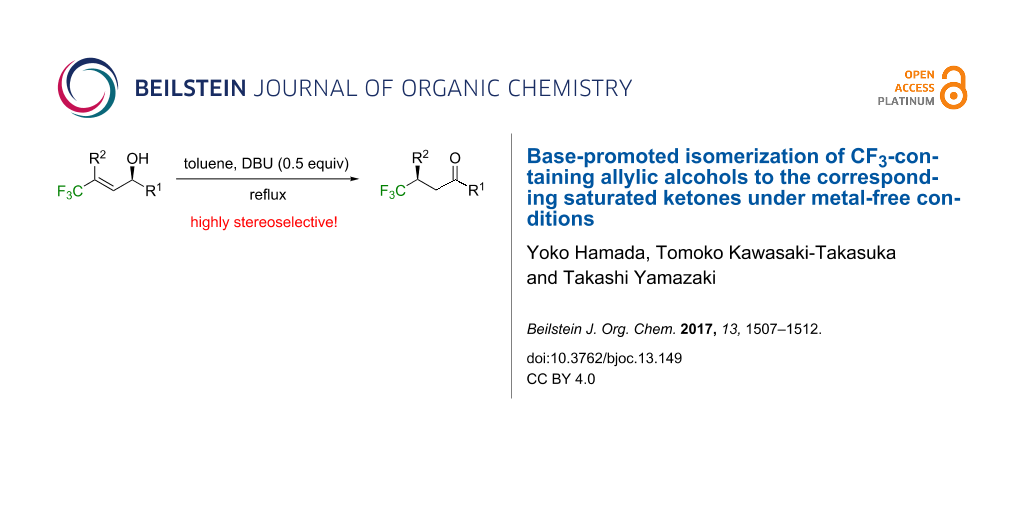
Abstract
Following to the computational expectation, our previously reported intriguing 1,3-proton shift of 4,4,4-trifluorobut-2-yn-1-ols was successfully extended to the 4,4,4-trifluorobut-2-en-1-ol system under metal-free conditions to afford the corresponding saturated ketones in high to excellent chemical yields using such a convenient and easy-to-handle base as DBU at the toluene refluxing temperature, and utilization of the corresponding optically active substrates unambiguously demonstrated that this transformation proceeded in a highly stereoselective fashion.

Graphical Abstract
Introduction
We have previously reported [1-3] an extremely efficient isomerization process of γ-trifluoromethylated propargylic alcohols 1F to the corresponding α,β-unsaturated ketones (E)-5 by the action of a weak base like Et3N under THF reflux conditions (Scheme 1). From the mechanistic point of view, deprotonation of the propargylic proton Ha was considered to be the initial step of this reaction and this step would be in competition with the proton abstraction from the OHb group in 1F. Thus, it was our first understanding that during the equilibration between 3F-O and 3F-C, the irreversible reprotonation by way of the latter intermediate would be occurred at the CF3-attached carbon atom to produce the corresponding allenyl type intermediate. Furthermore, its keto–enol interconversion would result in construction of the isomerized α,β-unsaturated ketones (E)-5 [4-6]. For a better comprehension of this interesting and efficient protocol, computation was performed [7] for obtaining the rough indication of the acidity of both protons Ha and Hb in 1F. Moreover, the corresponding allylic alcohols 2F as well as their non-fluorinated counterparts 1H and 2H were also employed for comparison whose results were summarized in Table 1.
![[1860-5397-13-149-i1]](/bjoc/content/inline/1860-5397-13-149-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Et3N-promoted isomerization of propargylic alcohols 1F.
Scheme 1: Et3N-promoted isomerization of propargylic alcohols 1F.
