Abstract
A lipase-catalyzed esterification of lignin model compounds in the ball mill was developed combining the advantages of enzyme catalysis and mechanochemistry. Under the described conditions, the primary aliphatic hydroxy groups present in the substrates were selectively modified by the biocatalyst to afford monoesterified products. Amongst the tested lipases, CALB proved to be the most effective biocatalyst for these transformations. Noteworthy, various acyl donors of different chain lengths were tolerated under the mechanochemical conditions.

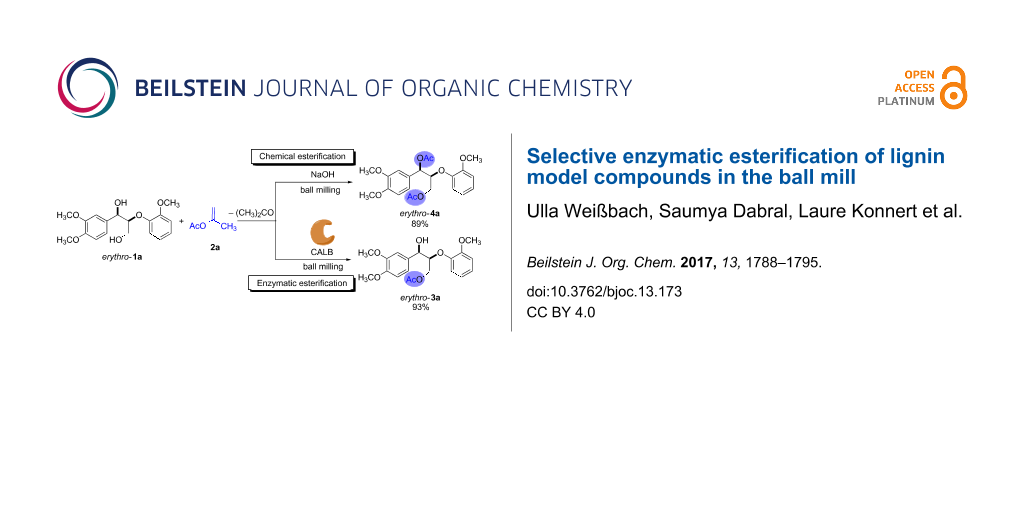
Graphical Abstract
Introduction
Mechanochemical reactions, particularly those carried out by ball milling, have recently attracted attention of a wider scientific community, owing to the many advantages the excellent mixing inside the ball mill can offer [1]. Besides avoiding or minimizing the use of organic solvents as reaction media, chemical transformations by ball milling very often take place more rapidly than their solution-based counterparts. Furthermore, mechanochemical reactions are known to afford products in higher yields with minimal formation of byproducts. In addition to this, mechanochemical activation has resulted in the discovery of otherwise inaccessible products or materials [2,3].
In organic chemistry, amino acids and short peptides are not only known for being stable under automated ball milling conditions during their preparation [4], but also when applied as catalysts to perform stereoselective transformations [5-7]. Encouraged by these facts, we recently investigated the resilience of enzymes under ball milling conditions. The results from these studies have shown that biocatalysts such as cysteine and serine proteases tolerated the milling conditions and catalyzed the mechanoenzymatic peptide and amide bond formation after short milling times (Scheme 1a) [8].
![[1860-5397-13-173-i1]](/bjoc/content/inline/1860-5397-13-173-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Enzymatic reactions under ball milling conditions.
Scheme 1: Enzymatic reactions under ball milling conditions.
Similarly, immobilized lipases (triglycerol acylhydrolases EC 3.1.1.3) such as Amano lipase PS-IM from Burkholderia cepacia immobilized on diatomaceous earth and lipase B from Candida antarctica (expressed in Aspergillus niger) adsorbed on polymethacrylate beads (ca. 400 μm–600 μm in diameter) [9], demonstrated to efficiently mediate the enzymatic kinetic resolution of secondary alcohols under solvent-free conditions in both mixer and planetary ball mills (Scheme 1b) [10]. Interestingly, this latter lipase (a commercial preparation known as Novozyme 345, hereinafter referred as CALB), showed the highest selectivity and could also be recycled by centrifugation and reused with little loss in stereoselectivity after four consecutive cycles [10].
