Abstract
Thiazolium carbene-catalyzed reactions of 1,2-diketones and 1,2,3-triketones with enones and ynones have been investigated. The diketones gave α,β-double acylation products via unique Breslow intermediates isolable as acid salts, whereas the triketones formed stable adducts with the NHC instead of the coupling products.

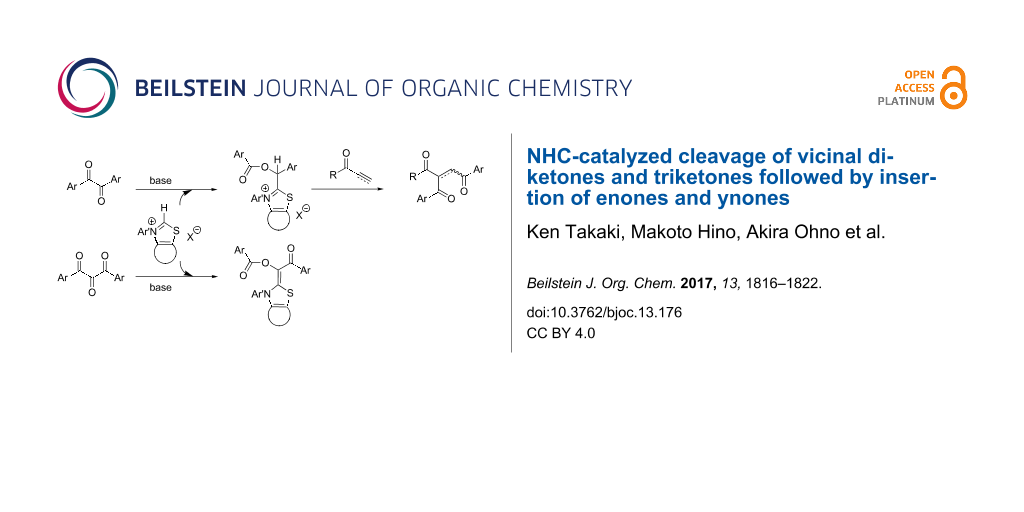
Graphical Abstract
Introduction
N-Heterocyclic carbenes (NHCs) have been indispensable catalysts for organic synthesis, particularly for umpolung of various functional groups [1-9]. In the Stetter reaction, NHCs convert aldehydes to nucleophilic species, which react with activated alkenes to yield hydroacylation products [10-14]. When the carbonyl compounds I other than aldehyde behave similarly, functionalized 1,4-diketones IV would be produced (Scheme 1). Previously, we reported that benzils I (G = C(O)R1) reacted with enones III in the presence of thiazolium carbene catalysts to give double acylation products IV in good yields [15]. If enones can be replaced by ynones III in the reaction with benzils, alkenes IV having three acyl moieties would be formed directly. Related products were recently obtained by the dimsyl anion-promoted double acylation of enones with benzils, followed by dehydrogenation of the resulting alkanes in one pot [16]. Moreover, if the reaction of cyclic 1,2-diketone I (G = C(O)R1) with activated alkenes may take place similarly, this reaction could be utilized as a ring-enlargement procedure to afford cyclic 1,4-diketones IV.
![[1860-5397-13-176-i1]](/bjoc/content/inline/1860-5397-13-176-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
With respect to the active species in the Stetter reaction, aminoenols II (G = H, Breslow intermediates) had been postulated as true nucleophiles for a long time and those generated from imidazolinium NHC were recently isolated in pure form [17,18]. In the reaction of benzils with thiazolium NHC, aminoenol esters II (G = C(O)R1) could be formed similarly, but this active species were found to exist as isolable acid salts unexpectedly. Moreover, the reactivity of 1,2,3-triketones was also investigated in comparison with that of 1,2-diketones. We would like to report herein these results.
