Abstract
Conversion of D-glucose to 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose, which is a key precursor for the synthesis of different types of bicyclic/spiro nucleosides, led to the formation of an inseparable 1:1 mixture of the desired product and 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-xylofuranose. A convenient environment friendly Novozyme®-435 catalyzed selective acetylation methodology has been developed for the separation of an epimeric mixture of ribo- and xylotrihydroxyfuranosides in quantitative yields. The structure of both the monoacetylated epimers, i.e., 5-O-acetyl-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribo- and xylofuranose obtained by enzymatic acetylation, has been confirmed by an X-ray study on their corresponding 4-C-p-toluenesulfonyloxymethyl derivatives. Furthermore, the two separated epimers were used for the convergent synthesis of two different types of bicyclic nucleosides, which confirms their synthetic utility.

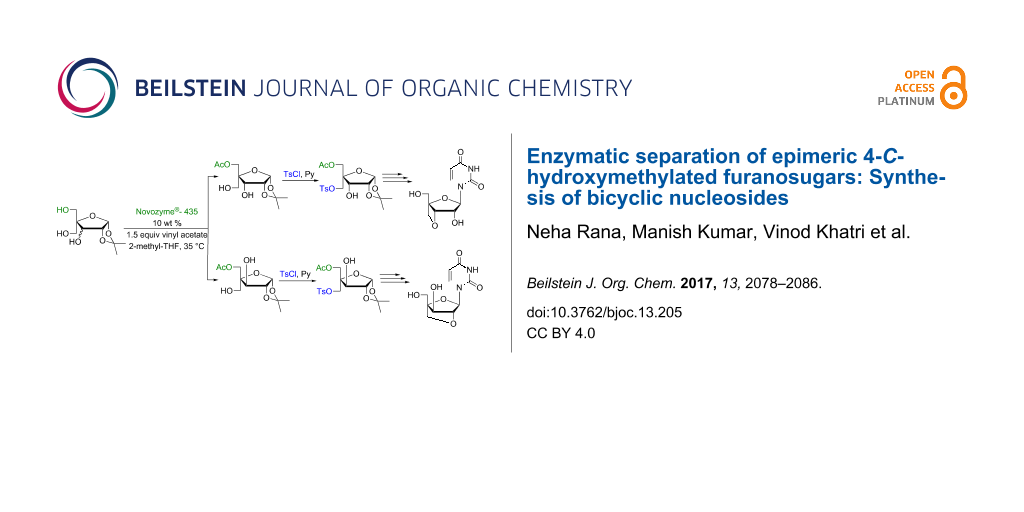
Graphical Abstract
Introduction
Sugar-modified bicyclic nucleosides have drawn the attention of synthetic chemists because of their effect on the conformational restriction of the furanose moiety of the nucleoside [1-9]. The conformational restriction has led to the enhancement in target selectivity and in vivo stability of the nucleoside-based drug candidates. One of the important precursors for the synthesis of different types of bicyclonucleosides is 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose. The synthesis of the ribo-trihydroxy sugar derivative starting from diacetone-D-glucose led to the formation of an inseparable 1:1 mixture of the required compound and its C-3 epimer, i.e., 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-xylofuranose [10].
Lipases have been used extensively for the selective manipulation of hydroxy groups present in different sugars and sugar moieties of synthetic or naturally occurring glycosides, nucleosides, etc. Gotor et al. [11] have reported a lipase-mediated acylation of an equimolecular mixture of D/L-thymidine with acetonoxime levulinate as acylating agent and Pseudomonas cepacia lipase as biocatalyst. Similar applications of lipases have been reported for the separation of mixtures of arabinofuranosyl and -pyranosyl nucleosides [12], O-aryl α,β-D-ribofuranosides, etc. [13-15]. We herein report for the first time the use of Novozyme®-435 for the separation of an epimeric mixture of xylo- and ribofuranosides. Separated epimers were further used as sugar precursors for the convergent synthesis of two different types of bicyclic nucleosides which are monomers of oxetano- and locked nucleic acids of medicinal importance [16].
Results and Discussion
4-C-Hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose (3a) can be obtained from D-glucose via diacetonylation followed by selective deprotection of 5,6-isopropylidene protection, sodium periodate oxidation of the vicinal diol and mixed aldol–Cannizaro reaction on the resulted aldehyde 2. However, this methodology always leads to the formation of an inseparable 1:1 mixture of 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribo/xylofuranose (3a,b, Scheme 1) [10].
![[1860-5397-13-205-i1]](/bjoc/content/inline/1860-5397-13-205-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of a 1:1 epimeric mixture of 3a and 3b.
Scheme 1: Formation of a 1:1 epimeric mixture of 3a and 3b.