Besides the above stated, one additional advantage of mechanochemistry includes the possibility to overcome solubility restrictions in chemical reactions involving reactants of poor solubility. This characteristic feature of mechanochemistry has proven fundamental while dealing with chemically induced cleavage of biomaterials such as lignin [11,12], cellulose [13-15] or chitin [16]. In regard to lignin chemistry, solution-based lignin depolymerization approaches or new applications of lignocellulose materials [17] often encounter solubility obstacles, forcing the alternate use of highly polar organic solvents, which thereby pose problems during metal-catalyzed transformations in the presence of strongly Lewis basic or donor solvents. In addition to this, miscibility and solubility of lignin samples in apolar matrices during the blending of lignin with polymeric materials is always a challenge.
To mitigate such solubility problems and to facilitate the utilization of lignin for various applications, efforts have been devoted to improve its lipophilicity, for instance through sulfation [18], silylation or esterification [19] of the aliphatic hydroxy and phenolic groups found in lignin. Chemical esterification of lignin [19-21] or its model compounds [22], using acetic anhydride in organic solvents such as DCM or pyridine have previously been reported to be effective in yielding new molecules and materials with higher hydrophobicity. However, controlling the degree of acetylation has not been an easy task, with the esterification process often resulting in a mixture of esters or fully esterified samples.
In this regard, enzymatic esterification thus can be an attractive alternative to specifically address one type of hydroxy groups in the complex lignin structure. This could not only allow a selective control over the degree of hydrophilicity in lignin samples, but would also help tailoring their potential applications. One interesting approach in this field of study involves the modification of lignins by selectively esterifying the primary alcohols present in the biopolymer (Figure 1a), leaving untouched the phenolic and secondary alcohol functionalities, given that these functional groups have been associated with the biopolymer’s antioxidant, antibacterial and sun protection properties [17,23,24]. Motivated by the aforementioned scenario and in line with our research interest on studying the compatibility of biocatalysts and mechanochemical milling, we decided to investigate the enzymatic esterification of lignin model compounds in the ball mill (Scheme 1c). The results of this proof-of-concept study are presented here.
![[1860-5397-13-173-1]](/bjoc/content/figures/1860-5397-13-173-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Molecular representation of lignin. (b) Lignin model compound erythro-1a.
Figure 1: (a) Molecular representation of lignin. (b) Lignin model compound erythro-1a.
Results and Discussion
Due to the high complexity of the lignin structure, which often presents a challenge during the product composition analysis, the use of lignin model compounds to monitor preliminary research advancements is a common practice [25-29]. Thus, for this investigation various dimeric compounds containing the β-O-4 linkage, primary and secondary hydroxy groups as well as several methoxy/phenolic moieties, were used. To begin with, we selected erythro-1a as a model compound to study the enzymatic esterification reactions in the ball mill (Scheme 2).
![[1860-5397-13-173-i2]](/bjoc/content/inline/1860-5397-13-173-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Chemical and enzymatic esterification of erythro-1a with isopropenyl acetate (2a) in the ball mill. Reaction conditions: erythro-1a (50 mg, 0.15 mmol), 2a (0.60 mmol), CALB (30 mg of immobilized enzyme) or NaOH (12 mg, 0.30 mmol), 10 mL ZrO2 milling jar, 6 ZrO2 milling balls (5 mm in diameter).
Scheme 2: Chemical and enzymatic esterification of erythro-1a with isopropenyl acetate (2a) in the ball mill....
Based on our previous work [10], isopropenyl acetate (2a), a non-reversible acyl donor, was chosen as the acetylating agent. Milling a mixture of erythro-1a and 2a for 2 h at 30 Hz did not afford any product, and only the reactants were observed by 1H NMR spectroscopy. Repeating the experiment in the presence of 30 mg of the immobilized lipase CALB led to a total conversion of the erythro-1a after just 1 h. Purification of the product by column chromatography afforded the monoacetylated erythro-3a in 93% yield (Scheme 2; right). To corroborate the role of the biocatalyst in the esterification, the experiment was repeated with sodium hydroxide in place of CALB. Consequently, after 2 h of milling the reaction only generated the diacetylated product erythro-4a (Scheme 2; left). These results reflect the high selectivity of the biocatalyst for primary hydroxy groups. In nature, lipases catalyze the hydrolysis of triglycerides, and are known for acting preferentially at the terminal position of triacylglycerol derivatives [30]. It is worth mentioning here that, even when erythro-1a was milled with an excess of acyl donor for longer time, CALB yielded exclusively the monoacetylated product erythro-3a.
Further screening of the reaction parameters revealed that lowering the amount of acyl donor was doable, although longer milling times were required. Similarly, the effect of the number of milling balls, frequency of milling, reaction time and additives was also investigated (Table S1 in Supporting Information File 1). In addition to this, the catalytic activity of a number of other lipases was studied (Table 1).
