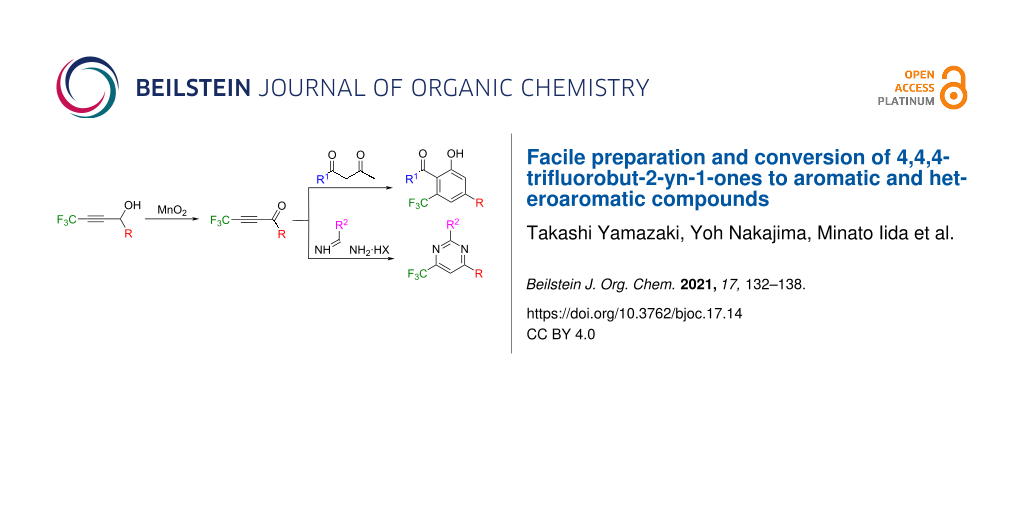
Abstract
The concise preparation of 4,4,4-trifluorobut-2-yn-1-ones by the oxidation of the readily accessible corresponding propargylic alcohols as well as their utilization as Michael acceptors for the construction of aromatic and heteroaromatic compounds are reported.

Graphical Abstract
Introduction
It is well known that the incorporation of a fluorine atom or a fluorine-containing group into organic compounds sometimes affects their original biological activities [1-3], and thus the development of methods for the construction of such molecules with this atom or group is an important task. During our research in the field of fluorine chemistry, we have previously developed methods to get convenient access to CF3-containing propargylic alcohols 1 using 2-bromo-3,3,3-trifluoropropene [4,5] as well as 1-chloro-3,3,3-trifluoropropene [6] as a substrate and to utilize 1 by way of a variety of routes [7-14]. Recently, we turned our attention to oxidized propargyl alcohols, namely ynones 2, because of the interesting structure with two strongly electron-withdrawing moieties, resulting in a high electrophilicity. However, only a few methods have been reported thus far for their preparation [15-20], including one that appeared in 2020 [21]. However, they suffer from problems, such as the formation of the desired product 2 only as regioisomeric mixtures [15,17] or the applicability to only one substrate without any extension to other related compounds [16,18-20]
Aromatic and heteroaromatic compounds are recognized as useful intermediates, and as pointed out above, the expected high electrophilicity of 2 was considered to allow the utilization as efficient Michael acceptors. Actually, this was the case, and the Sandford group clarified the high potency of these compounds for the conjugate addition of N- as well as O-nucleophiles [21]. On the basis of such an idea, we tried two routes to gain access to 1) 4-substituted 6-(trifluoromethyl)salicylate derivatives (C-nucleophiles) and 2) 6-substituted 4-(trifluoromethyl)pyrimidines (N-nucleophiles), both in a concise fashion, with the full details, including the preparation of 2, being reported herein.
Results and Discussion
Investigations on the reaction conditions were carried out for the oxidation of propargylic alcohols 1, which were readily accessible by our already reported procedure [4,19] as well as by the ones developed by other groups [22,23]. In spite of the previous unfavorable results for the oxidative transformation of 1a by MnO2 as well as other oxidants [18,23], MnO2 was selected due to the low cost and convenient handling. After a number of trials aimed at optimizing the reaction conditions, the unexpectedly smooth conversion of 1a to 2a was realized. Although it was reported [18,23] that only a small amount of 2a was confirmed after the reaction of 1a with 10 equiv of MnO2 at ambient temperature after 1 day, the same amount of oxidant actually led to the complete conversion after only 4 h at room temperature. In this instance, for the clean preparation of the desired compound 2a, the important point was to initiate this oxidation process at 0 °C, followed by warming to ambient temperature. With this intriguing result in hand, the oxidation of the other substrates 1 was performed under similar reaction condition, the results of which are summarized in Table 1.