Separation of the mixture of 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribo/xylofuranose (3a,b) has been achieved using a selective lipase catalyzed reaction. We screened two different lipases, i.e., Thermomyces lanuginosus lipase immobilized on silica (Lipozyme® TL IM) and Candida antarctica lipase-B immobilized on polyacrylate (Lewatit), commonly known as Novozyme®-435 in five different organic solvents, viz. 2-methyltetrahydofuran (2-methyl-THF), acetonitrile (MeCN), diisopropyl ether (DIPE), toluene and dioxane. We carried out all ten sets of reactions for diastereoselective acetylation of epimeric mixtures of ribo- and xylotrihydroxyfuranosides 3a,b by using vinyl acetate at 30, 35, 40 and 45 °C and at 250 rpm in an incubator shaker to evaluate the appropriate lipase and the reaction conditions. Among the two screened lipases, Novozyme®-435 (10% w/w of the substrate) in 2-methyl-THF and MeCN at 35 °C was found to exhibit exclusive selectivity for the transfer of the acetyl group from vinyl acetate to the C-5 position of 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose (3a) and 4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-xylofuranose (3b) to afford 5-O-acetyl-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose (4a) and 5-O-acetyl-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-xylofuranose (4b) in quantitative yields, respectively (Scheme 2 and Figure 1). Out of the two suitable solvents identified from the screening test, 2-methyl-THF as an environmentally benign solvent was used for further enzymatic separation reactions.
![[1860-5397-13-205-i2]](/bjoc/content/inline/1860-5397-13-205-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Lipase-catalysed separation of a mixture of ribo- and xylotrihydroxyfuranosides.
Scheme 2: Lipase-catalysed separation of a mixture of ribo- and xylotrihydroxyfuranosides.
![[1860-5397-13-205-1]](/bjoc/content/figures/1860-5397-13-205-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Screening of Novozyme®-435 and Lipozyme TL IM in different organic solvents at 35 °C for regioselective acetylation of trihydroxyribo/xylofuranose 3a and 3b (none of the reactions yielded any product when performed in the absence of the enzyme).
Figure 1: Screening of Novozyme®-435 and Lipozyme TL IM in different organic solvents at 35 °C for regioselec...
In a classical enzymatic reaction, a mixture of ribo- and xylotrihydroxyfuranosides 3a,b was incubated with Novozyme®-435 in 2-methyl-THF using vinyl acetate as acetyl donor in an incubator shaker at 250 rpm and at 35 °C. We followed the progress of the reaction by using analytical TLC. On complete conversion of the starting materials into the corresponding products, the enzyme was filtered off to quench the reaction and the filtrate was concentrated under reduced pressure to afford a colourless oil. The two products formed in the reaction had different polarity and were easily separated by column chromatography over silica gel in quantitative yields. The structure elucidation of the two products revealed that Novozyme®-435 exhibited exclusive selectivity for the acetylation of C-5 hydroxy group of the substrate 3a,b to afford 5-O-acetylated ribo- and xylofuranose derivatives 4a and 4b, respectively, in 42% and 46% yields, calculated on the basis of individual share of the trihydroxy furanoses in the mixture (Scheme 2).
The exclusive selectivity of the Novozyme®-435 for the acetylation of the C-5 hydroxy group of ribo-/xylotrihydroxyfuranoses 3a and 3b have been confirmed by X-ray diffraction studies on the single crystal of their corresponding 4-C-p-toluenesulfonyloxymethyl derivatives, i.e., 5-O-acetyl-1,2-O-isopropylidene-4-C-p-toluenesulfonyloxymethyl-α-D-ribofuranose (5) and 5-O-acetyl-1,2-O-isopropylidene-4-C-p-toluenesulphonyloxymethyl-α-D-xylofuranose (10, Figure 2). The tosyl derivatives 5 and 10 of dihydroxyfuranosides 4a and 4b were obtained by their tosylation with TsCl-pyridine in 94 and 95% yields, respectively (Scheme 3 and Scheme 4).
![[1860-5397-13-205-2]](/bjoc/content/figures/1860-5397-13-205-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram of tosylated sugar derivatives 5 and 10.
Figure 2: ORTEP diagram of tosylated sugar derivatives 5 and 10.
![[1860-5397-13-205-i3]](/bjoc/content/inline/1860-5397-13-205-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Convergent synthesis of 3′-O,4′-C-methyleneuridine.
Scheme 3: Convergent synthesis of 3′-O,4′-C-methyleneuridine.
![[1860-5397-13-205-i4]](/bjoc/content/inline/1860-5397-13-205-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Convergent synthesis of 2′-O,4′-C-methylene-xylouridine.
Scheme 4: Convergent synthesis of 2′-O,4′-C-methylene-xylouridine.
Two tosylated sugar derivatives 5 and 10 have been successfully used for the convergent synthesis of 3′-O,4′-C-methyleneuridine (9) and 2′-O,4′-C-methylene-xylouridine (14) to illustrate the usefulness of the trihydroxyribo-/xylofuranose sugar derivatives separated by an enzymatic acetylation methodology (Scheme 3 and Scheme 4). The acetylation of the lone hydroxy group in monotosylated sugar derivative 5 was carried out with acetic anhydride and 4-dimethylaminopyridine (DMAP) in dichloromethane (DCM) to give 3,5-di-O-acetyl-1,2-O-isopropylidene-4-C-p-toluenesulphonyloxymethyl-α-D-ribofuranose (6) in 98% yield. The glycosyl donor 7a,b was prepared from acetolysis of compound 6 with acetic acid/acetic anhydride/sulfuric acid (100:10:0.1) in 93% yield. The Vorbrüggen coupling [17] of 7a,b with uracil in the presence of N,O-bis(trimethylsilyl)acetamide (BSA) and trimethylsilyltrifluoromethane sulfonate (TMS-triflate) in acetonitrile yielded the triacetylated nucleoside 8 in 94% yield. Subsequently, deacetylation of acetoxy groups in nucleoside 8 with 2 M NaOH solution in water/dioxane (1:1) also led to the concomitant cylization between suitably placed C-3′-OH and C-1′′-tosyl groups to afford 3′-O,4′-C-methyleneuridine (9) in 82% yield (Scheme 3) in similar manner as described in our earlier report [18].