Results and Discussion
The reaction of benzil (1a) with various ynones 2 was carried out by use of thiazolium salt 3 under similar conditions to that with enones (Table 1) [15]. In the reaction of 1a with 1-phenylprop-2-yn-1-one (2a), tribenzoylethylene (4aa) was formed in 64% yield (Table 1, entry 1). Fortunately possible propargylic alcohols by cross-benzoin reaction were not detected [19]. The other products of 4 were obtained as inseparable mixtures of E- and Z-isomer, except for 4ae and 4ar, but their stereochemistry could not be determined by NMR. Only the major isomer of 4ai crystallized from the mixture and thus Z-stereochemistry of the two aroyl groups (ClC6H4CO and PhCO) was confirmed by X-ray analysis (Figure 1) [20]. Electron-donating substituents of ynones 2 gave slightly better yields than electron-withdrawing ones. The yields and isomer ratios were also affected by the position of the substituents on the aroyl groups, i.e., ortho-substituents decreased the product yields and increased the isomer ratios (Table 1, entries 2–4 and 7–9). Mesityl and aliphatic ynones 2e and 2r gave the single isomers 4ae and 4ar in low yields (Table 1, entries 5 and 18).
Table 1: Reaction of benzil (1a) with ynones 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-176-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Ynone 2 | R | Product 4 | Yield (%)b | Isomer ratioc |
|---|---|---|---|---|---|
| 1 | 2a | Ph | 4aa | 64 | – |
| 2 | 2b | 2-MeC6H4 | 4ab | 54 | 74:26 |
| 3 | 2c | 3-MeC6H4 | 4ac | 64 | 52:48 |
| 4 | 2d | 4-MeC6H4 | 4ad | 63 | 52:48 |
| 5 | 2e | 2,4,6-Me3C6H2 | 4ae | 32 | –d |
| 6 | 2f | 4-MeOC6H4 | 4af | 63 | 53:47 |
| 7 | 2g | 2-ClC6H4 | 4ag | 31 | 80:20 |
| 8 | 2h | 3-ClC6H4 | 4ah | 49 | 61:39 |
| 9 | 2i | 4-ClC6H4 | 4ai | 53 | 60:40e |
| 10 | 2j | 4-BrC6H4 | 4aj | 57 | 59:41 |
| 11 | 2k | 4-MeO2CC6H4 | 4ak | 44 | 59:41 |
| 12 | 2l | 4-NCC6H4 | 4al | 20 | 71:29 |
| 13 | 2m | 2-furyl | 4am | 41 | 65:35 |
| 14 | 2n | 2-thienyl | 4an | 54 | 67:33 |
| 15 | 2o | 3-thienyl | 4ao | 61 | 67:33 |
| 16 | 2p | 1-naphthyl | 4ap | 52 | 69:31 |
| 17 | 2q | 2-naphthyl | 4aq | 50 | 53:47 |
| 18 | 2r | Ph(CH2)2 | 4ar | 13 | –d |
aEquimolar amounts of 2 were used. bIsolated yields. cEstimated by NMR. dOne stereoisomer was formed. eE/Z = 40:60 determined by X-ray analysis.
![[1860-5397-13-176-1]](/bjoc/content/figures/1860-5397-13-176-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Then, the substituent effect of benzils 1 was investigated (Table 2). Electron-donating groups decreased the reaction efficiency slightly. The yields were improved by catalytic amounts of MgCl2 as reported previously [15], whereas the isomer ratios of 4 were nearly unchanged irrespective of the Lewis acid. When a mixture of benzils 1b and 1c (0.5 equiv each) was treated with 2a (1.0 equiv) under similar conditions, the products 4ba and 4ca were formed in 52% and 20% yields, respectively. However, cross products having three different aroyl groups were not detected at all. Thus, the present reaction was proved to take place by an intramolecular process.