Table 1: Destabilization energy ΔE of propargylic (1) and allylic (2) alcohols.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-149-i2.svg?max-width=637&scale=1.0)
|
|||||
| C–C bond | X | R | ΔE (kcal/mol) | ΔΔEc | |
|---|---|---|---|---|---|
| ΔECa | ΔEOb | (kcal/mol) | |||
| triple | F | CH3 (a) | 310.657 | 311.705 | –1.048 |
| triple | Ph (b) | 301.759 | 309.628 | –7.869 | |
| double | CH3 (a) | –d | 316.344 | –d | |
| double | Ph (b) | 313.332 | 313.653 | –0.321 | |
| triple | H | CH3 (a) | 338.596 | 319.647 | 18.949 |
| triple | Ph (b) | 324.957 | 317.154 | 7.803 | |
| double | CH3 (a) | 351.919 | 324.077 | 27.842 | |
| double | Ph (b) | 332.565 | 320.943 | 11.622 | |
aΔEC: E3X-C − E1X or E4X-C − E2X. bΔEO: E3X-O − E1X or E4X-O − E2X. cΔΔE: ΔEC − ΔEO. dThe stable 4F-Ca was failed to be located using the IEFPCM method.
For this purpose, evaluation was performed on the basis of the energetic difference between neutral compounds and the corresponding carbanion or alkoxide species: thus, in the former instance, ΔEC as E3X-C − E1X (or E4X-C − E2X) or the latter ΔEO as E3X-O − E1X (or E4X-O − E2X) in the case of propargylic (or allylic) alcohols, respectively, as the simple measure of destabilization. In the case of the non-fluorinated 1H and 2H series, as expected, a clear alkoxide preference was noticed irrespective of the carbon–carbon unsaturation pattern. For the corresponding fluorinated counterparts, in spite of the failure to locate the energy minimum for 4F-Ca, it was found out in other cases that ΔEC values were unanimously smaller than ΔEO which led to the definitive conclusion that protons at the propargylic and allylic positions should be more acidic than the ones at the corresponding OH groups. The strongly electron-withdrawing property of a CF3 group is considered to play a key role in stabilization of both 3F-C and 4F-C, and the phenyl group as well as the carbon–carbon multiple bonds would also provide the additional preferable effect by their efficient resonance. In the case of the propargylic alcohol 1Fa with a methyl moiety as R, its electron-donating ability unambiguously destabilized the carbanionic species 3F-Ca while the electron-withdrawal of a CF3 group nicely compensated this disadvantage, resulting in a better stability of 3F-Ca by 1 kcal/mol with respect to the alkoxide 3F-Oa. In the case of 1Fb, the phenyl group worked nicely for increase of the energetic preference of 3F-Cb to 3F-Ob of about 7.9 kcal/mol. For the allylic alcohol 2Fb (R = Ph), although the ΔΔE value was small, the carbanionic species 4F-Cb was calculated to be more (or at least almost equally) preferable to 4F-Ob. This result allowed us to similarly anticipate the successful isomerization of CF3-containing allylic alcohols 2F to the corresponding saturated ketones 7 when the substrates possessed appropriate aromatic substituents as R.
With reference to these computational results, we decided to employ the CF3-containing allylic alcohols (E)-6 at the γ-position as substrates for the amine-promoted isomerization, instead of the corresponding propargylic alcohols 1F. Although a similar Ru- [8-12] or Fe-catalyzed processes [13] have been previously demonstrated, our present work is considered to draw a clear line with these instances because of the apparently convenient metal-free process. Moreover, the same type of proton shift has also reported by two groups. For example, during the reaction of (E)-2-(trifluoromethyl)vinylsilane and benzaldehyde in the presence of an excess amount of CsF, the resultant product (E)-2Fb was further converted in situ to the corresponding saturated ketone (E)-5b (R = Ph). Confirmation of this process was performed by the action of DBU, resulting in 95% conversion of the starting allylic alcohol (E)-2Fb [14]. Quite recently [15], the same proton shift was published by using the bicyclic guanidine-type base, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), as a catalyst. In spite of these two preceding studies, we have also succeeded in attaining a similar level of chemical yields as well as stereoselectivity to the latter process by using far-less expensive DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), whose results are reported in this article in detail.