A similar sequence of reactions were performed for the synthesis of 2′-O,4′-C-methylene-xylouridine (14) from monotosylated sugar derivative 10 which, in turn, was obtained from enzymatic product 4b in 95% yield. Thus, the acetylation of the lone hydroxy group of 10 using acetic anhydride and DMAP in dichloromethane afforded 3,5-di-O-acetyl-1,2-O-isopropylidene-4-C-p-toluenesulfonyloxymethyl-α-D-xylofuranose (11) in 98% yield. Acetolysis of compound 11 yielded the glycosyl donor 12a,b in 95% yield, which on Vorbrüggen coupling with uracil under earlier used base-coupling conditions afforded the corresponding acetylated nucleoside 13 in 92% yield. Subsequent deacetylation of nucleosides 13 followed by concomitant cyclization with 2 M NaOH solution in water/dioxane (1:1) afforded 2′-O,4′-C-methylene-xylouridine (14) in 95% yield (Scheme 4) in a similar manner as described in our earlier report [18]. The structure of compound 2′-O,4′-C-methylene-xylouridine (14) was confirmed by X-ray diffraction studies on its single crystal which also confirms the possibility of restriction of ring puckering in bicyclic nucleoside and the sugar ring puckering is locked in N-type conformation (Figure 3).
![[1860-5397-13-205-3]](/bjoc/content/figures/1860-5397-13-205-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP diagram and preferential N-type sugar ring puckering of 2′-O,4′-C-methylene-xylouridine (14).
Figure 3: ORTEP diagram and preferential N-type sugar ring puckering of 2′-O,4′-C-methylene-xylouridine (14).
The structures of all the synthesized compounds, i.e., 4a, 4b, 5, 6, 7a,b, 8–11, 12a,b, 13 and 14 were unambiguously established on the basis of their spectral data (1H and 13C NMR spectra, IR spectra and HRMS) analysis. The structure of known compounds 9 [19] and 14 [20] were further confirmed on the basis of comparison of its physical and spectral data with those reported in the literature. The single crystal X-ray diffraction analysis has been performed on compounds 5, 10 and 14 and their detailed crystallographic data have been deposited in the Cambridge Crystallographic Data Centre with CCDC No. 1533725, 1533768 and 1532373, respectively.
Conclusion
A highly efficient and diastereoselective catalyzed Novozyme®-435-catalyzed acetylation methodology has been developed for the selective acetylation of one of the two diastereotopic primary hydroxymethyl groups present in the inseparable epimeric mixture of trihydroxyribo/xylofuranose. The biocatalytic selective acetylation of inseparable mixtures of ribo/xylofuranose derivatives has resulted in the easy separation of the 5-O-acetylated ribo/xylofuranose derivatives in quantitative yields. The sugar precursors separated by the biocatalytic methodology have been used for the convergent synthesis of the bicyclic nucleosides, 3′-O,4′-C-methyleneuridine and 2′-O,4′-C-methylene-xylouridine in 66% and 77% overall yields, respectively, from respective biaocatalytic monoacetylated product. The crystal structure of one of the synthesized bicyclic nucleosides, 2′-O,4′-C-methylene-xylouridine (14) revealed N-type puckering of the sugar ring of the compound. The developed biocatalytic methodology will have significant impact in the area of synthetic carbohydrate and nucleoside chemistry.
Experimental
The Candida antarctica lipase-B (CAL-B or Novozyme®-435) immobilized on polyacrylate was purchased from Sigma-Aldrich Co. (USA). Theremomyces lanuginosus lipase (Lipozyme TL IM) immobilized on silica was obtained as a gift from Novozymes Inc., Copenhagen, Denmark. For all the lipase-mediated reactions, AR grade organic solvents were used, which were purchased from SD Fine-Chem Ltd., Mumbai, India. The IR spectra were recorded by using thin film for oils and by making KBr discs for solid samples. The 1H and 13C NMR spectra were recorded on a JEOL Alpha-400 spectrometer at 400 and 100.6 MHz, respectively, using TMS as internal standard. The chemical shift values are on δ scale and the coupling constants (J) are in Hz. The HRMS analysis was done on a Q-TOF mass spectrometer using ESI positive mode. The optical rotations were measured using light of 589 nm wavelength. Analytical TLCs were performed on precoated silica-gel 60 F254 plates; the spots were detected either under UV light or by charring with a solution of 4% H2SO4 in ethanol. Silica gel (100–200 mesh) was used for column chromatography. All solvents were distilled before use. The single crystal X-ray diffraction data was collected with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at USIC, University of Delhi, Delhi.
General procedure for biocatalytic acetylation of ribo- and xylotrihydroxyfuranosides 3a,b: synthesis of compounds 4a,b. Similar as described in [18] to a solution of the mixture of compounds 3a,b (2 g, 9.08 mmol) in 2-methyl-THF (40 mL), vinyl acetate (1.26 mL, 13.6 mmol) was added followed by the addition of Novozyme®-435 (0.2 g, 10% w/w of the compound 3a,b). The reaction mixture was stirred at 35 °C in an incubator shaker for 1 h and the progress of the reaction was monitored periodically by TLC. On completion, the reaction was quenched by filtering off the enzyme, the solvent was removed under reduced pressure and the residue thus obtained was separated by column chromatography using methanol in chloroform as gradient solvent system to afford the two monoacetylated sugar derivatives 4a and 4b.
