Abstract
Herein, we present a new strategy for the preparation of a broad range of brassinosteroid biosynthetic precursors/metabolites differing by the ring A fragment. The protocol is based on the use of readily available phytohormones of this class bearing a 2α,3α-diol moiety (epibrassinolide or epicastasterone) as starting materials. The required functionalities (Δ2-, 2α,3α- and 2β,3β-epoxy-, 2α,3β-, 2β,3α-, and 2β,3β-dihydroxy-, 3-keto-, 3α- and 3β-hydroxy-, 2α-hydroxy-3-keto-) were synthesized from 2α,3α-diols in a few simple steps (Corey–Winter reaction, epoxidation, oxidation, hydride reduction, etc.).

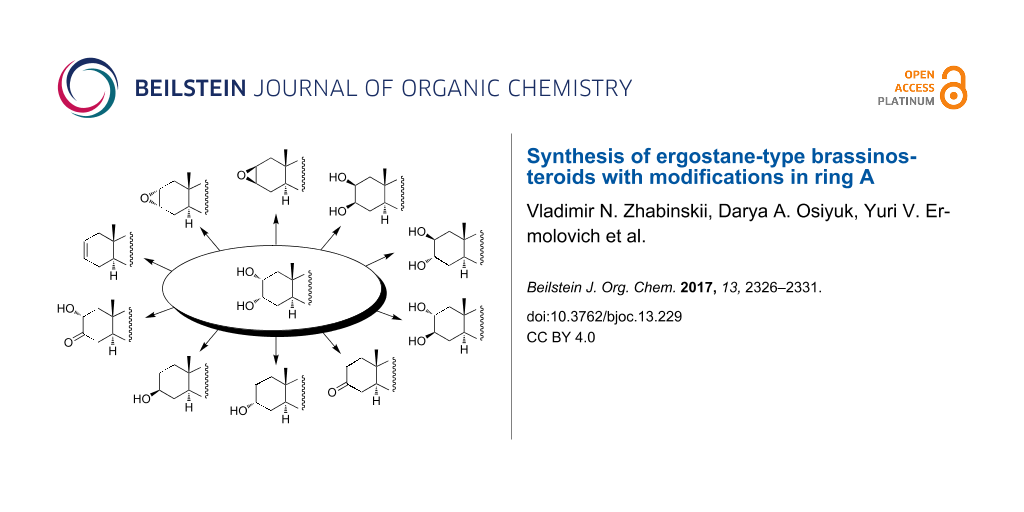
Graphical Abstract
Introduction
The group of steroid plant hormones called brassinosteroids (BS) currently comprises about 70 compounds [1]. It is generally accepted that only few of them (such as brassinolide, castasterone, epibrassinolide, etc.), possessing 2α,3α-, (22R,23R)-diol groups, B-lactone, and 6-ketone moieties, exhibit hormonal activity in plants, whereas other BS are considered to be either biosynthetic precursors or metabolites of the “real” phytohormones [2]. All these compounds are part of a multidimensional biosynthetic metabolic network, the functioning of which is still far from being completely understood. Evidently, the identification of as much as possible elements of this network would contribute to its better knowledge.
Because of the extremely low BS content in natural sources, studies on their identification almost always included a preliminary chemical synthesis of the compounds in question as standards for identification purposes. Typically, such syntheses were carried out by sequential introduction of functional groups into the starting molecule leading to more complex products [3]. This is a suitable approach for the preparation of one or a limited number of BS, but it rapidly becomes laborious if the synthesis of a large set of compounds is desired. Recently, we have proposed an alternative methodology to minor BS constituents that is based on the transformation of more complex compounds into simpler ones [4-7]. This strategy has now become even more attractive as some naturally occurring BS (e.g., epibrassinolide [8]) have found practical application in agriculture and thus are commercially available and a cheap source for further chemical modifications.