Table 1: Influence of various enzymes on the esterification of erythro-1a with isopropenyl acetate (2a) in the ball mill.a
| Entry | Enzyme | 1a:3a (%)b |
|---|---|---|
| 1c | CALB | 0:100 |
| 2 | CALA | 90:10 |
| 3 | PS-IM | 90:10 |
| 4 | lipase A | 100:0 |
aReaction conditions: erythro-1a (50 mg, 0.15 mmol), enzyme (30 mg), 2a (0.6 mmol), 10 mL ZrO2 milling jar, 6 ZrO2 milling balls (5 mm in diameter), milling time 2 h, milling frequency 30 Hz. bDetermined by 1H NMR spectroscopy. cMilling time 1 h. CALB (lipase B from Candida antarctica (expressed in Aspergillus niger) adsorbed on polymethacrylate beads, known also as Novozyme 345); CALA (lipase A from Candida antarctica, immobilized on Immobead 150, recombinant from Aspergillus oryzae); PS-IM (Amano lipase from Burkholderia cepacia immobilized on diatomaceous earth); Lipase A (Amano lipase A from Aspergillus niger).
Amongst the commercially available lipases, CALA (lipase A from Candida antarctica, immobilized on Immobead), immobilized lipase from Burkholderia cepacia (PS-IM) and lipase A from Aspergillus niger were tested. Firstly, hoping to find differences between the two hydrolases derived from Candida antarctica, an experiment using CALA was conducted. Despite CALB and CALA being produced by the same yeast, the latter proved less active at catalyzing the esterification of erythro-1a (Table 1, entry 2). This difference in reactivity between both of the lipases has been documented previously in the literature [31]. Comparably, lipase PS-IM, which has been reported to facilitate the acetylation of secondary β-nitro alcohols [32], and proved to be stable under ball milling conditions [10] exhibited lower catalytic activity than CALB (Table 1, entry 3). However, in both cases the alternative biocatalysts also afforded the monoacetylated dilignol derivative erythro-3a. Finally, lipase A showed no conversion of the substrate, which could be explained by its poor recognition of 1a (Table 1, entry 4). Furthermore, a possible reason could be the reduced stability of the non-immobilized lipase when subjected to mechanochemical stress.
In the preliminary results, isopropenyl acetate (2a) proved highly efficient for the enzyme-catalyzed selective esterification of the model compound erythro-1a, partly due to the non-reversibility of the reaction. However, isopropenyl esters of carboxylic acids are, in general, not readily available. Therefore, in order to find alternative acyl donors for the biocatalyst in the ball mill, a series of acylating agents was screened (Table 2) [33].
Table 2: Screening of acyl donors for the selective monoacetylation of dilignol erythro-1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-173-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | R | Milling time (min) | 1a:3a (%)b |
|---|---|---|---|
| 1 | isopropenyl (2a) | 120 | 0:100 |
| 2 | vinyl (2b) | 120 | 6:94 |
| 3 | phenyl (2c) | 120 | 7:93 |
| 4 | ethyl (2d) | 120 | 70:30 |
| 5 | isopropyl (2e) | 120 | 66:34 |
| 6 | allyl (2f) | 120 | 63:37 |
| 7 | tert-butyl (2g) | 120 | 98:2 |
| 8c | H (2h) | 90 | 100:0 |
aReaction conditions: erythro-1a (50 mg, 0.15 mmol), CALB (30 mg of immobilized enzyme), acyl donor (0.60 mmol), 10 mL ZrO2 milling jar, 6 ZrO2 milling balls (5 mm in diameter). bDetermined by 1H NMR spectroscopy. c10 equiv of 2h were used.
Out of all the acyl donors tested, vinyl acetate (2b) and phenyl acetate (2c) were recognized and transferred by the lipase CALB to the acceptor erythro-1a, affording selectively the product erythro-3a (Table 2, entries 2 and 3). Notably, ethyl acetate (2d), isopropyl acetate (2e) and allyl acetate (2f) were suitable for the enzymatic esterification of erythro-1a as well, although to a lesser extent (Table 2, entries 4–6). Finally, lower and no reactivity was observed using tert-butyl acetate (2g) and acetic acid (2h) as acyl donor, respectively (Table 2, entries 7 and 8).
Having determined the best reaction conditions for the selective enzymatic acetylation of the erythro-1a in the ball mill, the protocol was applied to other β-O-4 model compounds (Scheme 3).