5-O-Acetyl-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-ribofuranose (4a). It was obtained as colourless oil (1.0 g, 42% yield). Rf = 0.4 (5.0% methanol in chloroform); [α]D18 +9.16 (c 0.1, MeOH); IR (thin film) νmax: 3471, 2946, 1739, 1383, 1244, 1166, 1046 875 cm−1; 1H NMR (CDCl3, 400 MHz) δ 5.86 (d, J = 4.6 Hz, 1H), 4.73–4.70 (m, 1H), 4.26–4.14 (m, 3H), 3.84 (s, 2H), 3.19 (d, J = 8.4 Hz, 1H), 2.61 (s, 1H), 2.10 (s, 3H), 1.62 (s, 3H), 1.39 (s, 3H); 13C NMR (CDCl3, 100.6 MHz) δ 170.73, 113.57, 104.68, 86.94, 79.41, 72.86, 65.73, 62.25, 26.45, 26.22, 20.85; HR-ESI-TOF-MS m/z: [M + Na]+ calcd. for [C11H18O7Na]+ 285.0945, found: 285.0947,
5-O-Acetyl-4-C-hydroxymethyl-1,2-O-isopropylidene-α-D-xylofuranose (4b). It was obtained as colourless oil (1.10 g, 46% yield). Rf = 0.3 (5.0% methanol in chloroform); [α]D22 −13.85 (c 0.1, MeOH); IR (thin film) νmax: 3446, 2943, 1739, 1377, 1248, 1164, 1048, 862 cm−1; 1H NMR (CDCl3, 400 MHz) δ 5.93 (d, J = 4.4 Hz, 1H), 4.64–4.63 (m, 1H), 4.31–4.19 (m, 3H), 3.77 (d, J = 11.2 Hz, 1H), 3.63 (d, J = 4.8 Hz, 1H), 2.82 (s, 1H), 2.15 (s, 1H), 2.11 (s, 3H), 1.53 (s, 3H), 1.30 (s, 3H); 13C NMR (CDCl3, 100.6 MHz) δ 170.76, 113.56, 104.64, 86.87, 79.44, 72.84, 65.70, 62.23, 26.43, 26.20, 20.84; HR-ESI-TOF-MS m/z: [M + H]+ calcd. for [C11H19O7]+ 263.1125, found 263.1130.
General procedure for the tosylation of monoacetylated sugar derivatives 4a and 4b: synthesis of compounds 5 and 10. Similar as described in [18] to a stirred solution of compound 4a (2 g, 7.6 mmol) in pyridine (20 mL), p-toluenesulfonyl chloride (2.18 g, 11.4 mmol) was added at 0 °C. The progress of the reaction was monitored by TLC and on completion after 2 h, the reaction mixture was neutralized by 10% ice-cold hydrochloric acid solution (80 mL) and extracted with chloroform (3 × 100 mL). The combined organic extract was washed with saturated aqueous NaHCO3 (2 × 100 mL), water (2 × 100 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue thus obtained was purified by silica gel column chromatography using ethyl acetate in petroleum ether as gradient solvent system to afford the tosylated compound 5. The similar procedure has been followed for the synthesis of compound 10 using 4b as starting material.
5-O-Acetyl-1,2-O-isopropylidene-4-C-p-toluenesulfonyloxymethyl-α-D-ribofuranose (5). It was obtained as white solid (2.98 g, 94% yield). Rf = 0.6 (5.0% methanol in chloroform); mp: 94 °C; [α]D28 +11.98 (c 0.1, MeOH); IR (thin film) νmax: 3483, 2989, 1745, 1458, 1362, 1239, 1190, 1095, 985, 839 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.78 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 5.77 (d, J = 3.8 Hz, 1H), 4.64 (dd, J = 5.7 and 4.2 Hz, 1H), 4.38–4.02 (m, 5H), 2.71 (d, J = 6.9 Hz, 1H), 2.41 (s, 3H), 1.99 (s, 3H), 1.42 (s, 3H), 1.31 (s, 3H); 13C NMR (CDCl3, 100.6 MHz) δ 170.34, 144.92, 132.43, 129.79, 128.10, 113.81, 104.54, 84.37, 79.31, 73.06, 68.19, 64.75, 26.20, 21.60, 20.69; HR-ESI-TOF-MS m/z: [M + K]+, calcd. for [C18H24O9SK]+ 455.0773, found: 455.0768.
5-O-Acetyl-1,2-O-isopropylidene-4-C-p-toluenesulfonyloxymethyl-α-D-xylofuranose (10). It was obtained as white solid (3.01 g, 95% yield). Rf = 0.6 (5.0% methanol in chloroform); mp: 82 °C; [α]D24 −27.64 (c 0.1, MeOH); IR (thin film) νmax: 3464, 2928, 1742, 1363, 1176, 1037, 838 cm−1; 1H NMR (CDCl3, 400 MHz) δ 7.80 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 7.6 Hz, 2H), 5.91 (d, J = 4.0 Hz, 1H), 4.60 (d, J = 3.6 Hz, 1H), 4.30-4.12 (m, 5H), 2.45 (s, 3H), 2.10 (d, J = 5.2 Hz, 1H), 1.97 (s, 3H), 1.35 (s, 3H), 1.27 (s, 3H); 13C NMR (CDCl3, 100.6 MHz) δ 171.29, 145.11, 132.24, 129.88, 128.12, 112.59, 105.32, 86.95, 86.55, 75.88, 67.79, 62.48, 26.10, 25.66, 21.60, 20.68; HR-ESI-TOF-MS: m/z: [M + H]+, calcd. for [C18H25O9S]+ 417.1214, found: 417.1218.