Table 1: Oxidation of propargylic alcohols 1.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-14-i2.svg?max-width=637&scale=1.0)
|
|||||
| entry | R | T (°C) | t (h) | product | 19F NMR yieldb (%) |
| 1 | Ph | rt | 4 | 2a | 95 (100) |
| 2 | 4-(MeO)C6H4 | rt | 2.5 | 2b | 93 (100) |
| 3 | 4-MeC6H4 | rt | 3 | 2c | 90 (100) |
| 4 | 4-BrC6H4 | rt | 2 | 2d | 58 (93) |
| 5 | PhCH2CH2 | reflux | 20 | 2e | 35 (48) |
| 6c | PhCH2CH2 | reflux | 20 | 2e | 25 (80) |
a1 was added to a CH2Cl2 solution of MnO2 at 0 °C, and the mixture was stirred with gradually warming to a defined temperature for a defined reaction time, as indicated. bThe conversion of 1 as determined by 19F NMR spectroscopy is shown in parentheses. cCHCl3 was used instead of CH2Cl2.
We reported the yield as determined by 19F NMR spectroscopy rather than after purification because of the inherent instability of 2 on silica gel, causing partial decomposition. The crude mixture proved to be substantially pure, and thus 2c and 2d afforded appropriate analytical data, including NMR (1H, 13C, and 19F), IR, as well as high-resolution MS data. This process allowed us to obtain the required ynones 2 as long as the residue R was aromatic, while with an aliphatic substituent in this position, a full conversion was not observed even after a prolonged reaction period and at a higher temperature, and such conditions seemed to evoke the degradation of the products 2 to some extent. This was not the case for the Dess–Martin periodinane oxidation and the propargylic alcohols with C7H15 as the residue R1 by Hoye et al. [15] and C9H19 as the residue R1 by the Sandford group [21], who recorded 80% and 76% isolated yield, respectively, of the corresponding ketones. Because these compounds were previously isolated inconveniently by preparative GLC [15] or distillation [17,19], we decided to employ them without further purification.
The Michael addition reactions were then carried out under basic conditions for the ynone 2a thus obtained, which, to our astonishment, was found to be relatively rare even for the nonfluorinated counterparts (Table 2) [24-28].
Table 2: Michael addition of acetylacetone to 2a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-14-i3.svg?max-width=637&scale=1.0)
|
||||
| entry | base (equiv) | solvent | T (°C) | 19F NMR yielda (%) |
| 1 | NaH (1.5) | THF | 0 | 53 |
| 2 | NaH (1.5) | DMF | 0 | 34 |
| 3 | NaH (1.5) | Et2O | 0 | 76 (69) |
| 4 | NaH (1.5) | Et2O | −40 | 72 |
| 5 | NaH (1.5) | Et2O | rt | 61 |
| 6 | t-BuOK (1.5) | Et2O | 0 | 68 (67) |
| 7 | t-BuOK (0.1) | Et2O | 0 | 77 |
| 8 | t-BuOK (0.02) | Et2O | 0 | 94 (84) |
| 9 | t-BuOK (0.02) | t-BuOH | 30 | 78 |
| 10 | DBU (0.1) | Et2O | 0 | 33 |
| 11 | TMGb (0.1) | Et2O | 0 | 14 |
aThe isolated yield is shown in parentheses. b1,1,3,3-Tetramethylguanidine.
As expected for this model system, a very efficient addition was observed within a period of 0.5 h at 0 °C, leading to an isolated yield of 69% for the adduct 3aa when 1.5 equiv of acetylacetone and NaH in Et2O were used (Table 2, entries 1−3), which unambiguously demonstrated the high potency of 2a as a Michael acceptor. Stronger bases, such as NaH and t-BuOK seemed to be preferable (Table 2, entries 3 and 6), and amines such as DBU and 1,1,3,3-tetramethylguanidine (TMG) were found to be insufficient (Table 2, entries 10 and 11). Judging from the possible mechanism, this process was considered to proceed under the action of a catalytic amount of a base, and only 2 mol % of t-BuOK led to the formation of the desired adduct 3aa as a sole stereoisomer in 84% isolated yield under the same reaction conditions (Table 2, entries 6−8). The E-stereochemistry of 3aa was deduced from the NMR data of structurally similar compounds [29] on the basis of 1) comparison of the chemical shift of the CF3 group and 2) the absence of a coupling between the vinylic and allylic protons. In order to explore other nucleophiles, we employed 1,3-diketones, such as 5-methylhexane-1,3-dione and 5,5-dimethylhexane-1,3-dione (that is, one of the two methyl groups in acetylacetone was formally substituted for an isopropyl or a t-Bu moiety, respectively), but no reaction was observed at all. The conjugate addition of amines to this type of ynone has already been reported [21], where the authors pointed out the significant slowdown of the reaction rate when tert-butylamine was employed, which was considered to be a reflection of the sensitivity of ynones 2 towards steric hindrance.
Because appropriate conditions for the initial Michael addition reactions were thus determined, further conversion to the benzene derivative 4aa was also performed (Table 3).
Table 3: Investigation of the cyclization conditions for 3aa.
