinazolin-4(3H)-ones 7, would explain the differential behavior of N-substituted 3,4- and 1,4-dihydroquinazolines toward spontaneous oxidation.
By the same procedure used for 2a, a series of novel 1,4-dihydroquinazolines were synthesized. The results and optimized reaction conditions are reported in Table 4. Due to the different reactivity of each substrate, irradiation times and temperatures were individually adjusted.
Table 4: Synthesis of 1,4-dihydroquinazolines 2.
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-145-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Product 2 | R2 | R1 | Time (min) | Temperature (°C) | Yield (%)a |
|---|---|---|---|---|---|---|
| 1 | a | CH3 | CH2CH2CH3 | 11 | 80 | 67 |
| 2 | b | CH3 | CH2CH=CH2 | 11 | 90 | 75 |
| 3 | c | CH3 | CH2C6H5 | 11 | 90 | 72 |
| 4 | d | CH3 | CH(CH3)2 | 12 | 130 | 91 |
| 5 | e | CH(CH3)2 | CH2CH2CH3 | 21 | 110 | 79 |
| 6 | f | C(CH3)3 | CH2CH2CH3 | 20 | 130 | 68 |
| 7 | g | C(CH3)3 | CH2C6H5 | 25 | 130 | – |
| 8 | h | C(CH3)3 | CH(CH3)2 | 25 | 130 | – |
| 9 | i | C6H5 | CH2CH2CH3 | 13 | 90 | 25b |
| 10 | j | 2-CH3C6H4 | CH2CH2CH3 | 5 | 100 | 39b |
aYields correspond to pure compounds. bThe reported yield is approximate and was estimated by integration of suitable 1H NMR signals (see Supporting Information File 1).
Generally the yields for compounds 2 were good to excellent and the experimental conditions and reaction outcome varied depending on the electronic and steric features of the substituents R1 and R2 present in precursors 5. The cyclodehydration of acetamides 5a–c proceeded under mild reaction conditions (Table 4, entries 1–3), while acetamide 5d required a higher temperature probably due to the steric hindrance produced by the N-isopropyl moiety (Table 4, entry 4). A higher temperature and a longer reaction time were necessary in order to achieve cyclodehydration of isobutyramide 5e, and even harsher conditions were required for pivalamide 5f (Table 4, entries 5 and 6). In the case of precursors 5g,h, instead of the expected 1-substituted-2-tert-butyl-1,4-dihydroquinazolines, cyclodehydration led to 2-tert-butyl-3,4-dihydroquinazoline (1d, Table 4, entries 7 and 8), with loss of one N-substituent. A possible explanation is that the reaction conditions required in those cases due to steric hindrance bring about a competitive elimination reaction. Finally, as already mentioned, 1,4-dihydroquinazolines 2 are prone to spontaneous oxidation leading to 4(1H)-quinazolinones (Scheme 3a) and dihydroquinazolines 2i,j showed a comparatively higher lability toward this side reaction. For this reason, the cyclodehydration of the substrates 5i,j was carried out at lower temperatures in order to avoid benzylic oxidation. In spite of this, partial oxidation during workup and purification could not be avoided accounting for the lower yields obtained for the 2-aryl derivatives 2i,j (Table 4, entries 9 and 10). The higher sensitivity of compounds 2i,j towards oxidation may be a consequence of the enhanced resonance stabilization of the resulting 2-arylquinazolin-4(1H)-ones 6i,j (Scheme 3). The fact that the yield of the o-methylphenyl-substituted derivative 2j, in which this stabilization is reduced due to steric effects, was higher than for the phenyl-substituted derivative reinforces this hypothesis.
Conclusion
We have developed novel, general and straightforward strategies for the synthesis of 3,4-dihydroquinazolines with different substitution patterns, starting from 2-ABA as common precursor. An extension of the method allowed for the preparation of N-alkyl-1,4-dihydroquinazolines, a heterocyclic nucleus almost unexplored. To our knowledge, this is the first general method available for synthesis of these compounds. The products were synthesized by microwave-assisted ring closure of different aminoamides promoted by PPE. The yields and reaction times involved in the heterocyclization step compare favorably with the reported methodologies. The preparation of the required aminoamides involves N-acylation and N-alkylation reactions, which were optimized to achieve selective monofunctionalization thus avoiding additional protection/deprotection steps. A judicious combination of these steps into different sequences, allowed for the preparation of both 3,4- and 1,4-dihydroquinazolines with the desired N-substitution starting from a single precursor.