![[1860-5397-13-173-i3]](/bjoc/content/inline/1860-5397-13-173-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: CALB-catalyzed esterification of lignin model compounds in the ball mill.
Scheme 3: CALB-catalyzed esterification of lignin model compounds in the ball mill.
In general, all the substrates 1a–h generated the monoacetylated derivatives, and the reactions occurred regioselectively at the primary hydroxy group of the model compounds. The regioselectivity of the reaction was further confirmed after the milling of isopropenyl acetate (2a) and the monolignol 1i, only containing a benzylic alcohol. After the standard milling time, analysis of the reaction mixture by 1H NMR spectroscopy showed no product formation.
Moreover, under the standard reaction conditions, it was observed that the model compound threo-1b reacted slower in comparison to its diastereomer erythro-1a. After 2 h of milling, the product threo-3b was isolated in 45% yield (Scheme 3). These results highlight the importance of the stereochemistry of the substrates when interacting with the chiral biocatalyst. The reaction of the erythro-diastereoisomer 1c showed comparable reactivity to erythro-1a, and the corresponding monoacetylated product erythro-3c could be isolated in 89% yield (Scheme 3). On the other hand, its diastereomer threo-1d was much less reactive and only trace quantities of threo-3d could be isolated. This difference in reactivity, which follows the trend previously observed for the pair erythro-1a and threo-1b, could have stemmed from matched/mismatched interactions of the diastereomeric diols and the chiral biocatalyst. Similarly, the unsubstituted model compound 1e reacted smoothly to give 3e in 92% yield. Purification of 3e was done by filtration through a pad of celite, since it proved unstable towards standard purification procedures by column chromatography on silica gel.
Noteworthy is the low reactivity of the substrate 1f bearing a phenolic group in its structure. In this case, only trace quantities of the monoacetylated product 3f were observed after 2 h of milling and no esterification was seen to occur in the phenolic group. Initially, it was hypothesized that the presence of a phenolic functionality present in 1f could have inhibited the lipase activity or perhaps caused some degree of denaturation in the enzyme. To test this hypothesis, control experiments using erythro-1a, 2a and CALB in the presence of phenol (1.0 equiv) and phenol derivatives (guaiacol, 3-methoxyphenol, etc.) were carried out. In most cases, the presence of the additives had no negative effect on the performance of CALB (for details see Table S2 in Supporting Information File 1). Only the presence of 2,2’-biphenol seemed to have slowed down the acetylation of erythro-1a. A plausible explanation could be the nature of the 2,2’-biphenol moiety, which could have acted as a ligand interfering with the enzyme.
The resilience of CALB to phenols is in agreement with the high reactivity observed when phenyl acetate (2c), erythro-1a and CALB were milled (Table 2, entry 3), and formation of phenol was expected as a byproduct of the reaction. Hence, the lower reactivity of 1f could have been a consequence of aggregation of the substrate or possible changes in its conformation. This could have reduced the affinity of CALB for 1f compared to the non-phenolic counterparts. Additionally, milling experiments between 1f and 2a, where twice the amount of the enzyme was added in small portions, afforded the same negative result. Finally, the screening of the more hindered lignin model compounds 1g and 1h revealed that these substrates also reacted well in the ball mill, generating the monoacetylated derivatives 3g and 3h in 68% and 82% yield, respectively (Scheme 3).
To test the catalytic efficiency of CALB in the ball mill, we decided to evaluate the performance of the biocatalyst in the esterification of erythro-1a using saturated fatty esters as acyl donors (Scheme 4).
![[1860-5397-13-173-i4]](/bjoc/content/inline/1860-5397-13-173-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Selective esterification of erythro-1a using long-chain vinyl esters as acyl donors in the ball mill.
Scheme 4: Selective esterification of erythro-1a using long-chain vinyl esters as acyl donors in the ball mil...
Because of the excellent affinity of CALB for vinyl acetate (2b, Table 2, entry 2), and due to the commercial availability of vinyl esters in contrast to their isopropenyl analogues [33], vinyl esters 5a–c were chosen. Pleasingly, under the optimized milling reaction conditions (2 h, 30 Hz), erythro-1a and 5a afforded the monoacetylated dilignol derivative 6a in 74% yield (Scheme 4). On the other hand, lengthening the carbon chain of the acyl donor (e.g., 5b and 5c) resulted in slowing down the reaction speed. However, an increase in the milling time from 2 h to 6 h proved beneficial and both the long-chain fatty ester dilignol derivatives 6b,c were isolated in good yields (Scheme 4).