Results and Discussion
Preparation of substrates (E)-6 was carried out by way of our recently reported one-pot procedure [16]: thus, after reaction of an appropriate Grignard reagent and ethyl trifluoroacetate at −80 °C, a Horner–Wadsworth–Emmons reagent [17] activated by LiBr and Et3N [18-20] was introduced to this solution at room temperature, leading to the formation of (E)-6 after the NaBH4 reduction of the resultant products (see Supporting Information File 1 for the detailed procedure).
As shown in Table 2, we at first checked a type of bases suitable for this isomerization in THF under reflux for 3 h. Although Et3N was appropriate in the case of isomerization of propargylic alcohols 1F [1], this was not the case for the allylic alcohol (E)-6a, forming the desired 7a only in a trace amount with recovery of (E)-6a in a large amount (Table 2, entry 1). Because Et3N was proved to be inadequate, utilization of DBU with the higher basicity was tried at the next stage although this base gave detrimental results for the isomerization of 1F. Actually, DBU was found to work nicely in the present instance to afford the desired compound 7a in 67% isolated yield (Table 2, entry 2). The result that the stronger tertiary amine DBU worked more effectively than Et3N was a clear support of our computation at least from a qualitative point of view which estimated the lower acidity of the allylic proton rather than the one in the propargylic position (ΔΔE of −0.321 and −7.869 kcal/mol for the allylic and propargylic series, respectively). Due to the unacceptable efficiency of K2CO3 and NaOH (Table 2, entries 3–5), the solvent effect was surveyed at the next stage with fixing the base to DBU. Polarity seemed to affect the reaction significantly, and aprotic MeCN, DMF, and DMSO (Table 2, entries 9, 10, and 12, respectively) showed a similar potency to THF with respect to chemical yields of 7a. Further investigation clarified that toluene was the best solvent from the standpoint of the material balance, and with taking efficiency into consideration, we eventually decided that, as described in Table 2, entry 18, 3 h reflux in toluene with 0.5 equiv of DBU were the conditions of choice.
Table 2: Optimization of reaction conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-149-i3.svg?max-width=637&scale=1.0)
|
||||||
| Yielda (%) | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | Temp. (°C) | Time (h) | 7a | (E)-6a |
| 1 | THF | Et3N | reflux | 3 | 3 | [93] |
| 2 | THF | DBU | reflux | 3 | (67) | [20] |
| 3 | THF | K2CO3 | reflux | 3 | – | [99] |
| 4 | THF | NaOHb | reflux | 3 | – | [99] |
| 5 | THF | NaOHc | reflux | 3 | (45) | [4] |
| 6 | MeOH | NaOHb | reflux | 3 | (49) | [–] |
| 7 | MeOH | DBU | reflux | 3 | 4 | [84] |
| 8 | MTBE | DBU | reflux | 3 | 9 | [95] |
| 9 | MeCN | DBU | 70 | 3 | (61) | [1] |
| 10 | DMF | DBU | 70 | 3 | 64 | [29] |
| 11 | DMA | DBU | 70 | 3 | 54 | [11] |
| 12 | DMSO | DBU | 70 | 3 | 67 | [15] |
| 13 | DCM | DBU | reflux | 3 | – | [94] |
| 14 | toluene | DBU | 70 | 3 | (46) | [44] |
| 15 | THF | DBU | reflux | 24 | (87) | [0] |
| 16 | toluene | DBU | 70 | 24 | (93) | [0] |
| 17 | toluene | DBU | reflux | 3 | (91) | [0] |
| 18d | toluene | DBU | reflux | 3 | (91) | [0] |
| 19e | toluene | DBU | reflux | 24 | (95) | [0] |
| 20d | toluene | DABCO | reflux | 10 | 30 | [78] |
a All yields were determined by 19F NMR and in brackets were described the recovery of (E)-6a. In the parentheses were shown the isolated yields. bA 6 M aqueous solution was used. cSolid NaOH was used. d0.5 equiv of base was used. e0.1 equiv of base was used.