The protocols are operationally simple and involve easily available and inexpensive reagents. Due to their widespread use in organic synthesis, the selective N-functionalization reactions described herein may find application in many different molecular contexts.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization of new compounds. | ||
| Format: PDF | Size: 5.3 MB | Download |
References
-
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N. Beilstein J. Org. Chem. 2011, 7, 442–495. doi:10.3762/bjoc.7.57
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2013, 9, 2265–2319. doi:10.3762/bjoc.9.265
Return to citation in text: [1] -
Doyle, M. E.; Egan, J. M. Pharmacol. Rev. 2003, 55, 105–131. doi:10.1124/pr.55.1.7
Return to citation in text: [1] -
Szabo, B. Pharmacol. Ther. 2002, 93, 1–35. doi:10.1016/S0163-7258(01)00170-X
Return to citation in text: [1] -
Austin, W. C.; Courtney, W.; Danilewicz, J. C.; Morgan, D. H.; Conover, L. H.; Howes, H. L., Jr.; Lynch, J. E.; McFarland, J. W.; Cornwell, R. L.; Theodorides, V. J. Nature 1966, 212, 1273–1274. doi:10.1038/2121273b0
Return to citation in text: [1] -
Anand, N.; Sharma, S., Eds. Approaches to Design and Synthesis of Antiparasitic Drugs; Chapter 6, Vol. 25; Elsevier, 1997.
Return to citation in text: [1] -
Meyer, E. M.; Arendash, G. W.; Judkins, J. H.; Ying, L.; Wade, C.; Ken, W. R. J. Neurochem. 1987, 49, 1758–1762. doi:10.1111/j.1471-4159.1987.tb02433.x
Return to citation in text: [1] -
Weinhardt, K.; Wallach, M. B.; March, M. J. Med. Chem. 1985, 28, 694–698. doi:10.1021/jm00383a002
Return to citation in text: [1] -
Dunbar, P. G.; Durant, G. J.; Fang, Z.; Abuh, Y. F.; El-Assadi, A. A.; Ngur, D. O.; Periyasamy, S.; Hoss, W. P.; Messer, W. S., Jr. J. Med. Chem. 1993, 36, 842–847. doi:10.1021/jm00059a008
Return to citation in text: [1] -
Dunbar, P. G.; Durant, G. J.; Rho, T.; Ojo, B.; Huzl, J. J., III; Smith, D. A.; El-Assadi, A. A.; Sbeih, S.; Ngur, D. O. J. Med. Chem. 1994, 37, 2774–2782. doi:10.1021/jm00043a016
Return to citation in text: [1] -
Los, R.; Wesołowska-Trojanowska, M.; Malm, A.; Karpińska, M. M.; Matysiak, J.; Niewiadomy, A.; Głaszcz, U. Heteroat. Chem. 2012, 23, 265–275. doi:10.1002/hc.21012
Return to citation in text: [1] [2] -
Li, W.-J.; Li, Q.; Liu, D.-L.; Ding, M.-W. J. Agric. Food Chem. 2013, 61, 1419–1426. doi:10.1021/jf305355u
Return to citation in text: [1] [2] -
Lee, Y. S.; Lee, B. H.; Park, S. J.; Kang, S. B.; Rhim, H.; Park, J.-Y.; Lee, J.-H.; Jeong, S.-W.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2004, 14, 3379–3384. doi:10.1016/j.bmcl.2004.04.090
Return to citation in text: [1] [2] -
Heo, J. H.; Seo, H. N.; Choe, Y. J.; Kim, S.; Oh, C. R.; Kim, Y. D.; Rhim, H.; Choo, D. J.; Kim, J.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2008, 18, 3899–3901. doi:10.1016/j.bmcl.2008.06.034
Return to citation in text: [1] [2] -
Byun, J. S.; Sohn, J. M.; Leem, D. G.; Park, B.; Nam, J. H.; Shin, D. H.; Shin, J. S.; Kim, H. J.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 1073–1079. doi:10.1016/j.bmcl.2015.12.010
Return to citation in text: [1] [2] -