Table 2: Reaction of substituted benzils 1 with ynone 2aa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-176-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Benzil 1 | R | Product 4 | Yield (%)b,c | Isomer ratio |
|---|---|---|---|---|---|
| 1 | 1b | Me | 4ba | 48 (76) | 50:50 |
| 2 | 1c | OMe | 4ca | 16 (25) | 50:50 |
| 3 | 1d | Cl | 4da | 56 (74) | 53:47 |
| 4 | 1e | Br | 4ea | 42 (63) | 58:42 |
aEquimolar amounts of 2 were used. bIsolated yields. cYields in parentheses were observed in the presence of MgCl2 (20 mol %).
A reaction mechanism is proposed in Scheme 2, which is similar to that with enones [15]. The monoacylated Breslow intermediate C is formed by addition of thiazolium NHC A to benzil (1a), followed by migration of the benzoyl group. Then, this nucleophilic species C reacts with ynones 2 to generate the intermediate D. The second migration of the benzoyl group to the α-position of 2 and simultaneous elimination of A affords product 4.
![[1860-5397-13-176-i2]](/bjoc/content/inline/1860-5397-13-176-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
According to the intramolecular mechanism shown in Scheme 2, the two benzoyl groups of 1a should be delivered to the ynones 2 from the same side to yield E-products 4 exclusively. However, 4 were obtained as a mixture of E- and Z-isomer in the most cases. Since pure Z-isomer of the product 4ai was exceptionally isolated, its isomerization was tested in order to explain the stereochemistry (Scheme 3). When the Z-4ai was dissolved in CDCl3 and monitored by NMR, the ratio of E/Z changed slowly to 38:62 after 21 days. In contrast, the original ratio of 40:60 was quickly attained in the presence of the NHC catalyst A.
![[1860-5397-13-176-i3]](/bjoc/content/inline/1860-5397-13-176-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Isomerization of the stereochemistry of 4ai.
Scheme 3: Isomerization of the stereochemistry of 4ai.
We demonstrated previously the reaction of cyclohexane-1,2-dione (5a) with phenyl vinyl ketone (6a) to afford 2-benzoylcyclooctane-1,4-dione (7a) in 27% yield [15]. Since this reaction would be a unique ring enlargement by two carbons, an improvement of the reaction conditions has been tested. Although the yield increased to 45% by the use of diisopropylamine instead of DIPEA, the reaction efficiency was much lower than that of benzil (1a) (Scheme 4). The only isolated by-product in the reaction of 5a was bicyclo[3.2.1]octanone 8 (ca. 10%), which was formed by competitive tandem Michael–aldol reaction [21]. Cycloheptane-1,2-dione (5b) and cyclododecane-1,2-dione (5c) gave the 9- and 14-membered 1,4-diketones 7b and 7c at elevated temperature, respectively.
![[1860-5397-13-176-i4]](/bjoc/content/inline/1860-5397-13-176-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of cycloalkane-1,2-diones with phenyl vinyl ketone (6a).
Scheme 4: Reaction of cycloalkane-1,2-diones with phenyl vinyl ketone (6a).
Next, we investigated the NHC-catalyzed reaction of 1,2,3-triketone 9 with enone 6a. Since the central carbonyl group of 9 could be more electrophilic than the others at 1- and 3-positions, nucleophilic addition of NHC followed by migration of one neighboring acyl group to the central carbonyl oxygen would generate bisacylated Breslow intermediate 10 (Scheme 5). If this species behaves in a similar manner as the monoacylated intermediate C derived from 1,2-diketone 1, its reaction with enone 6a would be expected to yield tetraketone 11. However, when triketone 9 was treated with equimolar amounts of 6a in the presence of 3 and DIPEA (20 mol %, each), the predicted intermediate 10 was obtained in 19% yield based on 9, but any coupling products of 9 with 6a such as 11 were not formed. The yield of 10 increased to 83% in the equimolar reaction of 9 with 3. In addition, the product 10 was recovered unchanged after treatment with 6a irrespective of DIPEA. The nucleophilicity of 10 may be canceled by the two benzoyl groups. The product 10 was fairly stable to air and moisture, and thus E-structure was confirmed by X-ray analysis as shown in Figure 2 [20].