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-14-i4.svg?max-width=637&scale=1.0)
|
||||
| entry | base (equiv) | T (°C) | t (h) | 19F NMR yield of 4aaa (%) |
| 1 | t-BuOK (0.5) | 60 | 4 | 7 (49) |
| 2 | t-BuOK (1.0) | 60 | 4 | 13 (81)b {8} |
| 3 | t-BuOK (2.0) | 60 | 4 | 85 (<1) |
| 4 | t-BuOK (3.0) | 60 | 4 | >99 (0) |
| 5 | t-BuOK (3.0) | 60 | 2 | >99 (0) |
| 6 | t-BuOK (3.0) | 30 | 2 | >99 (0) {84} |
| 7c | t-BuOK (3.0) | 30 | 2 | >99 (0) |
| 8 | t-BuONa (3.0) | 30 | 2 | 58 (0) |
| 9 | t-BuOLi (3.0) | 30 | 3 | 66 (3) |
| 10d | EtONa (3.0) | 30 | 2 | 0 (0) |
| 11d | EtONa (3.0) | 60 | 6 | 18 (15) |
aThe 19F NMR yield of 5a and the isolated yield of 4aa are shown in parentheses and braces, respectively. bIsolated yield of 5a. cEt2O was used instead of t-BuOH. dEtOH was used instead of t-BuOH.
First of all, 3aa was introduced to a t-BuOH solution of 2 equiv of t-BuOK to afford the desired product 4aa in a high yield, and an increase in the amount of this base to 3 equiv led to the complete conversion of the substrate 3aa (Table 3, entries 3 and 4). This trend was followed even when the temperature was decreased to 30 °C and the reaction time was 2 h, furnished 4aa in a 84% yield (Table 3, entries 6 and 7). The use of the corresponding lithium and sodium tert-butoxides was found to afford a lower yield, which clarified the importance of the ionic character of the base (Table 3, entries 8 and 9).
For the present transformation, Hu and Guan already reported a similar type of ring closure of the substrate 3fa [30], which allowed the conversion to the phenolic product 5f as a result of the cyclization and was accompanied by deacetylation under the conditions depicted in Scheme 1 [31-33].
![[1860-5397-17-14-i1]](/bjoc/content/inline/1860-5397-17-14-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Deacetylative cyclization of 3fa.
Scheme 1: Deacetylative cyclization of 3fa.
In our case, it was quite interesting to note that the deacylated product 5a was also obtained as the major product when an equimolar or lesser amount of t-BuOK was employed (Table 3, entries 1 and 2), along with formation of a minor amount of 4aa. Moreover, when the isolated product 4aa was subjected to the conditions shown in Table 3, entry 4, no reaction was observed at all, with recovery of 4aa, which clearly demonstrated that the deacetylation step had occurred before the conversion to 4aa. In the present cyclization, the ionic character of the alkoxide seemed to be an important factor, and the corresponding lithium and sodium salts did not aid the reaction satisfactory. Moreover, EtONa was found to be inappropriate for this protocol (Table 3, entries 10 and 11).
Determination of the appropriate conditions both for Michael addition as well as cyclization/aromatization reactions allowed us to explore a brief scope and limitation of the present reaction by using four representative types of crude ynones 2 starting from the conjugate addition step either with acetylacetone or ethyl acetoacetate (Table 4).
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-14-i5.svg?max-width=637&scale=1.0)
In all instances, the desired products 4 were obtained in good to excellent yield without contamination by the deacetylated products 5.
With the success of the regiospecific formation of 4-substituted 6-(trifluoromethyl)salicylate derivatives 4, as shown in Table 4, the application of ynones 2 was demonstrated at the next stage for the construction of a variety of pyrimidine derivatives, 6, using amidines including guanidine [34]. First of all, various bases were employed for the reaction of the model substrate 2a. During initial testing, guanidine hydrochloride at 25 °C for 4 h in acetonitrile (Table 5, entries 1–7) as well as sodium carbonate were found to be adequate (Table 5, entry 5).
Table 5: Preparation of trifluoromethylated pyrimidine derivatives 6aa.
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-14-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | base | solvent | T (°C) | t (h) | 19F NMR yielda (%) | |
| 6aa | byproducts | |||||
| 1 | KOt-Bu | CH3CN | 25 | 4 | 8 | 54 |
| 2 | NaOEt | CH3CN | 25 | 4 | 15 | 9 |
| 3 | DBU | CH3CN | 25 | 4 | 3 | 17 |
| 4 | K2CO3 | CH3CN | 25 | 4 | 28 | 4 |
| 5 | Na2CO3 | CH3CN | 25 | 4 | 33 | 17 |
| 6 | CaCO3 | CH3CN | 25 | 4 | 25 | 39 |
| 7 | Li2CO3 | CH3CN | 25 | 4 | 27 | 29 |
| 8 | Na2CO3 | DME | 25 | 4 | 19 | 24 |
| 9 | Na2CO3 | DMF | 25 | 4 | 31 | 14 |
| 10 | Na2CO3 | DMSO | 25 | 4 | 24 | 9 |
| 11 | Na2CO3 | THF | 25 | 4 | 31 | 20 |
| 12 | Na2CO3 | CH3CN | 50 | 4 | 66 | 5 |
| 13 | Na2CO3 | CH3CN | 80 | 4 | 81 (57) | 2 |
| 14 | Na2CO3 | CH3CN | 80 | 8 | 83 (60) | 0 |
| 15 | Na2CO3 | CH3CN | 80 | 16 | 70 (56) | <1 |
aThe isolated yield is shown in parentheses.