Ghosh, A. K.; Pandey, S.; Gangarajula, S.; Kulkarni, S.; Xu, X.; Rao, K. V.; Huang, X.; Tang, J. Bioorg. Med. Chem. Lett. 2012, 22, 5460–5465. doi:10.1016/j.bmcl.2012.07.043
Return to citation in text: [1] [2] -
Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v
Return to citation in text: [1] [2] [3] [4] -
Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424
Return to citation in text: [1] [2] [3] -
Decker, M. Eur. J. Med. Chem. 2005, 40, 305–313. doi:10.1016/j.ejmech.2004.12.003
Return to citation in text: [1] [2] -
Jaén, J. C.; Gregor, V. E.; Lee, C.; Davis, R.; Emmerling, M. Bioorg. Med. Chem. Lett. 1996, 6, 737–742. doi:10.1016/0960-894X(96)00102-3
Return to citation in text: [1] -
Armarego, W. L. F. J. Chem. Soc. 1961, 2697–2701. doi:10.1039/jr9610002697
Return to citation in text: [1] [2] -
Armarego, W. L. F. Adv. Heterocycl. Chem. 1963, 11, 253–309. doi:10.1016/S0065-2725(08)60527-9
Return to citation in text: [1] [2] -
Smith, J. G.; Sheepy, J. M. J. Heterocycl. Chem. 1975, 12, 231–234. doi:10.1002/jhet.5570120203
Return to citation in text: [1] -
Richers, M. T.; Zhao, C.; Seidel, D. Beilstein J. Org. Chem. 2013, 9, 1194–1201. doi:10.3762/bjoc.9.135
Return to citation in text: [1] [2] -
He, K.-H.; Tan, F.-F.; Zhou, C.-Z.; Zhou, G.-J.; Yang, X.-L.; Li, Y. Angew. Chem. 2017, 56, 3080–3084. doi:10.1002/anie.201612486
Return to citation in text: [1] -
Burdick, B. A.; Benkovic, P. A.; Benkovic, S. J. J. Am. Chem. Soc. 1977, 99, 5716–5725. doi:10.1021/ja00459a032
Return to citation in text: [1] -
Papadopoulos, E. P.; George, B. J. Org. Chem. 1977, 42, 2530–2532. doi:10.1021/jo00434a049
Return to citation in text: [1] -
Grasso, S.; Micale, N.; Monforte, A.-M.; Monforte, P.; Polimeni, S.; Zappalà, M. Eur. J. Med. Chem. 2000, 35, 1115–1119. doi:10.1016/S0223-5234(00)01195-8
Return to citation in text: [1] -
Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/S0040-4039(01)01842-1
Return to citation in text: [1] [2] [3] -
Baxter, E. W.; Conway, K. A.; Kennis, L.; Bischoff, F.; Mercken, M. H.; De Winter, H. L.; Reynolds, C. H.; Tounge, B. A.; Luo, C.; Scott, M. K.; Huang, Y.; Braeken, M.; Pieters, S. M. A.; Berthelot, D. J. C.; Masure, S.; Bruinzeel, W. D.; Jordan, A. D.; Parker, M. H.; Boyd, R. E.; Qu, J.; Alexander, R. S.; Brenneman, D. E.; Reitz, A. B. J. Med. Chem. 2007, 50, 4261–4264. doi:10.1021/jm0705408
Return to citation in text: [1] -
Hsu, H.-Y.; Tseng, C.-C.; Matii, B.; Sun, C.-M. Mol. Diversity 2012, 16, 241–249. doi:10.1007/s11030-011-9350-1
Return to citation in text: [1] -
Mercan, D.; Çetinkaya, E.; Şahin, E. Inorg. Chim. Acta 2013, 400, 74–81. doi:10.1016/j.ica.2013.02.005
Return to citation in text: [1] -
Li, C.; An, S.; Zhu, Y.; Zhang, J.; Kang, Y.; Liu, P.; Wang, Y.; Li, J. RSC Adv. 2014, 4, 49888–49891. doi:10.1039/C4RA09240F
Return to citation in text: [1] -
Hati, S.; Sen, S. Synthesis 2016, 48, 1389–1398. doi:10.1055/s-0035-1560416
Return to citation in text: [1] -
Kumar, R. A.; Saidulu, G.; Sridhar, B.; Liu, S. T.; Reddy, K. R. J. Org. Chem. 2013, 78, 10240–10250. doi:10.1021/jo401622r
Return to citation in text: [1] -