Conclusion
In summary, the lipase-catalyzed esterification of lignin model compounds under mechanochemical conditions was investigated. Experimental parameters such as milling time, milling frequency, presence of additives and different acyl donors were studied in detail. Amongst the various biocatalysts tested, the lipase CALB proved superior in terms of catalytic activity and stability in the ball mill. The high catalytic activity of the enzyme facilitated the monoacetylation of β-O-4 lignin model compounds in good to high yields. Additionally, the biocatalyst exhibited higher preference for the aliphatic primary hydroxy group at the γ-position of the substrates. The enzymatic acetylation protocol was easily transferred to the esterification of the model substrate using long-chain fatty esters. This result is of high importance for introducing, in a controlled manner, various degrees of hydrophobicity to the substrates. This possibility is anticipated to be beneficial for future research initiatives employing lignin samples. Along these lines, it is important to comment on the lower reactivity towards the esterification of the substrate containing a phenolic substituent 1f. Although it is known that lignin samples contain units bearing aromatic phenols, these phenolic fragments are mostly located at the terminal sides of the biopolymer. Therefore, enzymatically addressing the centrally-located primary aliphatic hydroxyl content of lignins is still highly possible. This strategy is expected to allow the preservation of the phenolic and benzylic alcohol contents in modified lignins, in order to keep the antibacterial and antioxidant activities of this biopolymer.
Experimental
All reagents were obtained from commercial suppliers and used without further purification. All lignin model compounds were prepared following the reported procedures [25,34].
Analytical TLC was performed on silica gel plates, and the products were visualized by UV detection (wavelength 254 nm). Ball milling experiments were conducted using a Fritsch Mini-mill PULVERISETTE 23. NMR measurements were performed on Bruker AV 400 or AV 600 instruments. High-resolution mass spectra (HRMS) were measured using a Thermo Scientific LTQ Orbitrap XL with positive ion mode.
Enzymatic acetylation of erythro-1a with CALB in the ball mill
A mixture of erythro-1a (50 mg, 0.15 mmol), acyl donor 2 (0.60 mmol) and CALB (30 mg of the immobilized enzyme) was milled for 2 h to 6 h at 30 Hz in a 10 mL ZrO2 milling jar loaded with 6 ZrO2 milling balls (5 mm in diameter). After the milling was stopped, the reaction mixture was recovered from the milling jar, supported on silica gel and the product was purified by silica column chromatography.
Supporting Information
| Supporting Information File 1: Experimental procedures, optimization tables, characterization data and NMR spectra. | ||
| Format: PDF | Size: 1.9 MB | Download |
Acknowledgements
We thank the RWTH Aachen University for support from the Distinguished Professorship Program and the Cluster of Excellence “Tailor Made Fuels from Biomass” (TMFB), which are funded by the Excellence Initiative of the German federal and state governments. We are also grateful to the European Union (Marie Curie ITN ‘SuBiCat’ PITN-GA-2013-607044, S.D.) for financial support.
References
-
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/C1CS15171A
Return to citation in text: [1] -
Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887