A comparison of the results in Table 5, entries 8–11 vs entry 5 led to the determination that the adequate solvent was acetonitrile. After screening the reaction time and temperature (Table 5, entries 12–15 vs entry 5), we eventually selected the conditions as shown in Table 5, entry 14, which included heating the reaction mixture at 80 °C for 8 h in MeCN, furnishing the desired pyrimidine 6aa in 60% isolated yield.
Then, four types of ynones, 2a–d, were reacted with formamidine and acetamidine as well as guanidine under the optimized conditions outlined above, the results of which are summarized in Table 6. When the reactions were performed with acetamidine and guanidine, a good to high yield was recorded, while only a lower yield was obtained by the application of formamidine acetate, for which the reaction was not further elucidated at this point.
Table 6: Preparation of trifluoromethylated pyrimidine derivatives 6.
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-14-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | R1 | R2 | X | product | isolated yield (%) |
| 1 | Ph (2a) | NH2 | Cl | 6aa | 60 |
| 2 | 4-(MeO)C6H4 (2b) | NH2 | Cl | 6ab | 55 |
| 3 | 4-MeC6H4 (2c) | NH2 | Cl | 6ac | 52 |
| 4 | 4-BrC6H4 (2d) | NH2 | Cl | 6ad | 67 |
| 5 | Ph (2a) | Me | Cl | 6ba | 67 |
| 6 | 4-(MeO)C6H4 (2b) | Me | Cl | 6bb | 75 |
| 7 | 4-MeC6H4 (2c) | Me | Cl | 6bc | 53 |
| 8 | 4-BrC6H4 (2d) | Me | Cl | 6bd | 57 |
| 9 | Ph (2a) | H | AcO | 6ca | 21 |
| 10 | 4-(MeO)C6H4 (2b) | H | AcO | 6cb | 28 |
| 11 | 4-MeC6H4 (2c) | H | AcO | 6cc | 22 |
| 12 | 4-BrC6H4 (2d) | H | AcO | 6cd | 26 |
The unfavorable yield may be reasoned by the fact that there have been very limited reports for the syntheses of, for example, pyrimidine 6ca [35-38], with such drawbacks as the formation of a regioisomeric mixture [35] and the requirement of harsh conditions [38]. At the same time, for the use of a substrate similar to 2a where the CF3 and Ph groups were formally exchanged, an isolated yield of 84% has been recorded for 6ca [37]. Moreover, it is noteworthy that only one report dealt with the preparation of 6cb [37], and 6cc and 6cd have not been previously reported. Thus, including 6aa [37-40] and 6ba [37,38,41,42], it is noteworthy to realize the construction of relatively rare pyrimidines in a concise fashion via the ynones 2 with a CF3 group.
Conclusion
As described above, we succeeded in the construction of 4,4,4-trifluorobut-2-yn-1-ones 2 by way of a convenient MnO2 oxidation of readily accessible propargylic alcohols 1. In spite of the application of this method only to 1, with an aromatic substituent at the propargylic position and the requirement of an excess amount of this oxidant, the practical convenience, low cost, as well as the clean conversion are notable features of this method, which is worthy of attention. Furthermore, the high electrophilicity of ynones 2 obtained was nicely demonstrated through two representative reactions, namely 1) Michael additions of β-keto carbonyl compounds and 2) amidines and guanidine for the construction of aromatic and heteroaromatic products, which successfully expanded this unique synthetic approach to compounds with a limited precedent. Further work is ongoing for the application of ynones 2.
Supporting Information
The Supporting Information features the experimental part of this article.
