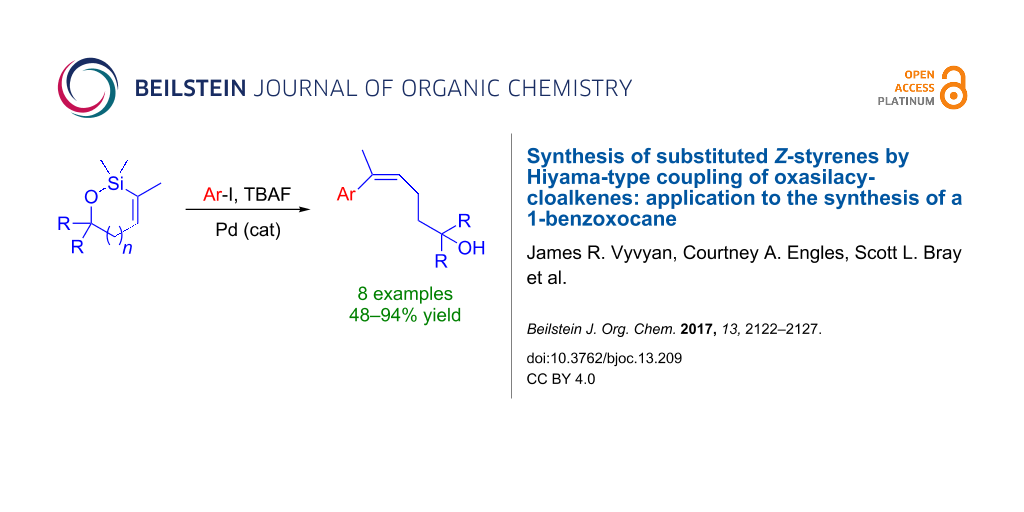
Abstract
Several Hiyama cross-coupling reactions of oxasilacycloalkenes and aryl iodides are described that produce trisubstituted Z-styrenes in moderate to excellent yields. Both electron-rich and electron-poor aryl iodides are tolerated in the cross-coupling reaction. The oxasilacycloalkene coupling partners were prepared by ruthenium-catalyzed intramolecular anti-hydrosilylation of alkynols. One of the cross-coupling products was converted to a 1-benzoxocane, albeit in low yield, using an intramolecular Buchwald–Hartwig etherification. The cyclic ether produced contains the carbon skeleton of heliannuol A.

Graphical Abstract
Introduction
The development of transition metal-catalyzed cross-coupling technologies over the last four decades revolutionized the synthetic chemistry. Indeed, the importance of Pd-catalyzed coupling was recognized with the 2010 Nobel Prize awarded to Heck, Negishi and Suzuki [1,2]. Developed somewhat later than the aforementioned methods and the couplings of organomagnesium and organotin reagents was the cross-coupling of silanes, pioneered by Hiyama [3,4] and siloxanes [5]. This versatile method has been extensively reviewed [6-8]. The cross-coupling of alkenyl silanols [9,10] and oxasilacycloalkenes (cyclic siloxanes, cf. Figure 1) [6-8] is an excellent method to prepare stereodefined alkenes. The cyclic siloxanes can be prepared in a number of ways: hydrosilylation of alkynes [11-13], semihydrogenation of silyl alkynes [14], ring-closing metathesis (RCM) [15-21] and enyne metathesis [22,23].
![[1860-5397-13-209-1]](/bjoc/content/figures/1860-5397-13-209-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Retrosynthetic analysis of heliannuol A.
Figure 1: Retrosynthetic analysis of heliannuol A.
We became interested in the cross-coupling of cyclic siloxanes in the context of preparing trisubstituted Z-styrenes for the synthesis of natural product targets [24]. Heliannuol A was the first member of a family of allelopathic [25-27] sesquiterpenoids isolated from the sunflower Helianthus annuus [28-33], and it contains an unusual benzoxocane moiety. Owing to their unusual structures and biological activity, the heliannuols have attracted significant attention from synthetic chemists. The various approaches to the heliannuols have recently been reviewed [34]. We have previously reported syntheses of heliannuols C, D, and E via intramolecular epoxide opening reactions [35,36], but this approach did not translate well to the synthesis of heliannuol A. One of our alternative strategies for the synthesis of heliannuol A was an intramolecular Buchwald–Hartwig etherification of a Z-styrene derivative to provide a conformational constraint to facilitate the formation of the eight-membered ring [37]. The presence of the alkene reduces the conformational degrees of freedom in 2, thereby partially offsetting the entropic penalty of forming the eight-membered ring. We endeavored to test the strategy through the preparation of a simplified model compound 1 (Figure 1). We envisaged the precursor to the cycloetherification, 2, would be prepared from the Hiyama-type cross-coupling of the appropriate aryl iodide 3 and the oxasilacycloalkene 4.