![[1860-5397-13-176-i5]](/bjoc/content/inline/1860-5397-13-176-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation and reactivity of the bisacylated Breslow intermediate 10.
Scheme 5: Preparation and reactivity of the bisacylated Breslow intermediate 10.
![[1860-5397-13-176-2]](/bjoc/content/figures/1860-5397-13-176-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Isolation of 10 prompted us to get the monoacylated Breslow intermediate C also as a stable compound. Treatment of benzil 1d with equimolar amounts of 3 and DIPEA at room temperature, followed by non-aqueous work-up and column chromatography gave crystalline product 12 in 92% yield (Scheme 6). Although 12 did not provide single crystals suitable for X-ray analysis, its structure was determined by NMR and elemental analysis. 1H NMR spectra of 12 showed eleven protons in the aromatic region, i.e., two ClC6H4, Me3C6H2, and one particular proton at δ 6.68. In 13C NMR spectra, one unknown signal appeared in the neighborhood of the ester carbon (δ 163.1 and 167.1). Moreover, an unusual signal appeared at δ 71.5. These data indicated definitely that the isolated product 12 was different from the expected intermediate C. Instead, the iminium salt 12, that is, the HClO4 salt of C, could account for all unusual signals. Furthermore, elemental analysis of 12 agreed very closely with its theoretical value and deviated much from that of C.
![[1860-5397-13-176-i6]](/bjoc/content/inline/1860-5397-13-176-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Preparation of the iminium salt 12 and its reactivity.
Scheme 6: Preparation of the iminium salt 12 and its reactivity.
After identification of the product 12, we noticed that Massi and his group obtained the similar adducts during their study on the multicomponent reaction of thiazolium carbenes, benzils and water to yield 1,4-thiazin-3-ones [22]. Although the initial process producing the salt 12 was similar, the total mode of the two reactions was quite different, that is, stoichiometric vs catalytic reaction with respect to the thiazolium carbenes. In the Massi reaction, monoacylated Breslow intermediate C was readily hydrolyzed by hydroxide, whereas Stetter reaction took place exclusively in our reaction. The difference would be caused by the reaction conditions, particularly by the solvent system.
Since the product 12 was obtained unexpectedly, its reactivity and role in the catalytic cycle were investigated. The stoichiometric reaction of 12 with phenyl vinyl ketone (6a) in the presence of DIPEA gave the product 13 in 94% yield, while no product was formed without the base. Moreover, the reaction of benzil 1d with enone 6a was catalyzed by 12 to give the product 13 in good yield, which are comparable results to the use of 3 [15]. These results suggested that compound 12 played a resting state of the monoacylated Breslow intermediate C as shown in Scheme 7. Accordingly, when enone 6a was present in the mixture, facile regeneration of C and its reaction with 6a took place predominantly. It seems surprising that compound 12 could be quantitatively generated from C and HClO4 regardless of equimolar amounts of DIPEA, while analogous intermediates derived from imidazole NHC were protonated by the acid alone [23].
![[1860-5397-13-176-i7]](/bjoc/content/inline/1860-5397-13-176-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Resting state of the monoacylated Breslow intermediate C.
Scheme 7: Resting state of the monoacylated Breslow intermediate C.
Conclusion
We have demonstrated the thiazolium NHC-catalyzed reaction of benzils with ynones to give triacylated alkenes in fairly good yields. When this reaction was applied to aliphatic cyclic diketones with enones, two carbon ring-enlarged products were formed, though in low yields. Moreover, aromatic 1,2,3-triketones reacted with NHCs to afford bisacylated Breslow intermediates in high yield. However, their nucleophilicity was so weak that they were recovered unchanged in the reaction with enones. It was also found that the monoacylated Breslow intermediates changed reversibly to the resting state of acid salts.