As a part of our programme aimed at the study of BS biosynthesis, we focussed in the present investigation on the preparation of a set of minor ergostane-type BS bearing A-ring structural units 3–12 (Scheme 1). The required functionalities were thought to be realized via a relatively short synthetic route starting from 2α,3α-diol fragment 13 of epicastasterone (1) or epibrassinolide (2).
![[1860-5397-13-229-i1]](/bjoc/content/inline/1860-5397-13-229-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structural features of epicastasterone (1), epibrassinolide (2) and A-ring units 3–12 of BS biosynthetic precursors/metabolites.
Scheme 1: Structural features of epicastasterone (1), epibrassinolide (2) and A-ring units 3–12 of BS biosynt...
Results and Discussion
Δ2-Steroids of type 3
The possible existence of Δ2-steroids of type 3 (Scheme 1) as biosynthetic intermediates of BS was proposed in 1981 [9], but it lasted until 2003 when the corresponding compound named secasterol was found in seedlings of Secale cereale [10]. In subsequent years, a number of related Δ2-6-keto-22,23-diols was synthesized and assessed for biological activities in non-plant models. Some of the studied compounds showed a marked cytotoxicity against human cancer cell lines MCF-7 and LNCaP [11-13]. We tested two approaches for the transformation of epicastasterone (1) and epibrassinolide (2) into the corresponding Δ2-steroids. The first route comprised the selective protection of the side chain diol in 1 and 2 through exhaustive acetylation followed by saponification of the intermediate tetraacetates under controlled conditions [14]. Next, a Corey–Winter reaction [15] of the cyclic thiocarbonate 15 as a key reaction step gave the expected Δ2-olefin 16 in an excellent yield (Scheme 2). Subsequent deacetylation of 16 then afforded 24-episecasterol (17).
![[1860-5397-13-229-i2]](/bjoc/content/inline/1860-5397-13-229-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: (a) Ac2O, Py, DMAP, 60 °C; (b) K2CO3, MeOH, 20 °C (97% over 2 steps); (c) TCDI, DMAP, THF, 65 °C (76%); (d) (EtO)3P, 150 °C (94%); (e) KOH, MeOH, 65 °C (89%).
Scheme 2: (a) Ac2O, Py, DMAP, 60 °C; (b) K2CO3, MeOH, 20 °C (97% over 2 steps); (c) TCDI, DMAP, THF, 65 °C (7...
An alternative procedure to Δ2-steroids was applied for the preparation of the B-ring lactone derivative 21 (Scheme 3). The mesylation of diol 18 produced dimesylate 19, which was treated with zinc dust and sodium iodide in refluxing DMF (Tipson–Cohen reaction [16]) to give, after deacetylation, B-lactone olefin 21 in 96% yield.
![[1860-5397-13-229-i3]](/bjoc/content/inline/1860-5397-13-229-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: (a) MsCl, Py, 20 °C (95%); (b) Zn, NaI, DMF, 150 °C (83%); (c) KOH, MeOH, 65 °C (96%).
Scheme 3: (a) MsCl, Py, 20 °C (95%); (b) Zn, NaI, DMF, 150 °C (83%); (c) KOH, MeOH, 65 °C (96%).
2α,3α- and 2β,3β-epoxides of type 4 and 5
Olefins of type 3 are evident intermediates for the preparation of BS 4 and 5 with a 2,3-epoxide moiety (Scheme 1). To date, only the corresponding 6-ketones were found in natural sources (2,3-diepisecasterone [10] with an α-oriented epoxide 4 and two BS with a β-oriented epoxide 5: secasterone [10,17] and 24-episecasterone [18]). The synthesis of both isomeric 2,3-epoxides 22 and 24 with a 7-membered B-ring lactone was accomplished starting from the olefin 21 (Scheme 4).
![[1860-5397-13-229-i4]](/bjoc/content/inline/1860-5397-13-229-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (a) MCPBA, CH2Cl2, 20 °C (90%); (b) NBS, DME, 20 °C; (c) KOH, MeOH, 20 °C (85% over 2 steps).
Scheme 4: (a) MCPBA, CH2Cl2, 20 °C (90%); (b) NBS, DME, 20 °C; (c) KOH, MeOH, 20 °C (85% over 2 steps).