Zhong, Y.; Wang, L.; Ding, M.-W. Tetrahedron 2011, 67, 3714–3723. doi:10.1016/j.tet.2011.03.056
Return to citation in text: [1] -
He, P.; Wu, J.; Nie, Y.-B.; Ding, M.-W. Eur. J. Org. Chem. 2010, 1088–1095. doi:10.1002/ejoc.200901287
Return to citation in text: [1] -
Charton, J.; Girault-Mizzi, S.; Debreu-Fontaine, M.-A.; Foufelle, F.; Hainault, I.; Bizot-Espiard, J.-G.; Caignard, D.-H.; Sergheraert, C. Bioorg. Med. Chem. 2006, 14, 4490–4518. doi:10.1016/j.bmc.2006.02.028
Return to citation in text: [1] [2] -
Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108
Return to citation in text: [1] [2] -
Nakagawa, Y.; Stevenst, R. V. J. Org. Chem. 1988, 53, 1873–1876. doi:10.1021/jo00244a007
Return to citation in text: [1] [2] -
Lv, Y.; Li, Y.; Xiong, T.; Pu, W.; Zhang, H.; Sun, K.; Liu, Q.; Zhang, Q. Chem. Commun. 2013, 49, 6439–6441. doi:10.1039/c3cc43129k
Return to citation in text: [1] -
Kreher, R.; Bergmann, U. Heterocycles 1981, 16, 1693–1696. doi:10.3987/R-1981-10-1693
Return to citation in text: [1] -
Hoang, L. T. M.; Ngo, L. H.; Nguyen, H. L.; Nguyen, H. T. H.; Nguyen, C. K.; Nguyen, B. T.; Ton, Q. T.; Nguyen, H. K. D.; Cordova, K. E.; Truong, T. Chem. Commun. 2015, 51, 17132–17135. doi:10.1039/C5CC05985B
Return to citation in text: [1] -
Yadav, D. K. T.; Bhanage, B. M. Synlett 2015, 26, 1862–1866. doi:10.1055/s-0034-1380811
Return to citation in text: [1] -
Pelagalli, R.; Chiarotto, I.; Feroci, M.; Vecchio, S. Green Chem. 2012, 14, 2251–2255. doi:10.1039/c2gc35485c
Return to citation in text: [1] -
Apfel, C.; Banner, D. W.; Bur, D.; Dietz, M.; Hubschwerlen, C.; Locher, H.; Marlin, F.; Masciadri, R.; Pirson, W.; Stalder, H. J. Med. Chem. 2001, 44, 1847–1852. doi:10.1021/jm000352g
Return to citation in text: [1] -
Rzasa, R. M.; Kaller, M. R.; Liu, G.; Magal, E.; Nguyen, T. T.; Osslund, T. D.; Powers, D.; Santora, V. J.; Viswanadhan, V. N.; Wang, H.-L.; Xiong, X.; Zhong, W.; Norman, M. H. Bioorg. Med. Chem. 2007, 15, 6574–6595. doi:10.1016/j.bmc.2007.07.005
Return to citation in text: [1] -
Bar-Haim, G.; Kol, M. Tetrahedron Lett. 1998, 39, 2643–2644. doi:10.1016/S0040-4039(98)00227-5
Return to citation in text: [1] -
Coyne, W. E.; Cusic, J. W. J. Med. Chem. 1968, 11, 1208–1213. doi:10.1021/jm00312a022
Return to citation in text: [1] -
Camacho, M. E.; Chayah, M.; García, M. E.; Fernández-Sáez, N.; Arias, F.; Gallo, M. A.; Carrión, M. D. Arch. Pharm. 2016, 349, 638–650. doi:10.1002/ardp.201600020
Return to citation in text: [1] -
Farhanullah; Samrin, F.; Ram, V. J. Tetrahedron 2009, 65, 1635–1638. doi:10.1016/j.tet.2008.12.041
Return to citation in text: [1] -
López, S. E.; Restrepo, J.; Salazar, J. J. Chem. Res. 2007, 497–502. doi:10.3184/030823407X245598
Return to citation in text: [1] [2] -
Kanaoka, Y.; Kuga, T.; Tanizawa, K. Chem. Pharm. Bull. 1970, 18, 397–399. doi:10.1248/cpb.18.397
Return to citation in text: [1] -
Kanaoka, Y.; Yonemitsu, O.; Sato, E.; Ban, Y. Chem. Pharm. Bull. 1968, 16, 280–284. doi:10.1248/cpb.16.280
Return to citation in text: [1] -
Van der Eicken, E.; Kappe, C. O., Eds. Microwave-Assisted Synthesis of Heterocycles; Distribution Center, GmbH, 2006.