| Supporting Information File 1: General information, synthetic procedures, and spectral data. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] -
Mei, H.; Han, J.; Fustero, S.; Medio‐Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/c39950000051
Return to citation in text: [1] [2] -
Yamazaki, T.; Mizutani, K.; Kitazume, T. J. Org. Chem. 1995, 60, 6046–6056. doi:10.1021/jo00124a013
Return to citation in text: [1] -
Miyagawa, A.; Naka, M.; Yamazaki, T.; Kawasaki-Takasuka, T. Eur. J. Org. Chem. 2009, 4395–4399. doi:10.1002/ejoc.200900370
Return to citation in text: [1] -
Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073–4076. doi:10.1021/ol048229x
Return to citation in text: [1] -
Yamazaki, T.; Yamamoto, T.; Ichihara, R. J. Org. Chem. 2006, 71, 6251–6253. doi:10.1021/jo060909l
Return to citation in text: [1] -
Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945–5948. doi:10.1016/j.tet.2009.05.087
Return to citation in text: [1] -
Watanabe, Y.; Yamazaki, T. Synlett 2009, 3352–3354. doi:10.1055/s-0029-1218383
Return to citation in text: [1] -
Watanabe, Y.; Yamazaki, T. J. Fluorine Chem. 2010, 131, 646–651. doi:10.1016/j.jfluchem.2010.01.003
Return to citation in text: [1] -
Watanabe, Y.; Yamazaki, T. J. Org. Chem. 2011, 76, 1957–1960. doi:10.1021/jo102503s
Return to citation in text: [1] -
Yamazaki, T.; Watanabe, Y.; Yoshida, N.; Kawasaki-Takasuka, T. Tetrahedron 2012, 68, 6665–6673. doi:10.1016/j.tet.2012.05.131
Return to citation in text: [1] -
Ichikawa, T.; Kawasaki-Takasuka, T.; Yamada, S.; Yamazaki, T.; Kubota, T. J. Fluorine Chem. 2013, 152, 38–45. doi:10.1016/j.jfluchem.2012.10.013
Return to citation in text: [1] -
Shen, Y.; Xin, Y.; Cen, W.; Huang, Y. Synthesis 1984, 35–37. doi:10.1055/s-1984-30723
Return to citation in text: [1] [2] [3] [4] -
Bumgardner, C. L.; Bunch, J. E.; Whangbo, M. H. J. Org. Chem. 1986, 51, 4082–4083. doi:10.1021/jo00371a037
Return to citation in text: [1] [2] -
Chechulin, P. I.; Filyakova, V. I.; Pashkevich, K. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1989, 38, 189–190. doi:10.1007/bf00953736
Return to citation in text: [1] [2] [3] -
Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/s0022-1139(00)80446-7
Return to citation in text: [1] [2] [3] [4] -
Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h
Return to citation in text: [1] [2] [3] [4] -
Wang, T.; Niu, D.; Hoye, T. R. J. Am. Chem. Soc. 2016, 138, 7832–7835. doi:10.1021/jacs.6b03786
Return to citation in text: [1] [2] -
Murray, B. J.; Marsh, T. G. F.; Yufit, D. S.; Fox, M. A.; Harsanyi, A.; Boulton, L. T.; Sandford, G. Eur. J. Org. Chem. 2020, 6236–6244. doi:10.1002/ejoc.202001071
Return to citation in text: [1] [2] [3] [4] -
Ishihara, T.; Yamasaki, Y.; Ando, T. Tetrahedron Lett. 1985, 26, 79–82. doi:10.1016/s0040-4039(00)98472-7
Return to citation in text: [1] -
Sibous, L.; Tipping, A. E. J. Fluorine Chem. 1993, 62, 39–49. doi:10.1016/s0022-1139(00)80079-2
Return to citation in text: [1] [2] [3] -
Fouli, F. A.; Basyouni, M. N. Acta Chim. Acad. Sci. Hung. 1981, 106, 297–302.
Return to citation in text: [1] -
Shankar, R.; Jha, A. K.; Singh, U. S.; Hajela, K. Tetrahedron Lett. 2006, 47, 3077–3079. doi:10.1016/j.tetlet.2006.03.008
Return to citation in text: [1] -
Ramachary, D. B.; Venkaiah, C.; Murali Krishna, P. Chem. Commun. 2012, 48, 2252–2254. doi:10.1039/c2cc17219d
Return to citation in text: [1] -
Tong, W.; Li, Q.-Y.; Xu, Y.-L.; Wang, H.-S.; Chen, Y.-Y.; Pan, Y.-M. Adv. Synth. Catal. 2017, 359, 4025–4035. doi:10.1002/adsc.201700830
Return to citation in text: [1] -
Yuan, Y.; Guo, Z.; Mu, Y.; Wang, Y.; Xu, M.; Li, Y. Adv. Synth. Catal. 2020, 362, 1298–1302. doi:10.1002/adsc.201901631
Return to citation in text: [1] -
Ramasamy, M.; Lin, H.-C.; Kuo, S.-C.; Hsieh, M.-T. Synlett 2019, 30, 356–360. doi:10.1055/s-0037-1611694
Return to citation in text: [1] -
Guan, H.-P.; Hu, C.-M. J. Fluorine Chem. 1996, 78, 101–102. doi:10.1016/0022-1139(96)03407-0
Return to citation in text: [1] -
Furuta, T.; Nakayama, M.; Suzuki, H.; Tajimi, H.; Inai, M.; Nukaya, H.; Wakimoto, T.; Kan, T. Org. Lett. 2009, 11, 2233–2236. doi:10.1021/ol900689m
Return to citation in text: [1] -
Wang, X.-j.; Zhang, L.; Byrne, D.; Nummy, L.; Weber, D.; Krishnamurthy, D.; Yee, N.; Senanayake, C. H. Org. Lett. 2014, 16, 4090–4093. doi:10.1021/ol501755h
Return to citation in text: [1] -
Matsuda, T.; Matsumoto, T. Org. Biomol. Chem. 2016, 14, 5023–5027. doi:10.1039/c6ob00734a
Return to citation in text: [1] -
Nenajdenko, V. G., Ed. Fluorine in Heterocyclic Chemistry; Springer: Berlin, Germany, 2014; Vol. 1 and 2.