The reaction of peracids with Δ2-steroids possessing a six-membered B ring is known to proceed from the less hindered side of the molecule and results in the formation of 2α,3α-epoxides [19]. The 7-membered B ring in 21 had no influence on the stereochemical outcome of the reaction with MCPBA which resulted in the formation of the α-epoxide 22. The same was true for the electrophilic addition of Br+ to the olefinic unit of 21. It produced the trans-diaxial bromohydrine 23 that was transformed, on treatment with KOH, into the epoxide 24. The structures of isomeric epoxides 22 and 24 were confirmed by their NOESY spectra as shown in Scheme 4. The obvious NOE correlation between H-3 and H-5 in 24 suggested the spatial vicinity of these two protons, thus the epoxide ring was β-oriented. On the contrary, no correlation was observed between the corresponding protons in compound 22, which is consistent with the α-orientation of the epoxide ring.
2β,3β-, 2β,3α- and 2α,3β-diols of type 6–8
Experiments on the identification of steroidal phytohormones in immature seeds of Phaseolus vulgaris revealed the existence of all possible configurations of vicinal hydroxy groups among the 2,3-stereoisomers of castasterone [20]. Minor BS constituents with structural fragments of types 6–8 (Scheme 1) showed reduced growth promoting biological activity when compared to castasterone, thus indicating that the epimerization of the hydroxy group at C-2/C-3 is the inactivation process contributing to a constant level of the active phytohormone. 2β,3β-Diols 6 are available through Woodward–Prévost cis-dihydroxylation of Δ2-steroids 3 [21]. The diaxial diols 7 can be easily synthesized by an acid-catalyzed opening of the 2α,3α-epoxides 4 [22-24] or 2β,3β-epoxides 5 [24]. The preparation of the diequatorial derivatives 8 requires more effort. The synthesis of 3,24-diepicastasterone (29), identified in immature seeds of Phaseolus vulgaris as a natural BS [25], started from the diol 14 (Scheme 5). The selective benzylation of its equatorial hydroxy group [14] followed by chlorochromate oxidation gave, after removal of the benzyl protecting group in 26, the diketone 27. Its reduction proceeded regio- and stereoselectively to afford the 2α,3β-diol 28. Finally, treatment of this compound with KOH in MeOH led to the deprotected tetraol 29.
![[1860-5397-13-229-i5]](/bjoc/content/inline/1860-5397-13-229-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: (a) BnBr, DMAP, Bu2SnO, TBAI, DIPEA, 110 °C (94%); (b) PCC, CH2Cl2, 20 °C (84%); (c) H2, Pd/C, 20 °C (99%); (d) NaBH4, EtOH, −25 °C (61%); (e) KOH, MeOH, 65 °C (96%).
Scheme 5: (a) BnBr, DMAP, Bu2SnO, TBAI, DIPEA, 110 °C (94%); (b) PCC, CH2Cl2, 20 °C (84%); (c) H2, Pd/C, 20 °...
3-Ketones of type 9
Three compounds of this type are known among natural BS: 3-dehydroteasterone [26,27], 3-dehydro-6-deoxoteasterone [28], and 3-dehydro-6-deoxo-28-norteasterone [29]. Our proposed approach to ketones of type 9 (Scheme 1) is based on a stereospecific C(3) → C(2)-hydride shift/elimination process [30]. So, tosylation of the diol 14 occurred regioselectively at the equatorial C-2 hydroxy group to give monotosylate 30, which upon heating in pyridine, yielded compound 31 (Scheme 6). Its treatment with KOH in methanol led to 24-epi-3-dehydroteasterone 32, which is unknown as a natural phytohormone.
![[1860-5397-13-229-i6]](/bjoc/content/inline/1860-5397-13-229-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: (a) TsCl, DMAP, Py, 30 °C (91%); (b) Py, 115 °C (65%); (c) KOH, MeOH, 20 °C (52%).