Because we have successfully determined the appropriate reaction conditions for the present intriguing isomerization, clarification of its scope and limitation was carried out whose results were summarized in Table 3. Entry 1 depicts the result already shown in entry 18 of Table 2 where the substrate (E)-6a was transformed into the saturated ketone 7a in 91% yield. A slower reaction was suspected for (E)-6b in Table 3, entry 2 because of the possible destabilizing effect of the partial anionic charge at the 3 position by the electron-donating 4-MeOC6H4 group as R2. However, the effect was only quite limited and isomerization of (E)-6b was occurred in the same 3 h period as entry 1 with recording 89% isolated yield. Moreover, as expected, the electron-withdrawing 4-fluorophenyl moiety worked nicely to quantitatively furnish the ketone 7c in shorter time (Table 3, entry 3). However, as shown in Table 3, entries 4 and 5, clear retardation of this process was noticed for substrates with alkyl groups as R2. Different from the case of R2, an electronic effect of the substituent on the benzene ring of R1 affected this transformation quite significantly, and the substrate (E)-6f with 4-MeOC6H4 and Ph(CH2)2- groups as R1 and R2, respectively, experienced “double retardation” to require 48 h for attainment of an adequate level of conversion. Because of such substituent sensitivity of R1, alkyl groups at this position completely inhibited the reaction (Table 3, entries 8 and 9). Entry 10 described the result for the non-fluorinated substrate (E)-6j possessing a CH3 group instead of a CF3 moiety, and its simple recovery was observed. Exchange of the strongly electron-withdrawing CF3 group to CH3 led to complete loss of the good stabilizing factor of the developing carbanionic species. As a result, the alkoxide was more energetically preferred and thus this proton shift became difficult.
Table 3: Transformation of allylic alcohols (E)-6 to the corresponding ketones 7.
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-149-i4.svg?max-width=637&scale=1.0)
|
|||||
| Yielda (%) | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | 7 | (E)-6 |
| 1 | Ph | Ph | 3 | 91 (a) | [–] |
| 2 | Ph | 4-MeOC6H4- | 3 | 89 (b) | [–] |
| 3 | Ph | 4-FC6H4- | 2 | >99 (c) | [–] |
| 4 | Ph | Et | 24 | 78 (d) | [3b] |
| 5 | Ph | Ph(CH2)2- | 24 | 93 (e) | [–] |
| 6 | 4-MeOC6H4- | Ph(CH2)2- | 48 | 76 (f) | [–] |
| 7 | 4-BrC6H4- | Ph(CH2)2- | 3 | 91 (g) | [–] |
| 8 | Ph(CH2)2- | Ph | 24 | – (h) | [75] |
| 9 | Et | Ph | 24 | – (i) | [89] |
| 10c | Ph | Ph | 3 | – (j) | [77] |
aIsolated yields were shown and in brackets were described the recovery of the starting materials (E)-6. bYield determined by 19F NMR. cThis substrate (E)-6j contains a CH3 group instead of a CF3 moiety with CH3 and Ph located at the opposite positions.