Return to citation in text: [1] -
Bougrin, K.; Loupy, A.; Soufiaoui, M. J. Photochem. Photobiol., C: Photochem. Rev. 2005, 6, 139–167. doi:10.1016/j.jphotochemrev.2005.07.001
Return to citation in text: [1] -
Sharma, A.; Appukkuttan, P.; Van der Eycken, E. Chem. Commun. 2012, 48, 1623–1637. doi:10.1039/C1CC15238F
Return to citation in text: [1] -
García, M. B.; Torres, R. A.; Orelli, L. R. Tetrahedron Lett. 2006, 47, 4857–4859. doi:10.1016/j.tetlet.2006.05.042
Return to citation in text: [1] -
Díaz, J. E.; Bisceglia, J. Á.; Mollo, M. C.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 1895–1897. doi:10.1016/j.tetlet.2011.02.042
Return to citation in text: [1] -
Díaz, J. E.; Gruber, N.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 6443–6445. doi:10.1016/j.tetlet.2011.09.097
Return to citation in text: [1] -
Díaz, J. E.; Mollo, M. C.; Orelli, L. R. Beilstein J. Org. Chem. 2016, 12, 2026–2031. doi:10.3762/bjoc.12.190
Return to citation in text: [1] -
Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122
Return to citation in text: [1] -
Link, N. P.; Díaz, J. E.; Orelli, L. R. Synlett 2009, 751–754. doi:10.1055/s-0028-1087817
Return to citation in text: [1] [2] -
Nivsarkar, M.; Kaushik, M. P. Catal. Commun. 2005, 6, 367–370. doi:10.1016/j.catcom.2005.02.012
Return to citation in text: [1] -
Pollmann, W.; Schramm, G. Biochim. Biophys. Acta 1964, 80, 1–7. doi:10.1016/0926-6550(64)90192-6
Return to citation in text: [1] -
Brain, K. R.; Thapa, B. B. J. Chromatogr. 1983, 258, 183–188. doi:10.1016/S0021-9673(00)96410-0
Return to citation in text: [1]
| 52. | López, S. E.; Restrepo, J.; Salazar, J. J. Chem. Res. 2007, 497–502. doi:10.3184/030823407X245598 |
| 53. | Kanaoka, Y.; Kuga, T.; Tanizawa, K. Chem. Pharm. Bull. 1970, 18, 397–399. doi:10.1248/cpb.18.397 |
| 52. | López, S. E.; Restrepo, J.; Salazar, J. J. Chem. Res. 2007, 497–502. doi:10.3184/030823407X245598 |
| 54. | Kanaoka, Y.; Yonemitsu, O.; Sato, E.; Ban, Y. Chem. Pharm. Bull. 1968, 16, 280–284. doi:10.1248/cpb.16.280 |
| 55. | Van der Eicken, E.; Kappe, C. O., Eds. Microwave-Assisted Synthesis of Heterocycles; Distribution Center, GmbH, 2006. |
| 56. | Bougrin, K.; Loupy, A.; Soufiaoui, M. J. Photochem. Photobiol., C: Photochem. Rev. 2005, 6, 139–167. doi:10.1016/j.jphotochemrev.2005.07.001 |
| 1. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Nikbin, N. Beilstein J. Org. Chem. 2011, 7, 442–495. doi:10.3762/bjoc.7.57 |
| 2. | Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2013, 9, 2265–2319. doi:10.3762/bjoc.9.265 |
| 8. | Weinhardt, K.; Wallach, M. B.; March, M. J. Med. Chem. 1985, 28, 694–698. doi:10.1021/jm00383a002 |
| 36. | Zhong, Y.; Wang, L.; Ding, M.-W. Tetrahedron 2011, 67, 3714–3723. doi:10.1016/j.tet.2011.03.056 |
| 37. | He, P.; Wu, J.; Nie, Y.-B.; Ding, M.-W. Eur. J. Org. Chem. 2010, 1088–1095. doi:10.1002/ejoc.200901287 |
| 63. | Link, N. P.; Díaz, J. E.; Orelli, L. R. Synlett 2009, 751–754. doi:10.1055/s-0028-1087817 |
| 7. | Meyer, E. M.; Arendash, G. W.; Judkins, J. H.; Ying, L.; Wade, C.; Ken, W. R. J. Neurochem. 1987, 49, 1758–1762. doi:10.1111/j.1471-4159.1987.tb02433.x |
| 17. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 18. | Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424 |