Return to citation in text: [1] -
Gupta, S.; Melanson, J. A.; Vaillancourt, L.; Nugent, W. A.; Tanoury, G. J.; Schatte, G.; Snieckus, V. Org. Lett. 2018, 20, 3745–3748. doi:10.1021/acs.orglett.8b01300
Return to citation in text: [1] [2] -
Geri, J. B.; Wade Wolfe, M. M.; Szymczak, N. K. Angew. Chem., Int. Ed. 2018, 57, 1381–1385. doi:10.1002/anie.201711316
Return to citation in text: [1] -
Romanov, A. R.; Rulev, A. Y.; Ushakov, I. A.; Muzalevskiy, V. M.; Nenajdenko, V. G. Eur. J. Org. Chem. 2017, 4121–4129. doi:10.1002/ejoc.201700727
Return to citation in text: [1] [2] [3] [4] [5] -
Saijo, R.; Watanabe, G.; Kurihara, K.-i.; Kawase, M. Heterocycles 2014, 89, 2334–2345. doi:10.3987/com-14-13075
Return to citation in text: [1] [2] [3] [4] -
Liu, C.; Cui, Z.; Yan, X.; Qi, Z.; Ji, M.; Li, X. Molecules 2016, 21, 828. doi:10.3390/molecules21070828
Return to citation in text: [1] -
Rawal, R. K.; Tripathi, R.; Katti, S. B.; Pannecouque, C.; De Clercq, E. Bioorg. Med. Chem. 2007, 15, 3134–3142. doi:10.1016/j.bmc.2007.02.044
Return to citation in text: [1] -
Funabiki, K.; Nakamura, H.; Matsui, M.; Shibata, K. Synlett 1999, 756–758. doi:10.1055/s-1999-2741
Return to citation in text: [1] -
Jeong, I. H.; Jeon, S. L.; Kim, M. S.; Kim, B. T. J. Fluorine Chem. 2004, 125, 1629–1638. doi:10.1016/j.jfluchem.2004.09.021
Return to citation in text: [1]
| 37. | Romanov, A. R.; Rulev, A. Y.; Ushakov, I. A.; Muzalevskiy, V. M.; Nenajdenko, V. G. Eur. J. Org. Chem. 2017, 4121–4129. doi:10.1002/ejoc.201700727 |
| 37. | Romanov, A. R.; Rulev, A. Y.; Ushakov, I. A.; Muzalevskiy, V. M.; Nenajdenko, V. G. Eur. J. Org. Chem. 2017, 4121–4129. doi:10.1002/ejoc.201700727 |
| 38. | Saijo, R.; Watanabe, G.; Kurihara, K.-i.; Kawase, M. Heterocycles 2014, 89, 2334–2345. doi:10.3987/com-14-13075 |
| 39. | Liu, C.; Cui, Z.; Yan, X.; Qi, Z.; Ji, M.; Li, X. Molecules 2016, 21, 828. doi:10.3390/molecules21070828 |
| 40. | Rawal, R. K.; Tripathi, R.; Katti, S. B.; Pannecouque, C.; De Clercq, E. Bioorg. Med. Chem. 2007, 15, 3134–3142. doi:10.1016/j.bmc.2007.02.044 |
| 37. | Romanov, A. R.; Rulev, A. Y.; Ushakov, I. A.; Muzalevskiy, V. M.; Nenajdenko, V. G. Eur. J. Org. Chem. 2017, 4121–4129. doi:10.1002/ejoc.201700727 |
| 38. | Saijo, R.; Watanabe, G.; Kurihara, K.-i.; Kawase, M. Heterocycles 2014, 89, 2334–2345. doi:10.3987/com-14-13075 |
| 41. | Funabiki, K.; Nakamura, H.; Matsui, M.; Shibata, K. Synlett 1999, 756–758. doi:10.1055/s-1999-2741 |
| 42. | Jeong, I. H.; Jeon, S. L.; Kim, M. S.; Kim, B. T. J. Fluorine Chem. 2004, 125, 1629–1638. doi:10.1016/j.jfluchem.2004.09.021 |
| 1. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 2. | Mei, H.; Han, J.; Fustero, S.; Medio‐Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840 |
| 3. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 15. | Shen, Y.; Xin, Y.; Cen, W.; Huang, Y. Synthesis 1984, 35–37. doi:10.1055/s-1984-30723 |
| 16. | Bumgardner, C. L.; Bunch, J. E.; Whangbo, M. H. J. Org. Chem. 1986, 51, 4082–4083. doi:10.1021/jo00371a037 |
| 17. | Chechulin, P. I.; Filyakova, V. I.; Pashkevich, K. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1989, 38, 189–190. doi:10.1007/bf00953736 |
| 18. | Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/s0022-1139(00)80446-7 |