Supporting Information
| Supporting Information File 1: Experimental procedure, characterization data and copies of 1H and 13C NMR spectra of the products. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307–9387. doi:10.1021/acs.chemrev.5b00060
Return to citation in text: [1] -
Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384
Return to citation in text: [1] -
Izquierdo, J.; Hutson, G. E.; Cohen, D. T.; Scheidt, K. A. Angew. Chem., Int. Ed. 2012, 51, 11686–11698. doi:10.1002/anie.201203704
Return to citation in text: [1] -
Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415
Return to citation in text: [1] -
Mahatthananchai, J.; Bode, J. W. Chem. Sci. 2012, 3, 192–197. doi:10.1039/C1SC00397F
Return to citation in text: [1] -
Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511–3522. doi:10.1039/c2cs15333e
Return to citation in text: [1] -
Nair, V.; Menon, R. S.; Biju, A. T.; Sinu, C. R.; Paul, R. R.; Jose, A.; Streekumar, V. Chem. Soc. Rev. 2011, 40, 5336–5346. doi:10.1039/c1cs15139h
Return to citation in text: [1] -
Biju, A. T.; Kuhl, N.; Glorius, F. Acc. Chem. Res. 2011, 44, 1182–1195. doi:10.1021/ar2000716
Return to citation in text: [1] -
Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z
Return to citation in text: [1] -
Schedler, M.; Wang, D.-S.; Glorius, F. Angew. Chem., Int. Ed. 2013, 52, 2585–2589. doi:10.1002/anie.201209291
Return to citation in text: [1] -
Christmann, M. Angew. Chem., Int. Ed. 2005, 44, 2632–2634. doi:10.1002/anie.200500761
Return to citation in text: [1] -
Stetter, H.; Kuhlmann, H. Org. React. 1991, 40, 407–496. doi:10.1002/0471264180.or040.04
Return to citation in text: [1] -
Stetter, H. Angew. Chem., Int. Ed. Engl. 1976, 15, 639–647. doi:10.1002/anie.197606391
Return to citation in text: [1] -
Stetter, H.; Schreckenberg, M. Angew. Chem., Int. Ed. Engl. 1973, 12, 81. doi:10.1002/anie.197300811
Return to citation in text: [1] -
Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ragno, D.; Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A. J. Org. Chem. 2015, 80, 1937–1945. doi:10.1021/jo502582e
Return to citation in text: [1] -
Berkessel, A.; Yatham, V. R.; Elfert, S.; Neudörft, J.-M. Angew. Chem., Int. Ed. 2013, 52, 11158–11162. doi:10.1002/anie.201303107
Return to citation in text: [1] -
Berkessel, A.; Elfert, S.; Yatham, V. R.; Neudörfl, J.-M.; Schlörer, N. E.; Teles, J. H. Angew. Chem., Int. Ed. 2012, 51, 12370–12374. doi:10.1002/anie.201205878
Return to citation in text: [1] -
Sánchez-Díez, E.; Fernández, M.; Uria, U.; Reyes, E.; Carrillo, L.; Vicario, J. L. Chem. – Eur. J. 2015, 21, 8384–8388. doi:10.1002/chem.201501044
Return to citation in text: [1] -
Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1506357 (4ai) and CCDC 1506358 (10). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0) 1223 336033 or e-mail: deposit@ccdc.cam.ac.uk].