Results and Discussion
The oxasilacycloalkenes used in this study were readily prepared by intramolecular anti-hydrosilylation of alkynols 5–7 using the method reported by Ball and Trost (Scheme 1) [12,13]. Alkynol 5 was commercially available, 6 was readily prepared following literature procedures (see Experimental section), and alkynol 7 was, surprisingly, a new compound. The hydrosilylation reactions of 5–7 can be carried out on gram scales and generally produce high yields of the siloxane products after distillation. Siloxane 8a was isolated in more modest yield, however, probably owing to its volatility.
![[1860-5397-13-209-i1]](/bjoc/content/inline/1860-5397-13-209-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The siloxanes 8 participated efficiently in Hiyama-type cross-couplings with aryl iodides in the presence of Pd2(dba)3 catalyst and tetrabutylammonium fluoride (TBAF, Table 1). Both electron-rich (Table 1, entries 1, 2, and 7) and electron-poor (Table 1, entries 5 and 6) iodides give coupled products in moderate to excellent yields. When multiple halogens were present, high selectivity for reaction at the aryl iodide was observed (Table 1, entries 3–5). Pyridyl iodide 22 also worked well in the cross-coupling (Table 1, entry 8).
Table 1: Pd-catalyzed couplings of oxasilacycloalkenes with aryl iodides.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-209-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Aryl iodide | Siloxane | Product | Yield (%) |
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-209-i4.svg?max-width=637&scale=1.0)
9 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-209-i5.svg?max-width=637&scale=1.0)
8a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-209-i6.svg?max-width=637&scale=1.0)
10 |
92a |
| 2 | 9 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-209-i7.svg?max-width=637&scale=1.0)
8b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-209-i8.svg?max-width=637&scale=1.0)
11 |
52 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-209-i9.svg?max-width=637&scale=1.0)
12 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-209-i10.svg?max-width=637&scale=1.0)
8c |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-209-i11.svg?max-width=637&scale=1.0)
13 |
87 |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-209-i12.svg?max-width=637&scale=1.0)
14 |
8c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-209-i13.svg?max-width=637&scale=1.0)
15 |
94 |
| 5 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-209-i14.svg?max-width=637&scale=1.0)
16 |
8c |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-209-i15.svg?max-width=637&scale=1.0)
17 |
80 |
| 6 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-209-i16.svg?max-width=637&scale=1.0)
18 |
8c |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-209-i17.svg?max-width=637&scale=1.0)
19 |
60 |
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-209-i18.svg?max-width=637&scale=1.0)
20 |
8c |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-209-i19.svg?max-width=637&scale=1.0)
21 |
48 |
| 8 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-209-i20.svg?max-width=637&scale=1.0)
22 |
8c |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-209-i21.svg?max-width=637&scale=1.0)
23 |
58 |
a10 mol % Pd2(dba)3 used in this trial.