| 21. | Armarego, W. L. F. J. Chem. Soc. 1961, 2697–2701. doi:10.1039/jr9610002697 |
| 22. | Armarego, W. L. F. Adv. Heterocycl. Chem. 1963, 11, 253–309. doi:10.1016/S0065-2725(08)60527-9 |
| 29. | Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/S0040-4039(01)01842-1 |
| 38. | Charton, J.; Girault-Mizzi, S.; Debreu-Fontaine, M.-A.; Foufelle, F.; Hainault, I.; Bizot-Espiard, J.-G.; Caignard, D.-H.; Sergheraert, C. Bioorg. Med. Chem. 2006, 14, 4490–4518. doi:10.1016/j.bmc.2006.02.028 |
| 39. | Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108 |
| 40. | Nakagawa, Y.; Stevenst, R. V. J. Org. Chem. 1988, 53, 1873–1876. doi:10.1021/jo00244a007 |
| 65. | Pollmann, W.; Schramm, G. Biochim. Biophys. Acta 1964, 80, 1–7. doi:10.1016/0926-6550(64)90192-6 |
| 5. | Austin, W. C.; Courtney, W.; Danilewicz, J. C.; Morgan, D. H.; Conover, L. H.; Howes, H. L., Jr.; Lynch, J. E.; McFarland, J. W.; Cornwell, R. L.; Theodorides, V. J. Nature 1966, 212, 1273–1274. doi:10.1038/2121273b0 |
| 6. | Anand, N.; Sharma, S., Eds. Approaches to Design and Synthesis of Antiparasitic Drugs; Chapter 6, Vol. 25; Elsevier, 1997. |
| 17. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 19. | Decker, M. Eur. J. Med. Chem. 2005, 40, 305–313. doi:10.1016/j.ejmech.2004.12.003 |
| 20. | Jaén, J. C.; Gregor, V. E.; Lee, C.; Davis, R.; Emmerling, M. Bioorg. Med. Chem. Lett. 1996, 6, 737–742. doi:10.1016/0960-894X(96)00102-3 |
| 21. | Armarego, W. L. F. J. Chem. Soc. 1961, 2697–2701. doi:10.1039/jr9610002697 |
| 22. | Armarego, W. L. F. Adv. Heterocycl. Chem. 1963, 11, 253–309. doi:10.1016/S0065-2725(08)60527-9 |
| 23. | Smith, J. G.; Sheepy, J. M. J. Heterocycl. Chem. 1975, 12, 231–234. doi:10.1002/jhet.5570120203 |
| 24. | Richers, M. T.; Zhao, C.; Seidel, D. Beilstein J. Org. Chem. 2013, 9, 1194–1201. doi:10.3762/bjoc.9.135 |
| 25. | He, K.-H.; Tan, F.-F.; Zhou, C.-Z.; Zhou, G.-J.; Yang, X.-L.; Li, Y. Angew. Chem. 2017, 56, 3080–3084. doi:10.1002/anie.201612486 |
| 63. | Link, N. P.; Díaz, J. E.; Orelli, L. R. Synlett 2009, 751–754. doi:10.1055/s-0028-1087817 |
| 3. | Doyle, M. E.; Egan, J. M. Pharmacol. Rev. 2003, 55, 105–131. doi:10.1124/pr.55.1.7 |
| 4. | Szabo, B. Pharmacol. Ther. 2002, 93, 1–35. doi:10.1016/S0163-7258(01)00170-X |
| 11. | Los, R.; Wesołowska-Trojanowska, M.; Malm, A.; Karpińska, M. M.; Matysiak, J.; Niewiadomy, A.; Głaszcz, U. Heteroat. Chem. 2012, 23, 265–275. doi:10.1002/hc.21012 |
| 16. | Ghosh, A. K.; Pandey, S.; Gangarajula, S.; Kulkarni, S.; Xu, X.; Rao, K. V.; Huang, X.; Tang, J. Bioorg. Med. Chem. Lett. 2012, 22, 5460–5465. doi:10.1016/j.bmcl.2012.07.043 |
| 26. | Burdick, B. A.; Benkovic, P. A.; Benkovic, S. J. J. Am. Chem. Soc. 1977, 99, 5716–5725. doi:10.1021/ja00459a032 |
| 27. | Papadopoulos, E. P.; George, B. J. Org. Chem. 1977, 42, 2530–2532. doi:10.1021/jo00434a049 |
| 28. | Grasso, S.; Micale, N.; Monforte, A.-M.; Monforte, P.; Polimeni, S.; Zappalà, M. Eur. J. Med. Chem. 2000, 35, 1115–1119. doi:10.1016/S0223-5234(00)01195-8 |
| 29. | Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/S0040-4039(01)01842-1 |
| 30. | Baxter, E. W.; Conway, K. A.; Kennis, L.; Bischoff, F.; Mercken, M. H.; De Winter, H. L.; Reynolds, C. H.; Tounge, B. A.; Luo, C.; Scott, M. K.; Huang, Y.; Braeken, M.; Pieters, S. M. A.; Berthelot, D. J. C.; Masure, S.; Bruinzeel, W. D.; Jordan, A. D.; Parker, M. H.; Boyd, R. E.; Qu, J.; Alexander, R. S.; Brenneman, D. E.; Reitz, A. B. J. Med. Chem. 2007, 50, 4261–4264. doi:10.1021/jm0705408 |