Scheme 6: (a) TsCl, DMAP, Py, 30 °C (91%); (b) Py, 115 °C (65%); (c) KOH, MeOH, 20 °C (52%).
3α- and 3β-alcohols of type 10 and 11
The hydride reduction of 3,6-diketones proceeds in a regioselective manner at the C-3 position [19]. Both 3α- and 3β-alcohols of type 10 and 11 (Scheme 1) can be obtained in this way depending on the reducing agent used. Thus, the reaction of 3,6-diketones with K-selectride was shown to afford 3α-hydroxy-6-ketones [31]. On the other hand, treatment of the diketone 31 with NaBH4 followed by acetate deprotection led to 24-epiteasterone (34) having a 3β-hydroxy group on the A-ring (Scheme 7).
![[1860-5397-13-229-i7]](/bjoc/content/inline/1860-5397-13-229-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: (a) NaBH4, EtOH, −25 °C (49%); (b) KOH, MeOH, 65 °C (85%).
Scheme 7: (a) NaBH4, EtOH, −25 °C (49%); (b) KOH, MeOH, 65 °C (85%).
2α-Hydroxy-3-ketones of type 12
The only natural compound of type 12 (Scheme 1) known to date is 3-dehydro-24-epicastasterone (38) which was identified as a metabolite of 24-epicastasterone (1) in cell suspension cultures of Lycopersicon esculentum [32]. All attempts to prepare 38 through the intermediate 27 gave poor results, probably due to the low stability of the α-ketol moiety [24,33,34] under the conditions employed for the removal of the acetates at C-22 and C-23. The solution to this problem was the p-methoxybenzylidene protection of the 22,23-diol in epicastasterone (1). Its reaction with anisaldehyde proceeded regioselectively at the diol group of the side chain and gave, after benzylation at position 2, the alcohol 36 (Scheme 8). The oxidation of the latter product afforded compound 37, which after deprotection, delivered the desired 3-dehydro-24-epicastasterone (38).
![[1860-5397-13-229-i8]](/bjoc/content/inline/1860-5397-13-229-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: (a) Anisaldehyde, TMSCl, MeOH, 20 °C; (b) BnBr, DMAP, Bu2SnO, TBAI, DIPEA, 110 °C (86% over 2 steps); (c) PCC, CH2Cl2, 20 °C (81%); (d) H2, Pd/C, 20 °C (80%).
Scheme 8: (a) Anisaldehyde, TMSCl, MeOH, 20 °C; (b) BnBr, DMAP, Bu2SnO, TBAI, DIPEA, 110 °C (86% over 2 steps...
Conclusion
In conclusion, we have developed a new strategy for synthesizing minor constituents of the class of BS phytohormones which is based on the use of readily available brassinosteroids of this class. Its advantage is that it allows preparing a full set of A-ring units (Δ2-, 2α,3α- and 2β,3β-epoxy-, 2α,3β-, 2β,3α-, and 2β,3β-dihydroxy-, 3-keto-, 3α- and 3β-hydroxy-, 2α-hydroxy-3-keto-) which are characteristic for biosynthetic precursors/metabolites of BS. The 2α,3α-diol function of ergostane BS was shown to be transformable into the required functionalities in only two to four standard chemical transformations.
Supporting Information
| Supporting Information File 1: General information, experimental details, characterization data and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 11.7 MB | Download |
Acknowledgements
The study was supported by grants from the Russian Science Foundation (No. 16-16-04057), the Belarusian Foundation for Fundamental Research (projects X16K-057 and X17PM-040), and Ministry of Education, Youth and Sports of the Czech Republic through the National Program of Sustainability I (grant LO1204).
References
-
Bajguz, A. In Brassinosteroids: A Class of Plant Hormone; Hayat, S.; Ahmad, A., Eds.; Springer: Dordrecht, 2011; pp 1–27. doi:10.1007/978-94-007-0189-2_1
Return to citation in text: [1] -
Fujioka, S.; Yokota, T. Annu. Rev. Plant Biol. 2003, 54, 137–164. doi:10.1146/annurev.arplant.54.031902.134921
Return to citation in text: [1] -
Khripach, V. A.; Zhabinskii, V. N.; de Groot, A. Brassinosteroids. A New Class of Plant Hormones; Academic Press: San Diego, 1999.