Having prepared a number of substituted Z-styrenes, we next focused on the intramolecular Buchwald–Hartwig etherification [38-40] of bromoalcohol 15 to prepare eight-membered cyclic ether 24 (Table 2). The use of Pd(II) catalyst precursors with BINAP ligands and carbonate bases in toluene [38,39] was ineffective, returning significant amounts of unreacted starting material (Table 2, entry 1) or debrominated material 25 (Table 2, entry 2). The use of a bis(diphenylphosphino)ferrocenyl (dppf) ligand and a stronger base to irreversibly deprotonate the alcohol was similarly ineffective in both toluene and dioxane (Table 2, entry 3). Based on Hartwig’s success with similar intramolecular etherifications to make five- and six-membered rings [40], and our own preparation of an analogous seven-membered cyclic ether [41], we examined Pd(0) catalyst precursors with the Q-Phos ligand. The reaction with Pd(dba)2 with Q-Phos and sodium tert-butoxide produced cyclic ether 24 along with debrominated 25 as the major product (Table 2, entry 4). As before, irreversibly deprotonating the alcohol with sodium hydride was not productive (Table 2, entry 5). Using Pd2(dba)3 as catalyst precursor increased the yield of 24 relative to the amount of 25 produced, but the overall yield of the cyclic ether was still low (Table 2, entry 6). Microwave heating was not helpful (Table 2, entry 7). The non-nucleophilic base DBU was not effective in promoting the cyclization, either (Table 2, entry 8). We observed small amounts of products arising from the dehydration of 15 in the 1H NMR spectra of the crude reaction mixtures from these experiments, so we were reluctant to increase the reaction temperature further.
Table 2: Buchwald–Hartwig etherifications of bromoalcohol 15.
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-209-i22.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Pd catalyst | Ligand | Base | Yield(%) | |
| 24 | 25 | ||||
| 1 | Pd(OAc)2 | tol-BINAP | K2CO3 | 0 | 0a |
| 2 | Pd(TFA)2 | tol-BINAP | Cs2CO3 | 0 | 21 |
| 3 | Pd(OAc)2 | dppf | NaH | 0 | 45b |
| 4 | Pd(dba)2 | Q-Phos | NaOt-Bu | 7 | 53 |
| 5 | Pd(dba)2 | Q-Phos | NaH | 0 | 40 |
| 6 | Pd2(dba)3 | Q-Phos | NaOt-Bu | 10 | 12 |
| 7c | Pd(dba)2 | Q-Phos | NaOt-Bu | 6 | 21d |
| 8 | Pd(dba)2 | Q-Phos | DBU | 0 | 0e |
a73% starting material recovered. bUsing dioxane as solvent gave a 32% yield of 25. cMicrowave heating, 80 °C, 18 h. d25% starting material recovered. e65% starting material recovered.
We also hydrogenated the conjugated olefin of 15 to increase the flexibility of the tether, in case the conformational constraint was actually hindering the cyclization, but no cyclic ether was observed when this material was subjected to Pd catalyst and base (not shown).
Hydrogenation of 24 over Pd/C gave 26, whose 1H NMR spectrum matches that of the benzoxocane prepared by Pettus and co-workers [42] (Scheme 2), which was shown by them to be the now-refuted structure of helianane.
![[1860-5397-13-209-i2]](/bjoc/content/inline/1860-5397-13-209-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Hydrogenation of benzoxocane 24.
Scheme 2: Hydrogenation of benzoxocane 24.
Conclusion
In summary, we have confirmed the Hiyama cross-coupling of cyclic siloxanes is an efficient route to Z-trisubstituted styrenes that are useful for the synthesis of natural product frameworks. Although yields are low under the conditions attempted, a proof of concept has been established in applying the Buchwald–Hartwig coupling to 15, producing the benzoxocane 24, which contains the carbon skeleton of heliannuol A.