For the purpose of obtaining mechanistic information on the present reaction, three representative optically active substrates (R,E)-6 were selected and submitted to the identical conditions as above (Table 4). Furthermore, in the case of (R,E)-6a, conditions in entries 6 (6 M NaOH aq in MeOH) and 19 (DABCO in toluene) in Table 2 were also tried for comparison (see entries 2 and 3, respectively). Like the case of the corresponding racemic compounds, the DBU-mediated proton shift was realized in a similar fashion to afford the ketones 7 in excellent isolated yields and, moreover, accomplished a quite high degree of chirality transmission (CT) on the basis of the chiral HPLC analysis (Table 4, entries 1, 4, and 5). Unanimous formation of (R)-stereoisomers at the 3 position of 7 from (R,E)-6 led to confirmation that the proton attached to C1 was migrated to C3 from its si face, thus from the same back side if (R,E)-6 possessed its conformation as shown in Table 4. Although DABCO attained the same level of CT (Table 4, entry 3) albeit a slower reaction rate, this is not the case for the conditions of 6 M NaOH aq in MeOH and only 21% CT was observed. The latter result would be because of the competing occurrence of intermolecular reprotonation by the solvent.
Table 4: DBU-promoted proton shift of the chiral allylic alcohols (R,E)-6.
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-149-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R3 | eeSa,b (% ee) | Yield (%) | eePa,b (% ee) | CTc (%) |
|---|---|---|---|---|---|
| 1 | H | 83 | 88 (a) | 85 | >99 |
| 2d | H | 86 | 63 (a) | 18 | 21 |
| 3e | H | 87 | 60 (a) | 86 | 99 |
| 4 | MeO | 80 | 85 (b) | 77 | 96 |
| 5 | F | 80 | 88 (c) | 78 | 98 |
aeeS and eeP are the enantiomeric excess values for (R,E)-6 and (R)-7, respectively. bDetermined by HPLC analysis using CHIRALPAK OD and AD columns for substrates and products, respectively. cCT: Chirality transmission. d6 M NaOH aq (6 equiv) in MeOH was used instead of DBU and toluene, and the reaction was continued for 24 h (30% of (R,E)-6a was isolated). eDABCO was used instead of DBU and the reaction was continued for 24 h (42% of (R,E)-6a was detected by 19F NMR).
The present interesting proton shift reaction was also computationally simulated [7] by employing (R,E)-6h with the substitution pattern of R1 = Ph and R2 = Me as the model substrate. For simplicity, 1,4-diazabicyclo[2.2.2]octane (DABCO) was employed as the representative base rather than DBU. Successful location of the transition state TS-8h at the B3LYP/6-311++G** level of theory led to clear analysis that TS-8h was 27.10 kcal/mol higher in energy than the combination of the substrate (R,E)-6h and the reactant DABCO under vacuum. This barrier became lowered to 24.71 kcal/mol after the single point calculation by consideration of the solvent effect of toluene by the conductor-like polarizable continuum model (CPCM). This energy barrier is considered to qualitatively correspond to the requirement of toluene reflux temperature (110–111 °C) for promotion of the desired proton shift. The cleaving C1···H and forming C3···H bond lengths in TS-8h were calculated to be 220.0 and 238.1 pm which were significantly elongated from the ones of the corresponding substrate (R,E)-6h and product (R)-7h of 109.5 and 109.3 pm, respectively (Figure 1). The partial charge at C3 became more negative (–0.338, –0.110, and –0.299 for TS-8h, (R,E)-6h, and (R)-7h, respectively) and H more positive (0.528, 0.172 and 0.244). The DABCO molecule with activating the proton was found to be situated right behind the C2 atom with the C2···H distance of 201.6 pm. The N···H distance of 104.4 pm was found to be only 2.4 pm longer than the same bond in protonated DABCO which would be one of the major reasons why the weaker base Et3N did not work for this reaction.
![[1860-5397-13-149-1]](/bjoc/content/figures/1860-5397-13-149-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Calculated transition state model TS-8h for the present proton shift starting from (R,E)-6h (some hydrogen atoms are omitted for clarity).
Figure 1: Calculated transition state model TS-8h for the present proton shift starting from (R,E)-6h (some h...