Return to citation in text: [1] -
Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277
Return to citation in text: [1] -
Métro, T.-X.; Colacino, E.; Martinez, J.; Lamaty, F. Amino Acids and Peptides in Ball Milling. Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; The Royal Society of Chemistry: Cambridge, 2015; pp 114–150. doi:10.1039/9781782621980-00114
Return to citation in text: [1] -
Rodríguez, B.; Bruckmann, A.; Bolm, C. Chem. – Eur. J. 2007, 13, 4710–4722. doi:10.1002/chem.200700188
Return to citation in text: [1] -
Hernández, J. G.; Juaristi, E. J. Org. Chem. 2011, 76, 1464–1467. doi:10.1021/jo1022469
Return to citation in text: [1] -
Hernández, J. G.; Avila-Ortiz, C. G.; Juaristi, E. Useful Chemical Activation Alternatives in Solvent-Free Organic Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Molander, G. A.; Knochel, P., Eds.; Elsevier BV, 2014; pp 287–314. doi:10.1016/B978-0-08-097742-3.00935-6
Return to citation in text: [1] -
Hernández, J. G.; Ardila-Fierro, K. J.; Crawford, D.; James, S. L.; Bolm, C. Green Chem. 2017, 19, 2620–2625. doi:10.1039/C7gc00615b
Return to citation in text: [1] -
Wiemann, L. O.; Nieguth, R.; Eckstein, M.; Naumann, M.; Thum, O.; Ansorge-Schumacher, M. B. ChemCatChem 2009, 1, 455–462. doi:10.1002/cctc.200900199
Return to citation in text: [1] -
Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455
Return to citation in text: [1] [2] [3] [4] -
Kleine, T.; Buendia, J.; Bolm, C. Green Chem. 2013, 15, 160–166. doi:10.1039/C2GC36456E
Return to citation in text: [1] -
Calvaruso, G.; Clough, M. T.; Rinaldi, R. Green Chem. 2017, 19, 2803–2811. doi:10.1039/c6gc03191a
Return to citation in text: [1] -
Hick, S. M.; Griebel, C.; Restrepo, D. T.; Truitt, J. H.; Buker, E. J.; Bylda, C.; Blair, R. G. Green Chem. 2010, 12, 468–474. doi:10.1039/B923079C
Return to citation in text: [1] -
Boissou, F.; Sayoud, N.; De Oliveira Vigier, K.; Barakat, A.; Marinkovic, S.; Estrine, B.; Jérôme, F. ChemSusChem 2015, 8, 3263–3269. doi:10.1002/cssc.201500700
Return to citation in text: [1] -
Rechulski, M. D. K.; Käldström, M.; Richter, U.; Schüth, F.; Rinaldi, R. Ind. Eng. Chem. Res. 2015, 54, 4581–4592. doi:10.1021/acs.iecr.5b00224
Return to citation in text: [1] -
Chen, X.; Yang, H.; Zhong, Z.; Yan, N. Green Chem. 2017, 19, 2783–2792. doi:10.1039/c7gc00089h
Return to citation in text: [1] -
Qian, Y.; Qiu, X.; Zhu, S. ACS Sustainable Chem. Eng. 2016, 4, 4029–4035. doi:10.1021/acssuschemeng.6b00934
Return to citation in text: [1] [2] -
Prinsen, P.; Narani, A.; Hartog, A. F.; Wever, R.; Rothenberg, G. ChemSusChem 2017, 10, 2267–2273. doi:10.1002/cssc.201700376
Return to citation in text: [1] -
Buono, P.; Duval, A.; Verge, P.; Averous, L.; Habibi, Y. ACS Sustainable Chem. Eng. 2016, 4, 5212–5222. doi:10.1021/acssuschemeng.6b00903
Return to citation in text: [1] [2] -
Zhao, X.; Huang, A.; Zhang, Y.; Yang, M.; Chen, D.; Huang, K.; Hu, H.; Huang, A.; Qin, X.; Feng, Z. J. Appl. Polym. Sci. 2017, 134, 44276–44289. doi:10.1002/app.44276
Return to citation in text: [1] -
Hulin, L.; Husson, E.; Bonnet, J.-P.; Stevanovic, T.; Sarazin, C. Molecules 2015, 20, 16334–16353. doi:10.3390/molecules200916334
Return to citation in text: [1] -
Lohr, T. L.; Li, Z.; Marks, T. J. ACS Catal. 2015, 5, 7004–7007. doi:10.1021/acscatal.5b01972
Return to citation in text: [1] -
Pan, X.; Kadla, J. F.; Ehara, K.; Gilkes, N.; Saddler, J. N. J. Agric. Food Chem. 2006, 54, 5806–5813. doi:10.1021/jf0605392
Return to citation in text: [1] -