Supporting Information
| Supporting Information File 1: Full experimental details and copies of 1H and 13C NMR spectra for all new compounds. | ||
| Format: PDF | Size: 2.4 MB | Download |
References
-
Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. doi:10.1002/anie.201107017
Return to citation in text: [1] -
de Meijere, A.; Bräse, S.; Oestreich, M., Eds. Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH: Weinheim, 2014. doi:10.1002/9783527655588
Return to citation in text: [1] -
Hatanaka, Y.; Hiyama, T. J. Org. Chem. 1988, 53, 918–920. doi:10.1021/jo00239a056
Return to citation in text: [1] -
Hiyama, T. J. Organomet. Chem. 2002, 653, 58–61. doi:10.1016/S0022-328X(02)01157-9
Return to citation in text: [1] -
Mowery, M. E.; DeShong, P. J. Org. Chem. 1999, 64, 1684–1688. doi:10.1021/jo982463h
Return to citation in text: [1] -
Denmark, S. E.; Sweis, R. F. Organosilicon Compounds in Cross-Coupling Reactions. In Metal-Catalyzed Cross-Coupling Reactions and More; de Meijere, A.; Bräse, S.; Oestreich, M., Eds.; Wiley-VCH: Weinheim, 2014; pp 475–532. doi:10.1002/9783527655588.ch7
Return to citation in text: [1] [2] -
Chang, W.-T.; Smith, R. C.; Regens, C. S.; Bailey, A. D.; Werner, N. S.; Denmark, S. E. Org. React. 2011, 75, 213–746. doi:10.1002/0471264180.or075.03
Return to citation in text: [1] [2] -
Sore, H. F.; Galloway, W. R. J. D.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 1845–1866. doi:10.1039/C1CS15181A
Return to citation in text: [1] [2] -
Denmark, S. E.; Sweis, R. F. Org. Lett. 2002, 4, 3771–3774. doi:10.1021/ol026900x
Return to citation in text: [1] -
Denmark, S. E.; Regens, C. S. Acc. Chem. Res. 2008, 41, 1486–1499. doi:10.1021/ar800037p
Return to citation in text: [1] -
Denmark, S. E.; Pan, W. Org. Lett. 2002, 4, 4163–4166. doi:10.1021/ol026933c
Return to citation in text: [1] -
Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2003, 125, 30–31. doi:10.1021/ja028766h
Return to citation in text: [1] [2] -
Trost, B. M.; Ball, Z. T.; Laemmerhold, K. M. J. Am. Chem. Soc. 2005, 127, 10028–10038. doi:10.1021/ja051578h
Return to citation in text: [1] [2] -
Elbert, B. L.; Lim, D. S. W.; Gudmundsson, H. G.; O’Hanlon, J. A.; Anderson, E. A. Chem. – Eur. J. 2014, 20, 8594–8598. doi:10.1002/chem.201403255
Return to citation in text: [1] -
Ahmed, M.; Barrett, A. G. M.; Beall, J. C.; Braddock, D. C.; Flack, K.; Gibson, V. C.; Procopiou, P. A.; Salter, M. M. Tetrahedron 1999, 55, 3219–3232. doi:10.1016/S0040-4020(98)01135-1
Return to citation in text: [1] -
Barrett, A. G. M.; Beall, J. C.; Braddock, D. C.; Flack, K.; Gibson, V. C.; Salter, M. M. J. Org. Chem. 2000, 65, 6508–6514. doi:10.1021/jo000690p
Return to citation in text: [1] -
Denmark, S. E.; Yang, S.-M. Org. Lett. 2001, 3, 1749–1752. doi:10.1021/ol015950j
Return to citation in text: [1] -
Denmark, S. E.; Yang, S.-M. Tetrahedron 2004, 60, 9695–9708. doi:10.1016/j.tet.2004.06.149
Return to citation in text: [1] -
Xie, Q.; Denton, R. W.; Parker, K. A. Org. Lett. 2008, 10, 5345–5348. doi:10.1021/ol802063h
Return to citation in text: [1] -
Volchkov, I.; Park, S.; Lee, D. Org. Lett. 2011, 13, 3530–3533. doi:10.1021/ol2013473
Return to citation in text: [1] -
Volchkov, I.; Lee, D. J. Am. Chem. Soc. 2013, 135, 5324–5327. doi:10.1021/ja401717b
Return to citation in text: [1] -
Miller, R. L.; Maifeld, S. V.; Lee, D. Org. Lett. 2004, 6, 2773–2776. doi:10.1021/ol049019n
Return to citation in text: [1] -
Kim, H.; Lee, C. J. Am. Chem. Soc. 2005, 127, 10180–10181. doi:10.1021/ja052775j
Return to citation in text: [1] -
Denmark, S. E.; Liu, J. H.-C. Angew. Chem., Int. Ed. 2010, 49, 2978–2986. doi:10.1002/anie.200905657
Return to citation in text: [1] -
Vyvyan, J. R. Tetrahedron 2002, 58, 1631–1646. doi:10.1016/S0040-4020(02)00052-2
Return to citation in text: [1] -
Macías, F. A.; Molinillo, J. M. G.; Chinchilla, D.; Galindo, J. C. G. Heliannanes–a structure-activity relationship (SAR) study. In Allelopathy: chemistry and mode of action of allelochemicals; Macías, F. A.; Galindo, J. C. G.; Molinillo, J. M. G.; Cutler, H. G., Eds.; CRC Press: Boca Raton, 2004; pp 103–124.