General procedure for acetylation of the lone hydroxy group in compounds 5 and 10: synthesis of compounds 6 and 11. To a solution of compound 5 (3 g, 7.2 mmol) in dichloromethane (30 mL) were added DMAP (176 mg, 1.44 mmol) and Ac2O (1.02 mL, 10.8 mmol) and the reaction mixture was stirred at 25–30 °C for 3 h. On completion, the mixture was diluted with cold water (25 mL) and extracted with ethyl acetate (3 × 100 mL). The combined organic layer was washed with cold water (2 × 50 mL), dried over sodium sulfate and concentrated under reduced pressure. The residue thus obtained was purified by column chromatography using ethyl acetate in petroleum ether as gradient solvent system to afford compound 6 as colourless oil in 98% yield. The similar procedure has been followed for the synthesis of compound 11, which was obtained as colourless oil using 4b as starting material in 98% yield. The spectral data and other details of compound 6 and 11 are given in Supporting Information File 1.
General procedure for the acetolysis of compounds 6 and 11: synthesis of tetraacetate compounds 7a,b and 12a,b. Similar as described in [18] acetic anhydride (6.2 mL, 65.43 mmol) and concentrated sulfuric acid (0.03 mL, 0.65 mmol) was added to a stirred solution of compound 6 (3 g, 6.5 mmol) in acetic acid (37.4 mL, 654.33 mmol) at 0 oC and mixture was stirred for 6 h at 30 °C. On completion, the reaction was quenched by addition of water (200 mL) and the product was extracted with chloroform (3 × 100 mL). The combined organic layer was washed with sodium bicarbonate solution (2 × 100 mL), with cold water (2 × 100 mL) and then dried over sodium sulfate. The solvent was removed under reduced pressure and the residue thus obtained was purified on silica gel column chromatography using ethyl acetate in petroleum ether as gradient solvent system to afford an anomeric mixture 7a,b as colourless viscous oil in 93% yield. A similar procedure has been followed for the synthesis of compound 12a,b as colourless viscous oil using 11 as starting material in 95% yield. Detailed spectral data and other details of compounds 7a,b and 12a,b have been given in Supporting Information File 1.
General procedure for the Vorbrüggen coupling of tetraacetylated sugar derivatives 7a,b and 12a,b: synthesis of acetylated nucleosides 8 and 13. Similar as described in [18] to a stirred solution of compound 7a,b (3.0 g, 5.97 mmol) and nucleobase uracil (1.0 g, 8.9 mmol) in anhydrous acetonitrile (25 mL), N,O-bis(trimethylsilyl)acetamide (5.9 mL, 23.88 mmol) was added dropwise. The reaction mixture was stirred at reflux for 1 h, and then cooled to 0 °C. Trimethylsilyltrifluoromethane sulfonate (1.8 mL, 10.14 mmol) was added dropwise into the cooled reaction mixture under stirring and the reaction mixture was refluxed for 4–6 h. The reaction was quenched with a cold saturated aqueous solution of sodium hydrogen carbonate (200 mL) and the reaction mixture was extracted with chloroform (3 × 100 mL). The combined organic phase was washed with saturated aqueous solutions of NaHCO3 (2 × 100 mL), brine (2 × 100 mL) and cold water (2 × 100 mL). The washed organic phase was then dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue thus obtained was purified by silica gel column chromatography using methanol in chloroform as gradient solvent system to afford nucleoside 8 as white solid in 94% yield. The similar procedure has been followed for the synthesis of compound 13, which was afforded as white solid using 12a,b as starting material in 92% yield. Detailed spectral data and other details of compounds 8 and 13 have been given in Supporting Information File 1.
General procedure for the synthesis of bicyclic nucleosides 9 and 14. Similar as described in [18] to a stirred solution of triacetylated nucleosides 8 (1 g, 1.8 mmol) in dioxane/water (1:1, 8 mL) was added 2 M NaOH solution (8 mL) and the reaction mixture was stirred at 30 °C for 2–10 h. On completion, the reaction mixture was neutralized with acetic acid and the solvent was removed under reduced pressure. The residue thus obtained was purified by silica gel column chromatography using methanol in chloroform as gradient solvent system to afford 9. The similar procedure has been followed for the synthesis of compound 14 using triacetylated nucleoside 13 as starting material.