Qian, Y.; Qiu, X.; Zhu, S. Green Chem. 2015, 17, 320–324. doi:10.1039/c4gc01333f
Return to citation in text: [1] -
Buendia, J.; Mottweiler, J.; Bolm, C. Chem. – Eur. J. 2011, 17, 13877–13882. doi:10.1002/chem.201101579
Return to citation in text: [1] [2] -
Dabral, S.; Mottweiler, J.; Rinesch, T.; Bolm, C. Green Chem. 2015, 17, 4908–4912. doi:10.1039/C5GC00186B
Return to citation in text: [1] -
Mottweiler, J.; Puche, M.; Räuber, C.; Schmidt, T.; Concepción, P.; Corma, A.; Bolm, C. ChemSusChem 2015, 8, 2106–2113. doi:10.1002/cssc.201500131
Return to citation in text: [1] -
Mottweiler, J.; Rinesch, T.; Besson, C.; Buendia, J.; Bolm, C. Green Chem. 2015, 17, 5001–5008. doi:10.1039/C5GC01306B
Return to citation in text: [1] -
Dabral, S.; Hernández, J. G.; Kamer, P. C. J.; Bolm, C. ChemSusChem 2017, 10, 2707–2713. doi:10.1002/cssc.201700703
Return to citation in text: [1] -
Stergiou, P.-Y.; Foukis, A.; Filippou, M.; Koukouritaki, M.; Parapouli, M.; Theodorou, L. G.; Hatziloukas, E.; Afendra, A.; Pandey, A.; Papamichael, E. M. Biotechnol. Adv. 2013, 31, 1846–1859. doi:10.1016/j.biotechadv.2013.08.006
Return to citation in text: [1] -
Kirk, O.; Christensen, M. W. Org. Process Res. Dev. 2002, 6, 446–451. doi:10.1021/op0200165
Return to citation in text: [1] -
Xu, F.; Wang, J.; Liu, B.; Wu, Q.; Lin, X. Green Chem. 2011, 13, 2359–2361. doi:10.1039/C1GC15417F
Return to citation in text: [1] -
Paravidino, M.; Hanefeld, U. Green Chem. 2011, 13, 2651–2657. doi:10.1039/c1gc15576h
Return to citation in text: [1] [2] -
Rahimi, A.; Azarpira, A.; Kim, H.; Ralph, J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 6415–6418. doi:10.1021/ja401793n
Return to citation in text: [1]
| 1. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/C1CS15171A |
| 8. | Hernández, J. G.; Ardila-Fierro, K. J.; Crawford, D.; James, S. L.; Bolm, C. Green Chem. 2017, 19, 2620–2625. doi:10.1039/C7gc00615b |
| 19. | Buono, P.; Duval, A.; Verge, P.; Averous, L.; Habibi, Y. ACS Sustainable Chem. Eng. 2016, 4, 5212–5222. doi:10.1021/acssuschemeng.6b00903 |
| 20. | Zhao, X.; Huang, A.; Zhang, Y.; Yang, M.; Chen, D.; Huang, K.; Hu, H.; Huang, A.; Qin, X.; Feng, Z. J. Appl. Polym. Sci. 2017, 134, 44276–44289. doi:10.1002/app.44276 |
| 21. | Hulin, L.; Husson, E.; Bonnet, J.-P.; Stevanovic, T.; Sarazin, C. Molecules 2015, 20, 16334–16353. doi:10.3390/molecules200916334 |
| 5. | Rodríguez, B.; Bruckmann, A.; Bolm, C. Chem. – Eur. J. 2007, 13, 4710–4722. doi:10.1002/chem.200700188 |
| 6. | Hernández, J. G.; Juaristi, E. J. Org. Chem. 2011, 76, 1464–1467. doi:10.1021/jo1022469 |
| 7. | Hernández, J. G.; Avila-Ortiz, C. G.; Juaristi, E. Useful Chemical Activation Alternatives in Solvent-Free Organic Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Molander, G. A.; Knochel, P., Eds.; Elsevier BV, 2014; pp 287–314. doi:10.1016/B978-0-08-097742-3.00935-6 |
| 22. | Lohr, T. L.; Li, Z.; Marks, T. J. ACS Catal. 2015, 5, 7004–7007. doi:10.1021/acscatal.5b01972 |
| 4. | Métro, T.-X.; Colacino, E.; Martinez, J.; Lamaty, F. Amino Acids and Peptides in Ball Milling. Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; The Royal Society of Chemistry: Cambridge, 2015; pp 114–150. doi:10.1039/9781782621980-00114 |
| 18. | Prinsen, P.; Narani, A.; Hartog, A. F.; Wever, R.; Rothenberg, G. ChemSusChem 2017, 10, 2267–2273. doi:10.1002/cssc.201700376 |
| 2. | Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887 |
| 3. | Do, J.-L.; Friščić, T. ACS Cent. Sci. 2017, 3, 13–19. doi:10.1021/acscentsci.6b00277 |