Return to citation in text: [1] -
Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. J. Chem. Ecol. 2000, 26, 2173–2186. doi:10.1023/A:1005524601019
Return to citation in text: [1] -
Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G.; Fronczek, F. R. Tetrahedron Lett. 1993, 34, 1999–2002. doi:10.1016/S0040-4039(00)91986-5
Return to citation in text: [1] -
Macías, F. A.; Molinillo, J. M. G.; Varela, R. M.; Torres, A.; Fronczek, F. R. J. Org. Chem. 1994, 59, 8261–8266. doi:10.1021/jo00105a052
Return to citation in text: [1] -
Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. Tetrahedron Lett. 1999, 40, 4725–4728. doi:10.1016/S0040-4039(99)00833-3
Return to citation in text: [1] -
Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. J. Nat. Prod. 1999, 62, 1636–1639. doi:10.1021/np990249y
Return to citation in text: [1] -
Macías, F. A.; Torres, A.; Galindo, J. L. G.; Varela, R. M.; Álvarez, J. A.; Molinillo, J. M. G. Phytochemistry 2002, 61, 687–692. doi:10.1016/S0031-9422(02)00370-9
Return to citation in text: [1] -
El Marsni, Z.; Torres, A.; Varela, R. M.; Molinillo, J. M. G.; Casas, L.; Mantell, C.; Martinez de la Ossa, E. J.; Macías, F. A. J. Agric. Food Chem. 2015, 63, 6410–6421. doi:10.1021/acs.jafc.5b02261
Return to citation in text: [1] -
Chen, K.; Li, Y.; Du, Z.; Tao, Z. Synth. Commun. 2015, 45, 663–691. doi:10.1080/00397911.2014.979948
Return to citation in text: [1] -
Vyvyan, J. R.; Looper, R. E. Tetrahedron Lett. 2000, 41, 1151–1154. doi:10.1016/S0040-4039(99)02284-4
Return to citation in text: [1] -
Vyvyan, J. R.; Oaksmith, J. M.; Parks, B. W.; Peterson, E. M. Tetrahedron Lett. 2005, 46, 2457–2460. doi:10.1016/j.tetlet.2005.02.053
Return to citation in text: [1] -
Werner, E. W. W. M. S. Ph.D. Thesis, Western Washington University, Bellingham, WA, 2007.
Return to citation in text: [1] -
Torraca, K. E.; Huang, X.; Parrish, C. A.; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 10770–10771. doi:10.1021/ja016863p
Return to citation in text: [1] [2] -
Vorogushin, A. V.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2005, 127, 8146–8149. doi:10.1021/ja050471r
Return to citation in text: [1] [2] -
Kataoka, N.; Shelby, Q.; Stambuli, J. P.; Hartwig, J. F. J. Org. Chem. 2002, 67, 5553–5566. doi:10.1021/jo025732j
Return to citation in text: [1] [2] -
Vyvyan, J. R.; Wold, E. D.; Bray, S. L. unpublished results.