Conclusion
As shown above, our original isomerization of CF3-containing propargylic alcohols 1F to the corresponding α,β-unsaturated ketones (E)-5 was successfully extended to the transformation of allylic alcohols (E)-6 to saturated ketones 7. Different from the previously reported transition metal-catalyzed protocols by Cahard [8,9,13] and Liu [10-12], our transformation nicely proceeded with such a convenient and tractable tertiary amine as DBU, thus under metal-free conditions like the cases of the Ando [14] and Martín-Matute groups [15]. Moreover, this intriguing proton shift was clarified to be applied for optically active allylic alcohols whose chirality was transmitted almost in a perfect fashion. These results should stem from the acidic nature of the allylic proton which was successfully estimated from our independent computation. Further utilization of this reaction is being studied in this laboratory and the results obtained will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C NMR spectra, HPLC charts for optically active compounds, and computational details. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945. doi:10.1016/j.tet.2009.05.087
Return to citation in text: [1] [2] -
Watanabe, Y.; Yamazaki, T. J. Org. Chem. 2011, 76, 1957. doi:10.1021/jo102503s
Return to citation in text: [1] -
Jiang, Z.-X.; Qing, F.-L. J. Fluorine Chem. 2003, 123, 57. doi:10.1016/S0022-1139(03)00136-2
Return to citation in text: [1] -
Tiruveedhula, V. V. N. P. B.; Witzigmann, C. M.; Verma, R.; Kabir, M. S.; Rott, M.; Schwan, W. R.; Medina-Bielski, S.; Lane, M.; Close, W.; Polanowski, R. L.; Sherman, D.; Monte, A.; Deschamps, J. R.; Cook, J. M. Bioorg. Med. Chem. 2013, 21, 7830. doi:10.1016/j.bmc.2013.10.011
Return to citation in text: [1] -
Wang, Y.-H.; Liu, H.; Zhu, L.-L.; Li, X.-X.; Chen, Z. Adv. Synth. Catal. 2011, 353, 707. doi:10.1002/adsc.201000833
Return to citation in text: [1] -
Chen, J.; Fan, G.; Liu, Y. Org. Biomol. Chem. 2010, 8, 4806. doi:10.1039/c0ob00344a
Return to citation in text: [1] -
Gaussian 09W, Rev. D.01; Gaussian: Wallingford, CT, 2009.
Full optimization of each species was carried out using the B3LYP/6-311++G** level of theory, followed by the single point calculation using the IEFPCM method for consideration of the solvent effect by THF.
Return to citation in text: [1] [2] -
Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. Angew. Chem., Int. Ed. 2012, 51, 6467. doi:10.1002/anie.201200827
Return to citation in text: [1] [2] -
Bizet, V.; Pannecouocke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56. doi:10.1016/j.jfluchem.2013.01.004
Return to citation in text: [1] [2] -
Wu, M.; Kong, L.; Wang, K.; Jin, R.; Cheng, T.; Liu, G. Catal. Sci. Technol. 2015, 5, 1750. doi:10.1039/c4cy01404a
Return to citation in text: [1] [2] -
Xia, X.; Wu, M.; Jin, R.; Cheng, T.; Liu, G. Green Chem. 2015, 17, 3916. doi:10.1039/C5GC00479A