| 31. | Hsu, H.-Y.; Tseng, C.-C.; Matii, B.; Sun, C.-M. Mol. Diversity 2012, 16, 241–249. doi:10.1007/s11030-011-9350-1 |
| 32. | Mercan, D.; Çetinkaya, E.; Şahin, E. Inorg. Chim. Acta 2013, 400, 74–81. doi:10.1016/j.ica.2013.02.005 |
| 33. | Li, C.; An, S.; Zhu, Y.; Zhang, J.; Kang, Y.; Liu, P.; Wang, Y.; Li, J. RSC Adv. 2014, 4, 49888–49891. doi:10.1039/C4RA09240F |
| 34. | Hati, S.; Sen, S. Synthesis 2016, 48, 1389–1398. doi:10.1055/s-0035-1560416 |
| 35. | Kumar, R. A.; Saidulu, G.; Sridhar, B.; Liu, S. T.; Reddy, K. R. J. Org. Chem. 2013, 78, 10240–10250. doi:10.1021/jo401622r |
| 64. | Nivsarkar, M.; Kaushik, M. P. Catal. Commun. 2005, 6, 367–370. doi:10.1016/j.catcom.2005.02.012 |
| 13. | Lee, Y. S.; Lee, B. H.; Park, S. J.; Kang, S. B.; Rhim, H.; Park, J.-Y.; Lee, J.-H.; Jeong, S.-W.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2004, 14, 3379–3384. doi:10.1016/j.bmcl.2004.04.090 |
| 14. | Heo, J. H.; Seo, H. N.; Choe, Y. J.; Kim, S.; Oh, C. R.; Kim, Y. D.; Rhim, H.; Choo, D. J.; Kim, J.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2008, 18, 3899–3901. doi:10.1016/j.bmcl.2008.06.034 |
| 15. | Byun, J. S.; Sohn, J. M.; Leem, D. G.; Park, B.; Nam, J. H.; Shin, D. H.; Shin, J. S.; Kim, H. J.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 1073–1079. doi:10.1016/j.bmcl.2015.12.010 |
| 17. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 61. | Díaz, J. E.; Mollo, M. C.; Orelli, L. R. Beilstein J. Org. Chem. 2016, 12, 2026–2031. doi:10.3762/bjoc.12.190 |
| 12. | Li, W.-J.; Li, Q.; Liu, D.-L.; Ding, M.-W. J. Agric. Food Chem. 2013, 61, 1419–1426. doi:10.1021/jf305355u |
| 18. | Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424 |
| 62. | Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122 |
| 11. | Los, R.; Wesołowska-Trojanowska, M.; Malm, A.; Karpińska, M. M.; Matysiak, J.; Niewiadomy, A.; Głaszcz, U. Heteroat. Chem. 2012, 23, 265–275. doi:10.1002/hc.21012 |
| 57. | Sharma, A.; Appukkuttan, P.; Van der Eycken, E. Chem. Commun. 2012, 48, 1623–1637. doi:10.1039/C1CC15238F |
| 9. | Dunbar, P. G.; Durant, G. J.; Fang, Z.; Abuh, Y. F.; El-Assadi, A. A.; Ngur, D. O.; Periyasamy, S.; Hoss, W. P.; Messer, W. S., Jr. J. Med. Chem. 1993, 36, 842–847. doi:10.1021/jm00059a008 |
| 10. | Dunbar, P. G.; Durant, G. J.; Rho, T.; Ojo, B.; Huzl, J. J., III; Smith, D. A.; El-Assadi, A. A.; Sbeih, S.; Ngur, D. O. J. Med. Chem. 1994, 37, 2774–2782. doi:10.1021/jm00043a016 |
| 16. | Ghosh, A. K.; Pandey, S.; Gangarajula, S.; Kulkarni, S.; Xu, X.; Rao, K. V.; Huang, X.; Tang, J. Bioorg. Med. Chem. Lett. 2012, 22, 5460–5465. doi:10.1016/j.bmcl.2012.07.043 |
| 58. | García, M. B.; Torres, R. A.; Orelli, L. R. Tetrahedron Lett. 2006, 47, 4857–4859. doi:10.1016/j.tetlet.2006.05.042 |
| 59. | Díaz, J. E.; Bisceglia, J. Á.; Mollo, M. C.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 1895–1897. doi:10.1016/j.tetlet.2011.02.042 |
| 60. | Díaz, J. E.; Gruber, N.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 6443–6445. doi:10.1016/j.tetlet.2011.09.097 |
| 42. | Kreher, R.; Bergmann, U. Heterocycles 1981, 16, 1693–1696. doi:10.3987/R-1981-10-1693 |