Return to citation in text: [1] -
Green, J. C.; Jiménez-Alonso, S.; Brown, E. R.; Pettus, T. R. R. Org. Lett. 2011, 13, 5500–5503. doi:10.1021/ol2022214
Return to citation in text: [1]
| 38. | Torraca, K. E.; Huang, X.; Parrish, C. A.; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 10770–10771. doi:10.1021/ja016863p |
| 39. | Vorogushin, A. V.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2005, 127, 8146–8149. doi:10.1021/ja050471r |
| 12. | Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2003, 125, 30–31. doi:10.1021/ja028766h |
| 13. | Trost, B. M.; Ball, Z. T.; Laemmerhold, K. M. J. Am. Chem. Soc. 2005, 127, 10028–10038. doi:10.1021/ja051578h |
| 38. | Torraca, K. E.; Huang, X.; Parrish, C. A.; Buchwald, S. L. J. Am. Chem. Soc. 2001, 123, 10770–10771. doi:10.1021/ja016863p |
| 39. | Vorogushin, A. V.; Huang, X.; Buchwald, S. L. J. Am. Chem. Soc. 2005, 127, 8146–8149. doi:10.1021/ja050471r |
| 40. | Kataoka, N.; Shelby, Q.; Stambuli, J. P.; Hartwig, J. F. J. Org. Chem. 2002, 67, 5553–5566. doi:10.1021/jo025732j |
| 1. | Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. doi:10.1002/anie.201107017 |
| 2. | de Meijere, A.; Bräse, S.; Oestreich, M., Eds. Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH: Weinheim, 2014. doi:10.1002/9783527655588 |
| 9. | Denmark, S. E.; Sweis, R. F. Org. Lett. 2002, 4, 3771–3774. doi:10.1021/ol026900x |
| 10. | Denmark, S. E.; Regens, C. S. Acc. Chem. Res. 2008, 41, 1486–1499. doi:10.1021/ar800037p |
| 35. | Vyvyan, J. R.; Looper, R. E. Tetrahedron Lett. 2000, 41, 1151–1154. doi:10.1016/S0040-4039(99)02284-4 |
| 36. | Vyvyan, J. R.; Oaksmith, J. M.; Parks, B. W.; Peterson, E. M. Tetrahedron Lett. 2005, 46, 2457–2460. doi:10.1016/j.tetlet.2005.02.053 |
| 6. | Denmark, S. E.; Sweis, R. F. Organosilicon Compounds in Cross-Coupling Reactions. In Metal-Catalyzed Cross-Coupling Reactions and More; de Meijere, A.; Bräse, S.; Oestreich, M., Eds.; Wiley-VCH: Weinheim, 2014; pp 475–532. doi:10.1002/9783527655588.ch7 |
| 7. | Chang, W.-T.; Smith, R. C.; Regens, C. S.; Bailey, A. D.; Werner, N. S.; Denmark, S. E. Org. React. 2011, 75, 213–746. doi:10.1002/0471264180.or075.03 |
| 8. | Sore, H. F.; Galloway, W. R. J. D.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 1845–1866. doi:10.1039/C1CS15181A |
| 37. | Werner, E. W. W. M. S. Ph.D. Thesis, Western Washington University, Bellingham, WA, 2007. |
| 5. | Mowery, M. E.; DeShong, P. J. Org. Chem. 1999, 64, 1684–1688. doi:10.1021/jo982463h |
| 28. | Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G.; Fronczek, F. R. Tetrahedron Lett. 1993, 34, 1999–2002. doi:10.1016/S0040-4039(00)91986-5 |
| 29. | Macías, F. A.; Molinillo, J. M. G.; Varela, R. M.; Torres, A.; Fronczek, F. R. J. Org. Chem. 1994, 59, 8261–8266. doi:10.1021/jo00105a052 |
| 30. | Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. Tetrahedron Lett. 1999, 40, 4725–4728. doi:10.1016/S0040-4039(99)00833-3 |
| 31. | Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. J. Nat. Prod. 1999, 62, 1636–1639. doi:10.1021/np990249y |
| 32. | Macías, F. A.; Torres, A.; Galindo, J. L. G.; Varela, R. M.; Álvarez, J. A.; Molinillo, J. M. G. Phytochemistry 2002, 61, 687–692. doi:10.1016/S0031-9422(02)00370-9 |
| 33. | El Marsni, Z.; Torres, A.; Varela, R. M.; Molinillo, J. M. G.; Casas, L.; Mantell, C.; Martinez de la Ossa, E. J.; Macías, F. A. J. Agric. Food Chem. 2015, 63, 6410–6421. doi:10.1021/acs.jafc.5b02261 |