Return to citation in text: [1] [2] -
Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A.; Pacifico, S. Org. Biomol. Chem. 2011, 9, 8437–8444. doi:10.1039/c1ob06480k
Return to citation in text: [1] -
Bertorasi, V.; Bortolini, O.; Donvito, A.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A.; Pacifico, S. Org. Biomol. Chem. 2012, 10, 6579–6586. doi:10.1039/c2ob25928a
Return to citation in text: [1] -
Pignataro, L.; Papalia, T.; Slawin, A. M. Z.; Goldup, S. M. Org. Lett. 2009, 11, 1643–1646. doi:10.1021/ol900257t
Return to citation in text: [1]
| 1. | Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307–9387. doi:10.1021/acs.chemrev.5b00060 |
| 2. | Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384 |
| 3. | Izquierdo, J.; Hutson, G. E.; Cohen, D. T.; Scheidt, K. A. Angew. Chem., Int. Ed. 2012, 51, 11686–11698. doi:10.1002/anie.201203704 |
| 4. | Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415 |
| 5. | Mahatthananchai, J.; Bode, J. W. Chem. Sci. 2012, 3, 192–197. doi:10.1039/C1SC00397F |
| 6. | Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511–3522. doi:10.1039/c2cs15333e |
| 7. | Nair, V.; Menon, R. S.; Biju, A. T.; Sinu, C. R.; Paul, R. R.; Jose, A.; Streekumar, V. Chem. Soc. Rev. 2011, 40, 5336–5346. doi:10.1039/c1cs15139h |
| 8. | Biju, A. T.; Kuhl, N.; Glorius, F. Acc. Chem. Res. 2011, 44, 1182–1195. doi:10.1021/ar2000716 |
| 9. | Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z |
| 17. | Berkessel, A.; Yatham, V. R.; Elfert, S.; Neudörft, J.-M. Angew. Chem., Int. Ed. 2013, 52, 11158–11162. doi:10.1002/anie.201303107 |
| 18. | Berkessel, A.; Elfert, S.; Yatham, V. R.; Neudörfl, J.-M.; Schlörer, N. E.; Teles, J. H. Angew. Chem., Int. Ed. 2012, 51, 12370–12374. doi:10.1002/anie.201205878 |
| 15. | Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a |
| 16. | Ragno, D.; Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A. J. Org. Chem. 2015, 80, 1937–1945. doi:10.1021/jo502582e |
| 23. | Pignataro, L.; Papalia, T.; Slawin, A. M. Z.; Goldup, S. M. Org. Lett. 2009, 11, 1643–1646. doi:10.1021/ol900257t |
| 15. | Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a |
| 20. | Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1506357 (4ai) and CCDC 1506358 (10). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0) 1223 336033 or e-mail: deposit@ccdc.cam.ac.uk]. |
| 10. | Schedler, M.; Wang, D.-S.; Glorius, F. Angew. Chem., Int. Ed. 2013, 52, 2585–2589. doi:10.1002/anie.201209291 |
| 11. | Christmann, M. Angew. Chem., Int. Ed. 2005, 44, 2632–2634. doi:10.1002/anie.200500761 |
| 12. | Stetter, H.; Kuhlmann, H. Org. React. 1991, 40, 407–496. doi:10.1002/0471264180.or040.04 |
| 13. | Stetter, H. Angew. Chem., Int. Ed. Engl. 1976, 15, 639–647. doi:10.1002/anie.197606391 |
| 14. | Stetter, H.; Schreckenberg, M. Angew. Chem., Int. Ed. Engl. 1973, 12, 81. doi:10.1002/anie.197300811 |
| 22. | Bertorasi, V.; Bortolini, O.; Donvito, A.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A.; Pacifico, S. Org. Biomol. Chem. 2012, 10, 6579–6586. doi:10.1039/c2ob25928a |
| 15. | Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a |
| 15. | Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a |
| 20. | Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1506357 (4ai) and CCDC 1506358 (10). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0) 1223 336033 or e-mail: deposit@ccdc.cam.ac.uk]. |
| 21. | Bortolini, O.; Fantin, G.; Fogagnolo, M.; Giovannini, P. P.; Massi, A.; Pacifico, S. Org. Biomol. Chem. 2011, 9, 8437–8444. doi:10.1039/c1ob06480k |
| 19. | Sánchez-Díez, E.; Fernández, M.; Uria, U.; Reyes, E.; Carrillo, L.; Vicario, J. L. Chem. – Eur. J. 2015, 21, 8384–8388. doi:10.1002/chem.201501044 |
| 15. | Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a |
| 15. | Takaki, K.; Ohno, A.; Hino, M.; Shitaoka, T.; Komeyama, K.; Yoshida, H. Chem. Commun. 2014, 50, 12285–12288. doi:10.1039/c4cc05436a |
© 2017 Takaki et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)