Return to citation in text: [1] [2] -
Wu, M.; Kong, L.; Wang, K.; Jin, R.; Cheng, T.; Liu, G. Catal. Sci. Technol. 2015, 5, 1750. doi:10.1039/C4CY01404A
Return to citation in text: [1] [2] -
Cahard, D.; Bizet, V.; Dai, X.; Gaillard, S.; Renaud, J.-L. J. Fluorine Chem. 2013, 155, 78. doi:10.1016/j.jfluchem.2013.05.028
Return to citation in text: [1] [2] -
Ikeda, A.; Omote, M.; Nomura, S.; Tanaka, M.; Tarui, A.; Sato, K.; Ando, A. Beilstein J. Org. Chem. 2013, 9, 2417. doi:10.3762/bjoc.9.279
Return to citation in text: [1] [2] -
Martinez-Erro, S.; Sanz-Marco, A.; Gómez, A. B.; Vázquez-Romero, A.; Ahlquist, M. S. G.; Martín-Matute, B. J. Am. Chem. Soc. 2016, 138, 13408. doi:10.1021/jacs.6b08350
Return to citation in text: [1] [2] -
Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Tetrahedron 2015, 71, 8059. doi:10.1016/j.tet.2015.08.048
Return to citation in text: [1] -
Milburn, R. R.; McRae, K.; Chan, J.; Tedrow, J.; Larsen, R.; Faul, M. Tetrahedron Lett. 2009, 50, 870. doi:10.1016/j.tetlet.2008.11.112
Return to citation in text: [1] -
Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Masamune, S.; Roush, W. R.; Sakai, T. Tetrahedron Lett. 1984, 25, 2183. doi:10.1016/S0040-4039(01)80205-7
Return to citation in text: [1] -
Rathke, M. W.; Nowak, M. J. Org. Chem. 1985, 50, 2624. doi:10.1021/jo00215a004
Return to citation in text: [1] -
Takacs, J. M.; Helle, M. A.; Seely, F. L. Tetrahedron Lett. 1986, 27, 1257. doi:10.1016/S0040-4039(00)84232-X
Return to citation in text: [1]
| 1. | Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945. doi:10.1016/j.tet.2009.05.087 |
| 2. | Watanabe, Y.; Yamazaki, T. J. Org. Chem. 2011, 76, 1957. doi:10.1021/jo102503s |
| 3. | Jiang, Z.-X.; Qing, F.-L. J. Fluorine Chem. 2003, 123, 57. doi:10.1016/S0022-1139(03)00136-2 |
| 13. | Cahard, D.; Bizet, V.; Dai, X.; Gaillard, S.; Renaud, J.-L. J. Fluorine Chem. 2013, 155, 78. doi:10.1016/j.jfluchem.2013.05.028 |
| 14. | Ikeda, A.; Omote, M.; Nomura, S.; Tanaka, M.; Tarui, A.; Sato, K.; Ando, A. Beilstein J. Org. Chem. 2013, 9, 2417. doi:10.3762/bjoc.9.279 |
| 8. | Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. Angew. Chem., Int. Ed. 2012, 51, 6467. doi:10.1002/anie.201200827 |
| 9. | Bizet, V.; Pannecouocke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56. doi:10.1016/j.jfluchem.2013.01.004 |
| 10. | Wu, M.; Kong, L.; Wang, K.; Jin, R.; Cheng, T.; Liu, G. Catal. Sci. Technol. 2015, 5, 1750. doi:10.1039/c4cy01404a |
| 11. | Xia, X.; Wu, M.; Jin, R.; Cheng, T.; Liu, G. Green Chem. 2015, 17, 3916. doi:10.1039/C5GC00479A |
| 12. | Wu, M.; Kong, L.; Wang, K.; Jin, R.; Cheng, T.; Liu, G. Catal. Sci. Technol. 2015, 5, 1750. doi:10.1039/C4CY01404A |
| 15. | Martinez-Erro, S.; Sanz-Marco, A.; Gómez, A. B.; Vázquez-Romero, A.; Ahlquist, M. S. G.; Martín-Matute, B. J. Am. Chem. Soc. 2016, 138, 13408. doi:10.1021/jacs.6b08350 |
| 7. |
Gaussian 09W, Rev. D.01; Gaussian: Wallingford, CT, 2009.