| 3. | Hatanaka, Y.; Hiyama, T. J. Org. Chem. 1988, 53, 918–920. doi:10.1021/jo00239a056 |
| 4. | Hiyama, T. J. Organomet. Chem. 2002, 653, 58–61. doi:10.1016/S0022-328X(02)01157-9 |
| 34. | Chen, K.; Li, Y.; Du, Z.; Tao, Z. Synth. Commun. 2015, 45, 663–691. doi:10.1080/00397911.2014.979948 |
| 15. | Ahmed, M.; Barrett, A. G. M.; Beall, J. C.; Braddock, D. C.; Flack, K.; Gibson, V. C.; Procopiou, P. A.; Salter, M. M. Tetrahedron 1999, 55, 3219–3232. doi:10.1016/S0040-4020(98)01135-1 |
| 16. | Barrett, A. G. M.; Beall, J. C.; Braddock, D. C.; Flack, K.; Gibson, V. C.; Salter, M. M. J. Org. Chem. 2000, 65, 6508–6514. doi:10.1021/jo000690p |
| 17. | Denmark, S. E.; Yang, S.-M. Org. Lett. 2001, 3, 1749–1752. doi:10.1021/ol015950j |
| 18. | Denmark, S. E.; Yang, S.-M. Tetrahedron 2004, 60, 9695–9708. doi:10.1016/j.tet.2004.06.149 |
| 19. | Xie, Q.; Denton, R. W.; Parker, K. A. Org. Lett. 2008, 10, 5345–5348. doi:10.1021/ol802063h |
| 20. | Volchkov, I.; Park, S.; Lee, D. Org. Lett. 2011, 13, 3530–3533. doi:10.1021/ol2013473 |
| 21. | Volchkov, I.; Lee, D. J. Am. Chem. Soc. 2013, 135, 5324–5327. doi:10.1021/ja401717b |
| 24. | Denmark, S. E.; Liu, J. H.-C. Angew. Chem., Int. Ed. 2010, 49, 2978–2986. doi:10.1002/anie.200905657 |
| 42. | Green, J. C.; Jiménez-Alonso, S.; Brown, E. R.; Pettus, T. R. R. Org. Lett. 2011, 13, 5500–5503. doi:10.1021/ol2022214 |
| 14. | Elbert, B. L.; Lim, D. S. W.; Gudmundsson, H. G.; O’Hanlon, J. A.; Anderson, E. A. Chem. – Eur. J. 2014, 20, 8594–8598. doi:10.1002/chem.201403255 |
| 25. | Vyvyan, J. R. Tetrahedron 2002, 58, 1631–1646. doi:10.1016/S0040-4020(02)00052-2 |
| 26. | Macías, F. A.; Molinillo, J. M. G.; Chinchilla, D.; Galindo, J. C. G. Heliannanes–a structure-activity relationship (SAR) study. In Allelopathy: chemistry and mode of action of allelochemicals; Macías, F. A.; Galindo, J. C. G.; Molinillo, J. M. G.; Cutler, H. G., Eds.; CRC Press: Boca Raton, 2004; pp 103–124. |
| 27. | Macías, F. A.; Varela, R. M.; Torres, A.; Molinillo, J. M. G. J. Chem. Ecol. 2000, 26, 2173–2186. doi:10.1023/A:1005524601019 |
| 11. | Denmark, S. E.; Pan, W. Org. Lett. 2002, 4, 4163–4166. doi:10.1021/ol026933c |
| 12. | Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2003, 125, 30–31. doi:10.1021/ja028766h |
| 13. | Trost, B. M.; Ball, Z. T.; Laemmerhold, K. M. J. Am. Chem. Soc. 2005, 127, 10028–10038. doi:10.1021/ja051578h |
| 40. | Kataoka, N.; Shelby, Q.; Stambuli, J. P.; Hartwig, J. F. J. Org. Chem. 2002, 67, 5553–5566. doi:10.1021/jo025732j |
| 6. | Denmark, S. E.; Sweis, R. F. Organosilicon Compounds in Cross-Coupling Reactions. In Metal-Catalyzed Cross-Coupling Reactions and More; de Meijere, A.; Bräse, S.; Oestreich, M., Eds.; Wiley-VCH: Weinheim, 2014; pp 475–532. doi:10.1002/9783527655588.ch7 |
| 7. | Chang, W.-T.; Smith, R. C.; Regens, C. S.; Bailey, A. D.; Werner, N. S.; Denmark, S. E. Org. React. 2011, 75, 213–746. doi:10.1002/0471264180.or075.03 |
| 8. | Sore, H. F.; Galloway, W. R. J. D.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 1845–1866. doi:10.1039/C1CS15181A |
| 22. | Miller, R. L.; Maifeld, S. V.; Lee, D. Org. Lett. 2004, 6, 2773–2776. doi:10.1021/ol049019n |
| 23. | Kim, H.; Lee, C. J. Am. Chem. Soc. 2005, 127, 10180–10181. doi:10.1021/ja052775j |
© 2017 Vyvyan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)