3′-O,4′-C-Methyleneuridine (9). It was obtained as white solid (0.38 g, 82% yield). Rf = 0.5 (10% methanol in chloroform); mp: 215–218 °C; [α]D30 –17.50 (c 0.1, MeOH); IR (thin film) νmax: 3371, 2832, 1685, 1461, 1262, 1025, 816 cm−1; 1H NMR (DMSO-d6, 400 MHz) δ 11.44 (s, 1H), 7.95 (d, J = 8.4 Hz, 1H), 6.14 (d, J = 3.1 Hz, 1H), 5.98 (s, 1H), 5.73 (d, J = 8.4 Hz, 1H), 5.37 (s, 1H), 4.90 (s, 1H), 4.62 (d, J = 7.6 Hz, 1H), 4.50 (d, J = 2.3 Hz, 1H), 4.13 (d, J = 6.9 Hz, 1H) 3.53 (s, 2H); 13C NMR (DMSO-d6, 100.6 MHz) δ 163.72, 151.26, 141.64, 102.90, 94.81, 91.28, 90.21, 79.22, 75.88, 61.26; HR-ESI-TOF-MS: m/z: [M + H]+, calcd. for [C10H13N2O6]+ 257.0768, found: 257.0760.
2′-O,4′-C-Methylene-xylouridine (14). It was obtained as white solid (0.43 g, 95% yield). Rf = 0.5 (10% methanol in chloroform); mp: 117–120 °C; [α]D20 +36.86 (c 0.1, MeOH); IR (thin film) νmax: 3370, 2946, 1680, 1460, 1271, 1022, 755 cm−1; 1H NMR (DMSO-d6, 400 MHz) δ 11.27 (s, 1H), 7.67 (d, J = 8.2 Hz, 1H), 5.69 (d, J = 1.8 Hz, 1H), 5.51–5.48 (m, 2H), 5.01 (t, J = 5.3 Hz, 1H), 4.21 (s, 1H), 4.06 (s, 1H), 3.96 (d, J = 8.2 Hz, 1H), 3.83–3.76 (m, 2H), 3.73 (d, J = 8.2 Hz, 1H); 13C NMR (DMSO-d6, 100.6 MHz) δ 163.56, 150.26, 141.43, 98.85, 89.72, 88.33, 77.52, 72.86, 72.00 56.86; HR-ESI-TOF-MS: m/z: [M + H]+, calcd. for [C10H13N2O6]+ 257.0768, found: 257.0769.
X-ray diffraction studies on tosylated sugar derivatives 5 and 10 and bicyclic nucleoside 2′-O,4′-C-methylene-xylouridine (14). Single crystal suitable for X-ray diffraction studies were grown by dissolving the tosylated sugar derivative 5 in toluene and the other tosylated sugar derivative 10 and 2′-O,4′-C-methylene-xylouridine (14) in methanol/chloroform and allowing slow evaporation of the solutions at room temperature. The X-ray diffraction data was collected with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at a temperature of 293 K. The structures were solved by direct methods using SHELXS-97 and refined by full-matrix least-squqres method on F2 (SHELXL-97) [20]. All calculations were carried out using the WinGX package of the crystallographic programs [21]. For the molecular graphics, the programs DIAMOND-2 [22] and Mercury [23,24] were used. Molecular structures have been drawn using ORTEP as software as given in Figure 2 and Figure 3. The selected bond lengths, bond angles, etc. are given in Table 1.
Table 1: Single crystal X-ray diffraction data of tosylated sugar derivatives 5, 10 and 2′-O,4′-C-methylene-xylouridine (14).
| compound 5 | compound 10 | compound 14 | |
|---|---|---|---|
| empirical formula | C18H24O9S | C18H24O9S | C10H12N2O6 |
| formula weight | 416.43 | 416.43 | 256.22 |
| crystal system | monoclinic | monoclinic | monoclinic |
| space group | P21 | P21 | P21 |
| unit cell dimensions |
a = 8.3342(4) Å
α = 90° |
a = 11.354 Å
α = 90° |
a = 6.1313(3) Å
α = 90° |
|
b = 11.6972(5) Å
β = 95.660(4)° |
b = 9.964 Å
β = 98.66° |
b = 7.3638(3) Å
β = 93.213(4)° |
|
|
c = 10.4684(4) Å
γ = 90° |
c = 18.512 Å
γ = 90° |
c = 11.7021(6) Å
γ = 90° |
|
| volume | 1015.56(8) Å3 | 2070.4 Å3 | 527.52(4) Å3 |
| Z | 2 | 2 | 2 |
| density | 1.362 mg/m3 | 1.362 mg/m3 | 1.613 mg/m3 |
| absorption coefficient | 0.206 mm−1 | 0.205 mm−1 | 0.135 mm−1 |
| F(000) | 440 | 896 | 268 |
| index ranges |
−9<=h<=6,
−13<=k<=13, −12<=l<=12 |
−13<=h<=13,
−11<=k<=11, −19<=l<=22 |
−6<=h<=7,
−8<=k<=8, −10<=l<=13 |
| R(int) | 0.0130 | 0.0256 | 0.0140 |
| GOF on F2 | 1.030 | 1.022 | 1.030 |
| final R indices | R1 = 0.0311 | R1 = 0.0500 | R1 = 0.0288 |
| I>2sigma(I) | wR2 = 0.0720 | wR2 = 0.1070 | wR2 = 0.0691 |
| R indices | R1 = 0.0348 | R1 = 0.0647 | R1 = 0.0298 |
| all data | wR2 = 0.0738 | wR2 = 0.1150 | wR2 = 0.0701 |
| CCDC | 1533725 | 1533768 | 1532373 |
Supporting Information
| Supporting Information File 1: Additional analytical data and NMR spectra. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
We are grateful to the University of Delhi for providing financial support under DU-DST Purse Grant and under scheme to strengthen research and development. We are also thankful to CIF-USIC University of Delhi, Delhi, for providing crystallographic data and NMR spectral recording facility. N. R. and M. K. thank CSIR for the award of Senior and Junior/Senior Research Fellowships, respectively.