| 19. | Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h |
| 20. | Wang, T.; Niu, D.; Hoye, T. R. J. Am. Chem. Soc. 2016, 138, 7832–7835. doi:10.1021/jacs.6b03786 |
| 21. | Murray, B. J.; Marsh, T. G. F.; Yufit, D. S.; Fox, M. A.; Harsanyi, A.; Boulton, L. T.; Sandford, G. Eur. J. Org. Chem. 2020, 6236–6244. doi:10.1002/ejoc.202001071 |
| 7. | Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073–4076. doi:10.1021/ol048229x |
| 8. | Yamazaki, T.; Yamamoto, T.; Ichihara, R. J. Org. Chem. 2006, 71, 6251–6253. doi:10.1021/jo060909l |
| 9. | Yamazaki, T.; Kawasaki-Takasuka, T.; Furuta, A.; Sakamoto, S. Tetrahedron 2009, 65, 5945–5948. doi:10.1016/j.tet.2009.05.087 |
| 10. | Watanabe, Y.; Yamazaki, T. Synlett 2009, 3352–3354. doi:10.1055/s-0029-1218383 |
| 11. | Watanabe, Y.; Yamazaki, T. J. Fluorine Chem. 2010, 131, 646–651. doi:10.1016/j.jfluchem.2010.01.003 |
| 12. | Watanabe, Y.; Yamazaki, T. J. Org. Chem. 2011, 76, 1957–1960. doi:10.1021/jo102503s |
| 13. | Yamazaki, T.; Watanabe, Y.; Yoshida, N.; Kawasaki-Takasuka, T. Tetrahedron 2012, 68, 6665–6673. doi:10.1016/j.tet.2012.05.131 |
| 14. | Ichikawa, T.; Kawasaki-Takasuka, T.; Yamada, S.; Yamazaki, T.; Kubota, T. J. Fluorine Chem. 2013, 152, 38–45. doi:10.1016/j.jfluchem.2012.10.013 |
| 15. | Shen, Y.; Xin, Y.; Cen, W.; Huang, Y. Synthesis 1984, 35–37. doi:10.1055/s-1984-30723 |
| 6. | Miyagawa, A.; Naka, M.; Yamazaki, T.; Kawasaki-Takasuka, T. Eur. J. Org. Chem. 2009, 4395–4399. doi:10.1002/ejoc.200900370 |
| 18. | Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/s0022-1139(00)80446-7 |
| 23. | Sibous, L.; Tipping, A. E. J. Fluorine Chem. 1993, 62, 39–49. doi:10.1016/s0022-1139(00)80079-2 |
| 4. | Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/c39950000051 |
| 5. | Yamazaki, T.; Mizutani, K.; Kitazume, T. J. Org. Chem. 1995, 60, 6046–6056. doi:10.1021/jo00124a013 |
| 15. | Shen, Y.; Xin, Y.; Cen, W.; Huang, Y. Synthesis 1984, 35–37. doi:10.1055/s-1984-30723 |
| 21. | Murray, B. J.; Marsh, T. G. F.; Yufit, D. S.; Fox, M. A.; Harsanyi, A.; Boulton, L. T.; Sandford, G. Eur. J. Org. Chem. 2020, 6236–6244. doi:10.1002/ejoc.202001071 |
| 22. | Ishihara, T.; Yamasaki, Y.; Ando, T. Tetrahedron Lett. 1985, 26, 79–82. doi:10.1016/s0040-4039(00)98472-7 |
| 23. | Sibous, L.; Tipping, A. E. J. Fluorine Chem. 1993, 62, 39–49. doi:10.1016/s0022-1139(00)80079-2 |
| 16. | Bumgardner, C. L.; Bunch, J. E.; Whangbo, M. H. J. Org. Chem. 1986, 51, 4082–4083. doi:10.1021/jo00371a037 |
| 18. | Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/s0022-1139(00)80446-7 |
| 19. | Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h |
| 20. | Wang, T.; Niu, D.; Hoye, T. R. J. Am. Chem. Soc. 2016, 138, 7832–7835. doi:10.1021/jacs.6b03786 |
| 18. | Tajammal, S.; Tipping, A. E. J. Fluorine Chem. 1990, 47, 45–57. doi:10.1016/s0022-1139(00)80446-7 |
| 23. | Sibous, L.; Tipping, A. E. J. Fluorine Chem. 1993, 62, 39–49. doi:10.1016/s0022-1139(00)80079-2 |
| 15. | Shen, Y.; Xin, Y.; Cen, W.; Huang, Y. Synthesis 1984, 35–37. doi:10.1055/s-1984-30723 |
| 17. | Chechulin, P. I.; Filyakova, V. I.; Pashkevich, K. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1989, 38, 189–190. doi:10.1007/bf00953736 |