Full optimization of each species was carried out using the B3LYP/6-311++G** level of theory, followed by the single point calculation using the IEFPCM method for consideration of the solvent effect by THF. |
| 8. | Bizet, V.; Pannecoucke, X.; Renaud, J.-L.; Cahard, D. Angew. Chem., Int. Ed. 2012, 51, 6467. doi:10.1002/anie.201200827 |
| 9. | Bizet, V.; Pannecouocke, X.; Renaud, J.-L.; Cahard, D. J. Fluorine Chem. 2013, 152, 56. doi:10.1016/j.jfluchem.2013.01.004 |
| 13. | Cahard, D.; Bizet, V.; Dai, X.; Gaillard, S.; Renaud, J.-L. J. Fluorine Chem. 2013, 155, 78. doi:10.1016/j.jfluchem.2013.05.028 |
| 4. | Tiruveedhula, V. V. N. P. B.; Witzigmann, C. M.; Verma, R.; Kabir, M. S.; Rott, M.; Schwan, W. R.; Medina-Bielski, S.; Lane, M.; Close, W.; Polanowski, R. L.; Sherman, D.; Monte, A.; Deschamps, J. R.; Cook, J. M. Bioorg. Med. Chem. 2013, 21, 7830. doi:10.1016/j.bmc.2013.10.011 |
| 5. | Wang, Y.-H.; Liu, H.; Zhu, L.-L.; Li, X.-X.; Chen, Z. Adv. Synth. Catal. 2011, 353, 707. doi:10.1002/adsc.201000833 |
| 6. | Chen, J.; Fan, G.; Liu, Y. Org. Biomol. Chem. 2010, 8, 4806. doi:10.1039/c0ob00344a |
| 10. | Wu, M.; Kong, L.; Wang, K.; Jin, R.; Cheng, T.; Liu, G. Catal. Sci. Technol. 2015, 5, 1750. doi:10.1039/c4cy01404a |
| 11. | Xia, X.; Wu, M.; Jin, R.; Cheng, T.; Liu, G. Green Chem. 2015, 17, 3916. doi:10.1039/C5GC00479A |
| 12. | Wu, M.; Kong, L.; Wang, K.; Jin, R.; Cheng, T.; Liu, G. Catal. Sci. Technol. 2015, 5, 1750. doi:10.1039/C4CY01404A |
| 17. | Milburn, R. R.; McRae, K.; Chan, J.; Tedrow, J.; Larsen, R.; Faul, M. Tetrahedron Lett. 2009, 50, 870. doi:10.1016/j.tetlet.2008.11.112 |
| 1. | Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945. doi:10.1016/j.tet.2009.05.087 |
| 16. | Yamazaki, T.; Mano, N.; Hikage, R.; Kaneko, T.; Kawasaki-Takasuka, T.; Yamada, S. Tetrahedron 2015, 71, 8059. doi:10.1016/j.tet.2015.08.048 |
| 7. |
Gaussian 09W, Rev. D.01; Gaussian: Wallingford, CT, 2009.
Full optimization of each species was carried out using the B3LYP/6-311++G** level of theory, followed by the single point calculation using the IEFPCM method for consideration of the solvent effect by THF. |
| 15. | Martinez-Erro, S.; Sanz-Marco, A.; Gómez, A. B.; Vázquez-Romero, A.; Ahlquist, M. S. G.; Martín-Matute, B. J. Am. Chem. Soc. 2016, 138, 13408. doi:10.1021/jacs.6b08350 |
| 14. | Ikeda, A.; Omote, M.; Nomura, S.; Tanaka, M.; Tarui, A.; Sato, K.; Ando, A. Beilstein J. Org. Chem. 2013, 9, 2417. doi:10.3762/bjoc.9.279 |
| 18. | Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Masamune, S.; Roush, W. R.; Sakai, T. Tetrahedron Lett. 1984, 25, 2183. doi:10.1016/S0040-4039(01)80205-7 |
| 19. | Rathke, M. W.; Nowak, M. J. Org. Chem. 1985, 50, 2624. doi:10.1021/jo00215a004 |
| 20. | Takacs, J. M.; Helle, M. A.; Seely, F. L. Tetrahedron Lett. 1986, 27, 1257. doi:10.1016/S0040-4039(00)84232-X |
© 2017 Hamada et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)