| 19. | Buono, P.; Duval, A.; Verge, P.; Averous, L.; Habibi, Y. ACS Sustainable Chem. Eng. 2016, 4, 5212–5222. doi:10.1021/acssuschemeng.6b00903 |
| 11. | Kleine, T.; Buendia, J.; Bolm, C. Green Chem. 2013, 15, 160–166. doi:10.1039/C2GC36456E |
| 12. | Calvaruso, G.; Clough, M. T.; Rinaldi, R. Green Chem. 2017, 19, 2803–2811. doi:10.1039/c6gc03191a |
| 16. | Chen, X.; Yang, H.; Zhong, Z.; Yan, N. Green Chem. 2017, 19, 2783–2792. doi:10.1039/c7gc00089h |
| 10. | Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455 |
| 17. | Qian, Y.; Qiu, X.; Zhu, S. ACS Sustainable Chem. Eng. 2016, 4, 4029–4035. doi:10.1021/acssuschemeng.6b00934 |
| 10. | Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455 |
| 9. | Wiemann, L. O.; Nieguth, R.; Eckstein, M.; Naumann, M.; Thum, O.; Ansorge-Schumacher, M. B. ChemCatChem 2009, 1, 455–462. doi:10.1002/cctc.200900199 |
| 13. | Hick, S. M.; Griebel, C.; Restrepo, D. T.; Truitt, J. H.; Buker, E. J.; Bylda, C.; Blair, R. G. Green Chem. 2010, 12, 468–474. doi:10.1039/B923079C |
| 14. | Boissou, F.; Sayoud, N.; De Oliveira Vigier, K.; Barakat, A.; Marinkovic, S.; Estrine, B.; Jérôme, F. ChemSusChem 2015, 8, 3263–3269. doi:10.1002/cssc.201500700 |
| 15. | Rechulski, M. D. K.; Käldström, M.; Richter, U.; Schüth, F.; Rinaldi, R. Ind. Eng. Chem. Res. 2015, 54, 4581–4592. doi:10.1021/acs.iecr.5b00224 |
| 10. | Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455 |
| 17. | Qian, Y.; Qiu, X.; Zhu, S. ACS Sustainable Chem. Eng. 2016, 4, 4029–4035. doi:10.1021/acssuschemeng.6b00934 |
| 23. | Pan, X.; Kadla, J. F.; Ehara, K.; Gilkes, N.; Saddler, J. N. J. Agric. Food Chem. 2006, 54, 5806–5813. doi:10.1021/jf0605392 |
| 24. | Qian, Y.; Qiu, X.; Zhu, S. Green Chem. 2015, 17, 320–324. doi:10.1039/c4gc01333f |
| 25. | Buendia, J.; Mottweiler, J.; Bolm, C. Chem. – Eur. J. 2011, 17, 13877–13882. doi:10.1002/chem.201101579 |
| 26. | Dabral, S.; Mottweiler, J.; Rinesch, T.; Bolm, C. Green Chem. 2015, 17, 4908–4912. doi:10.1039/C5GC00186B |
| 27. | Mottweiler, J.; Puche, M.; Räuber, C.; Schmidt, T.; Concepción, P.; Corma, A.; Bolm, C. ChemSusChem 2015, 8, 2106–2113. doi:10.1002/cssc.201500131 |
| 28. | Mottweiler, J.; Rinesch, T.; Besson, C.; Buendia, J.; Bolm, C. Green Chem. 2015, 17, 5001–5008. doi:10.1039/C5GC01306B |
| 29. | Dabral, S.; Hernández, J. G.; Kamer, P. C. J.; Bolm, C. ChemSusChem 2017, 10, 2707–2713. doi:10.1002/cssc.201700703 |
| 25. | Buendia, J.; Mottweiler, J.; Bolm, C. Chem. – Eur. J. 2011, 17, 13877–13882. doi:10.1002/chem.201101579 |
| 34. | Rahimi, A.; Azarpira, A.; Kim, H.; Ralph, J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 6415–6418. doi:10.1021/ja401793n |
| 33. | Paravidino, M.; Hanefeld, U. Green Chem. 2011, 13, 2651–2657. doi:10.1039/c1gc15576h |
| 33. | Paravidino, M.; Hanefeld, U. Green Chem. 2011, 13, 2651–2657. doi:10.1039/c1gc15576h |
| 32. | Xu, F.; Wang, J.; Liu, B.; Wu, Q.; Lin, X. Green Chem. 2011, 13, 2359–2361. doi:10.1039/C1GC15417F |
| 10. | Hernández, J. G.; Frings, M.; Bolm, C. ChemCatChem 2016, 8, 1769–1772. doi:10.1002/cctc.201600455 |
| 30. | Stergiou, P.-Y.; Foukis, A.; Filippou, M.; Koukouritaki, M.; Parapouli, M.; Theodorou, L. G.; Hatziloukas, E.; Afendra, A.; Pandey, A.; Papamichael, E. M. Biotechnol. Adv. 2013, 31, 1846–1859. doi:10.1016/j.biotechadv.2013.08.006 |
| 31. | Kirk, O.; Christensen, M. W. Org. Process Res. Dev. 2002, 6, 446–451. doi:10.1021/op0200165 |
© 2017 Weißbach et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)