| 12. | Li, W.-J.; Li, Q.; Liu, D.-L.; Ding, M.-W. J. Agric. Food Chem. 2013, 61, 1419–1426. doi:10.1021/jf305355u |
| 13. | Lee, Y. S.; Lee, B. H.; Park, S. J.; Kang, S. B.; Rhim, H.; Park, J.-Y.; Lee, J.-H.; Jeong, S.-W.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2004, 14, 3379–3384. doi:10.1016/j.bmcl.2004.04.090 |
| 14. | Heo, J. H.; Seo, H. N.; Choe, Y. J.; Kim, S.; Oh, C. R.; Kim, Y. D.; Rhim, H.; Choo, D. J.; Kim, J.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2008, 18, 3899–3901. doi:10.1016/j.bmcl.2008.06.034 |
| 15. | Byun, J. S.; Sohn, J. M.; Leem, D. G.; Park, B.; Nam, J. H.; Shin, D. H.; Shin, J. S.; Kim, H. J.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 1073–1079. doi:10.1016/j.bmcl.2015.12.010 |
| 19. | Decker, M. Eur. J. Med. Chem. 2005, 40, 305–313. doi:10.1016/j.ejmech.2004.12.003 |
| 24. | Richers, M. T.; Zhao, C.; Seidel, D. Beilstein J. Org. Chem. 2013, 9, 1194–1201. doi:10.3762/bjoc.9.135 |
| 40. | Nakagawa, Y.; Stevenst, R. V. J. Org. Chem. 1988, 53, 1873–1876. doi:10.1021/jo00244a007 |
| 66. | Brain, K. R.; Thapa, B. B. J. Chromatogr. 1983, 258, 183–188. doi:10.1016/S0021-9673(00)96410-0 |
| 41. | Lv, Y.; Li, Y.; Xiong, T.; Pu, W.; Zhang, H.; Sun, K.; Liu, Q.; Zhang, Q. Chem. Commun. 2013, 49, 6439–6441. doi:10.1039/c3cc43129k |
| 39. | Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108 |
| 17. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 18. | Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424 |
| 29. | Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/S0040-4039(01)01842-1 |
| 49. | Coyne, W. E.; Cusic, J. W. J. Med. Chem. 1968, 11, 1208–1213. doi:10.1021/jm00312a022 |
| 50. | Camacho, M. E.; Chayah, M.; García, M. E.; Fernández-Sáez, N.; Arias, F.; Gallo, M. A.; Carrión, M. D. Arch. Pharm. 2016, 349, 638–650. doi:10.1002/ardp.201600020 |
| 51. | Farhanullah; Samrin, F.; Ram, V. J. Tetrahedron 2009, 65, 1635–1638. doi:10.1016/j.tet.2008.12.041 |
| 46. | Apfel, C.; Banner, D. W.; Bur, D.; Dietz, M.; Hubschwerlen, C.; Locher, H.; Marlin, F.; Masciadri, R.; Pirson, W.; Stalder, H. J. Med. Chem. 2001, 44, 1847–1852. doi:10.1021/jm000352g |
| 47. | Rzasa, R. M.; Kaller, M. R.; Liu, G.; Magal, E.; Nguyen, T. T.; Osslund, T. D.; Powers, D.; Santora, V. J.; Viswanadhan, V. N.; Wang, H.-L.; Xiong, X.; Zhong, W.; Norman, M. H. Bioorg. Med. Chem. 2007, 15, 6574–6595. doi:10.1016/j.bmc.2007.07.005 |
| 48. | Bar-Haim, G.; Kol, M. Tetrahedron Lett. 1998, 39, 2643–2644. doi:10.1016/S0040-4039(98)00227-5 |
| 45. | Pelagalli, R.; Chiarotto, I.; Feroci, M.; Vecchio, S. Green Chem. 2012, 14, 2251–2255. doi:10.1039/c2gc35485c |
| 38. | Charton, J.; Girault-Mizzi, S.; Debreu-Fontaine, M.-A.; Foufelle, F.; Hainault, I.; Bizot-Espiard, J.-G.; Caignard, D.-H.; Sergheraert, C. Bioorg. Med. Chem. 2006, 14, 4490–4518. doi:10.1016/j.bmc.2006.02.028 |
| 43. | Hoang, L. T. M.; Ngo, L. H.; Nguyen, H. L.; Nguyen, H. T. H.; Nguyen, C. K.; Nguyen, B. T.; Ton, Q. T.; Nguyen, H. K. D.; Cordova, K. E.; Truong, T. Chem. Commun. 2015, 51, 17132–17135. doi:10.1039/C5CC05985B |
| 44. | Yadav, D. K. T.; Bhanage, B. M. Synlett 2015, 26, 1862–1866. doi:10.1055/s-0034-1380811 |
© 2017 Díaz et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)