| 21. | Murray, B. J.; Marsh, T. G. F.; Yufit, D. S.; Fox, M. A.; Harsanyi, A.; Boulton, L. T.; Sandford, G. Eur. J. Org. Chem. 2020, 6236–6244. doi:10.1002/ejoc.202001071 |
| 4. | Mizutani, K.; Yamazaki, T.; Kitazume, T. J. Chem. Soc., Chem. Commun. 1995, 51–52. doi:10.1039/c39950000051 |
| 19. | Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h |
| 29. | Ramasamy, M.; Lin, H.-C.; Kuo, S.-C.; Hsieh, M.-T. Synlett 2019, 30, 356–360. doi:10.1055/s-0037-1611694 |
| 17. | Chechulin, P. I.; Filyakova, V. I.; Pashkevich, K. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1989, 38, 189–190. doi:10.1007/bf00953736 |
| 19. | Brisdon, A. K.; Crossley, I. R. Chem. Commun. 2002, 2420–2421. doi:10.1039/b207979h |
| 24. | Fouli, F. A.; Basyouni, M. N. Acta Chim. Acad. Sci. Hung. 1981, 106, 297–302. |
| 25. | Shankar, R.; Jha, A. K.; Singh, U. S.; Hajela, K. Tetrahedron Lett. 2006, 47, 3077–3079. doi:10.1016/j.tetlet.2006.03.008 |
| 26. | Ramachary, D. B.; Venkaiah, C.; Murali Krishna, P. Chem. Commun. 2012, 48, 2252–2254. doi:10.1039/c2cc17219d |
| 27. | Tong, W.; Li, Q.-Y.; Xu, Y.-L.; Wang, H.-S.; Chen, Y.-Y.; Pan, Y.-M. Adv. Synth. Catal. 2017, 359, 4025–4035. doi:10.1002/adsc.201700830 |
| 28. | Yuan, Y.; Guo, Z.; Mu, Y.; Wang, Y.; Xu, M.; Li, Y. Adv. Synth. Catal. 2020, 362, 1298–1302. doi:10.1002/adsc.201901631 |
| 38. | Saijo, R.; Watanabe, G.; Kurihara, K.-i.; Kawase, M. Heterocycles 2014, 89, 2334–2345. doi:10.3987/com-14-13075 |
| 37. | Romanov, A. R.; Rulev, A. Y.; Ushakov, I. A.; Muzalevskiy, V. M.; Nenajdenko, V. G. Eur. J. Org. Chem. 2017, 4121–4129. doi:10.1002/ejoc.201700727 |
| 35. | Gupta, S.; Melanson, J. A.; Vaillancourt, L.; Nugent, W. A.; Tanoury, G. J.; Schatte, G.; Snieckus, V. Org. Lett. 2018, 20, 3745–3748. doi:10.1021/acs.orglett.8b01300 |
| 36. | Geri, J. B.; Wade Wolfe, M. M.; Szymczak, N. K. Angew. Chem., Int. Ed. 2018, 57, 1381–1385. doi:10.1002/anie.201711316 |
| 37. | Romanov, A. R.; Rulev, A. Y.; Ushakov, I. A.; Muzalevskiy, V. M.; Nenajdenko, V. G. Eur. J. Org. Chem. 2017, 4121–4129. doi:10.1002/ejoc.201700727 |
| 38. | Saijo, R.; Watanabe, G.; Kurihara, K.-i.; Kawase, M. Heterocycles 2014, 89, 2334–2345. doi:10.3987/com-14-13075 |
| 35. | Gupta, S.; Melanson, J. A.; Vaillancourt, L.; Nugent, W. A.; Tanoury, G. J.; Schatte, G.; Snieckus, V. Org. Lett. 2018, 20, 3745–3748. doi:10.1021/acs.orglett.8b01300 |
| 31. | Furuta, T.; Nakayama, M.; Suzuki, H.; Tajimi, H.; Inai, M.; Nukaya, H.; Wakimoto, T.; Kan, T. Org. Lett. 2009, 11, 2233–2236. doi:10.1021/ol900689m |
| 32. | Wang, X.-j.; Zhang, L.; Byrne, D.; Nummy, L.; Weber, D.; Krishnamurthy, D.; Yee, N.; Senanayake, C. H. Org. Lett. 2014, 16, 4090–4093. doi:10.1021/ol501755h |
| 33. | Matsuda, T.; Matsumoto, T. Org. Biomol. Chem. 2016, 14, 5023–5027. doi:10.1039/c6ob00734a |
| 34. | Nenajdenko, V. G., Ed. Fluorine in Heterocyclic Chemistry; Springer: Berlin, Germany, 2014; Vol. 1 and 2. |
| 21. | Murray, B. J.; Marsh, T. G. F.; Yufit, D. S.; Fox, M. A.; Harsanyi, A.; Boulton, L. T.; Sandford, G. Eur. J. Org. Chem. 2020, 6236–6244. doi:10.1002/ejoc.202001071 |
| 30. | Guan, H.-P.; Hu, C.-M. J. Fluorine Chem. 1996, 78, 101–102. doi:10.1016/0022-1139(96)03407-0 |
© 2021 Yamazaki et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)