References
-
Merki, E.; Graham, M. J.; Mullick, A. E.; Miller, E. R.; Crooke, R. M.; Pitas, R. E.; Witztum, J. L.; Tsimikas, S. Circulation 2008, 118, 743–753. doi:10.1161/CIRCULATIONAHA.108.786822
Return to citation in text: [1] -
Seth, P. P.; Jazayeri, A.; Yu, J.; Allerson, C. R.; Bhat, B.; Swayze, E. E. Molecular Therapy–Nucleic Acids 2012, 1, e47. doi:10.1038/mtna.2012.34
Return to citation in text: [1] -
Obika, S. Chem. Pharm. Bull. 2004, 52, 1399–1404. doi:10.1248/cpb.52.1399
Return to citation in text: [1] -
Wengel, J. Acc. Chem. Res. 1999, 32, 301–310. doi:10.1021/ar980051p
Return to citation in text: [1] -
Evers, M. M.; Toonen, L. J. A.; van Roon-Mom, W. M. C. Adv. Drug Delivery Rev. 2015, 87, 90–103. doi:10.1016/j.addr.2015.03.008
Return to citation in text: [1] -
Prakash, T. P. Chem. Biodiversity 2011, 8, 1616–1641. doi:10.1002/cbdv.201100081
Return to citation in text: [1] -
Mitsuoka, Y.; Yamanoto, T.; Kugimiya, A.; Waki, R.; Wada, F.; Tahara, S.; Sawamura, M.; Noda, M.; Fujimura, Y.; Kato, Y.; Hari, Y.; Obika, S. J. Org. Chem. 2017, 82, 12–24. doi:10.1021/acs.joc.6b02417
Return to citation in text: [1] -
Istrate, A.; Medvecky, M.; Leumann, C. J. Org. Lett. 2015, 17, 1950–1953. doi:10.1021/acs.orglett.5b00662
Return to citation in text: [1] -
Medvecky, M.; Istrate, A.; Leumann, C. J. J. Org. Chem. 2015, 80, 3556–3565. doi:10.1021/acs.joc.5b00184
Return to citation in text: [1] -
Youssefyeh, R. D.; Verheyden, J. P. H.; Moffatt, J. G. J. Org. Chem. 1979, 44, 1301–1309. doi:10.1021/jo01322a024
Return to citation in text: [1] [2] -
García, J.; Fernández, S.; Ferrero, M.; Sanghvi, Y. S.; Gotor, V. Org. Lett. 2004, 6, 3759–3762. doi:10.1021/ol048502v
Return to citation in text: [1] -
Maity, J.; Shakya, G.; Singh, S. K.; Ravikumar, V. T.; Parmar, V. S.; Prasad, A. K. J. Org. Chem. 2008, 73, 5629–5632. doi:10.1021/jo800731u
Return to citation in text: [1] -
Singh, S. K.; Sharma, V. K.; Olsen, C. E.; Wengel, J.; Parmar, V. S.; Prasad, A. K. J. Org. Chem. 2010, 75, 7932–7935. doi:10.1021/jo101565e
Return to citation in text: [1] -
Sharma, R. K.; Singh, S.; Tiwari, R.; Mandal, D.; Olsen, C. E.; Parmar, V. S.; Parang, K.; Prasad, A. K. Bioorg. Med. Chem. 2012, 20, 6821–6830. doi:10.1016/j.bmc.2012.09.057
Return to citation in text: [1] -
Sharma, R. K.; Aggarwal, N.; Arya, A.; Olsen, C. E.; Parmar, V. S.; Prasad, A. K. Indian J. Chem., Sect. B 2009, 48B, 1727–1731.
Return to citation in text: [1] -
Astakhova, I. K.; Wengel, J. Acc. Chem. Res. 2014, 47, 1768–1777. doi:10.1021/ar500014g
Return to citation in text: [1] -
Vorbrüggen, H.; Lagoja, I. M.; Herdewijn, P. Curr. Protoc. Nucleic Acid Chem. 2007, 27, 1.13.1–1.13.16. doi:10.1002/0471142700.nc0113s27
Return to citation in text: [1] -
Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Obika, S.; Morio, K.-i.; Nanbu, D.; Hari, Y.; Itoh, H.; Imanishi, T. Tetrahedron 2002, 58, 3039–3049. doi:10.1016/S0040-4020(02)00227-2
Return to citation in text: [1] -
Singh, S. K.; Sharma, V. K.; Bohra, K.; Olsen, C. E.; Prasad, A. K. J. Org. Chem. 2011, 76, 7556–7562. doi:10.1021/jo201060t
Return to citation in text: [1] [2] -
Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/S0021889899006020
Return to citation in text: [1] -
Pennington, W. T. J. Appl. Crystallogr. 1999, 32, 1028–1029. doi:10.1107/S0021889899011486
Return to citation in text: [1] -
Macrae, C. F.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Shields, G. P.; Taylor, R.; Towler, M.; van de Streek, J. J. Appl. Crystallogr. 2006, 39, 453–457. doi:10.1107/S002188980600731X
Return to citation in text: [1]
| 21. | Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112–122. doi:10.1107/S0108767307043930 |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 20. | Singh, S. K.; Sharma, V. K.; Bohra, K.; Olsen, C. E.; Prasad, A. K. J. Org. Chem. 2011, 76, 7556–7562. doi:10.1021/jo201060t |
| 1. | Merki, E.; Graham, M. J.; Mullick, A. E.; Miller, E. R.; Crooke, R. M.; Pitas, R. E.; Witztum, J. L.; Tsimikas, S. Circulation 2008, 118, 743–753. doi:10.1161/CIRCULATIONAHA.108.786822 |
| 2. | Seth, P. P.; Jazayeri, A.; Yu, J.; Allerson, C. R.; Bhat, B.; Swayze, E. E. Molecular Therapy–Nucleic Acids 2012, 1, e47. doi:10.1038/mtna.2012.34 |
| 3. | Obika, S. Chem. Pharm. Bull. 2004, 52, 1399–1404. doi:10.1248/cpb.52.1399 |
| 4. | Wengel, J. Acc. Chem. Res. 1999, 32, 301–310. doi:10.1021/ar980051p |
| 5. | Evers, M. M.; Toonen, L. J. A.; van Roon-Mom, W. M. C. Adv. Drug Delivery Rev. 2015, 87, 90–103. doi:10.1016/j.addr.2015.03.008 |
| 6. | Prakash, T. P. Chem. Biodiversity 2011, 8, 1616–1641. doi:10.1002/cbdv.201100081 |
| 7. | Mitsuoka, Y.; Yamanoto, T.; Kugimiya, A.; Waki, R.; Wada, F.; Tahara, S.; Sawamura, M.; Noda, M.; Fujimura, Y.; Kato, Y.; Hari, Y.; Obika, S. J. Org. Chem. 2017, 82, 12–24. doi:10.1021/acs.joc.6b02417 |
| 8. | Istrate, A.; Medvecky, M.; Leumann, C. J. Org. Lett. 2015, 17, 1950–1953. doi:10.1021/acs.orglett.5b00662 |
| 9. | Medvecky, M.; Istrate, A.; Leumann, C. J. J. Org. Chem. 2015, 80, 3556–3565. doi:10.1021/acs.joc.5b00184 |
| 13. | Singh, S. K.; Sharma, V. K.; Olsen, C. E.; Wengel, J.; Parmar, V. S.; Prasad, A. K. J. Org. Chem. 2010, 75, 7932–7935. doi:10.1021/jo101565e |
| 14. | Sharma, R. K.; Singh, S.; Tiwari, R.; Mandal, D.; Olsen, C. E.; Parmar, V. S.; Parang, K.; Prasad, A. K. Bioorg. Med. Chem. 2012, 20, 6821–6830. doi:10.1016/j.bmc.2012.09.057 |
| 15. | Sharma, R. K.; Aggarwal, N.; Arya, A.; Olsen, C. E.; Parmar, V. S.; Prasad, A. K. Indian J. Chem., Sect. B 2009, 48B, 1727–1731. |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 12. | Maity, J.; Shakya, G.; Singh, S. K.; Ravikumar, V. T.; Parmar, V. S.; Prasad, A. K. J. Org. Chem. 2008, 73, 5629–5632. doi:10.1021/jo800731u |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 11. | García, J.; Fernández, S.; Ferrero, M.; Sanghvi, Y. S.; Gotor, V. Org. Lett. 2004, 6, 3759–3762. doi:10.1021/ol048502v |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 10. | Youssefyeh, R. D.; Verheyden, J. P. H.; Moffatt, J. G. J. Org. Chem. 1979, 44, 1301–1309. doi:10.1021/jo01322a024 |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 19. | Obika, S.; Morio, K.-i.; Nanbu, D.; Hari, Y.; Itoh, H.; Imanishi, T. Tetrahedron 2002, 58, 3039–3049. doi:10.1016/S0040-4020(02)00227-2 |
| 17. | Vorbrüggen, H.; Lagoja, I. M.; Herdewijn, P. Curr. Protoc. Nucleic Acid Chem. 2007, 27, 1.13.1–1.13.16. doi:10.1002/0471142700.nc0113s27 |
| 20. | Singh, S. K.; Sharma, V. K.; Bohra, K.; Olsen, C. E.; Prasad, A. K. J. Org. Chem. 2011, 76, 7556–7562. doi:10.1021/jo201060t |
| 10. | Youssefyeh, R. D.; Verheyden, J. P. H.; Moffatt, J. G. J. Org. Chem. 1979, 44, 1301–1309. doi:10.1021/jo01322a024 |
| 22. | Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/S0021889899006020 |
| 16. | Astakhova, I. K.; Wengel, J. Acc. Chem. Res. 2014, 47, 1768–1777. doi:10.1021/ar500014g |
| 18. | Kumar, M.; Kumar, R.; Rana, N.; Prasad, A. K. RSC Adv. 2016, 6, 17713–17719. doi:10.1039/C5RA25222A |
| 23. | Pennington, W. T. J. Appl. Crystallogr. 1999, 32, 1028–1029. doi:10.1107/S0021889899011486 |
| 24. | Macrae, C. F.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Shields, G. P.; Taylor, R.; Towler, M.; van de Streek, J. J. Appl. Crystallogr. 2006, 39, 453–457. doi:10.1107/S002188980600731X |
© 2017 Rana et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)