Return to citation in text: [1] -
Alshakova, I. D.; Ermolovich, Y. V.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2015, 97, 72–77. doi:10.1016/j.steroids.2014.08.020
Return to citation in text: [1] -
Baradzenka, A. G.; Barysau, B. M.; Hurski, A. L.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2015, 101, 90–95. doi:10.1016/j.steroids.2015.06.004
Return to citation in text: [1] -
Hurski, A. L.; Ermolovich, Y. V.; Zhabinskii, V. N.; Khripach, V. A. Org. Biomol. Chem. 2015, 13, 1446–1452. doi:10.1039/c4ob02197e
Return to citation in text: [1] -
Zhabinskii, V. N.; Gulyakevich, O. V.; Kurman, P. V.; Shabunya, P. S.; Fatykhava, S. A.; Khripach, V. A. J. Labelled Compd. Radiopharm. 2015, 58, 469–472. doi:10.1002/jlcr.3353
Return to citation in text: [1] -
Khripach, V. A.; Zhabinskii, V. N.; Khripach, N. B. New Practical Aspects of Brassinosteroids and Results of Their Ten-Year Agricultural Use in Russia and Belarus. In Brassinosteroids; Hayat, S.; Ahmad, A., Eds.; Springer: Dordrecht, 2003; pp 189–230. doi:10.1007/978-94-017-0948-4_9
Return to citation in text: [1] -
Wada, K.; Marumo, S. Agric. Biol. Chem. 1981, 45, 2579–2585. doi:10.1080/00021369.1981.10864908
Return to citation in text: [1] -
Antonchick, A. P.; Schneider, B.; Zhabinskii, V. N.; Konstantinova, O. V.; Khripach, V. A. Phytochemistry 2003, 63, 771–776. doi:10.1016/S0031-9422(03)00354-6
Return to citation in text: [1] [2] [3] -
Misharin, A. Y.; Mehtiev, A. R.; Zhabinskii, V. N.; Khripach, V. A.; Timofeev, V. P.; Tkachev, Y. V. Steroids 2010, 75, 287–294. doi:10.1016/j.steroids.2010.01.006
Return to citation in text: [1] -
Khripach, V. A.; Zhabinskii, V. N.; Gulyakevich, O. V.; Konstantinova, O. V.; Misharin, A. Y.; Mekhtiev, A. R.; Timofeev, V. P.; Tkachev, Y. V. Russ. J. Bioorg. Chem. 2010, 36, 746–754. doi:10.1134/S1068162010060117
Return to citation in text: [1] -
Khripach, V. A.; Zhabinskii, V. N.; Ermolovich, Y. V.; Gulyakevich, O. V.; Mekhtiev, A. R.; Karalkin, P. A. Russ. J. Bioorg. Chem. 2012, 38, 438–446. doi:10.1134/S1068162012040097
Return to citation in text: [1] -
Zhylitskaya, H. A.; Chashchina, N. M.; Litvinovskaya, R. P.; Zavadskaya, M. I.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2017, 117, 2–10. doi:10.1016/j.steroids.2016.06.006
Return to citation in text: [1] [2] -
Corey, E. J.; Winter, R. A. E. J. Am. Chem. Soc. 1963, 85, 2677–2678. doi:10.1021/ja00900a043
Return to citation in text: [1] -
Tipson, R. S.; Cohen, A. Carbohydr. Res. 1965, 1, 338–340. doi:10.1016/S0008-6215(00)81770-X
Return to citation in text: [1] -
Schmidt, J.; Spengler, B.; Yokota, T.; Nakayama, M.; Takatsuto, S.; Voigt, B.; Adam, G. Phytochemistry 1995, 38, 1095–1097. doi:10.1016/0031-9422(94)00797-W
Return to citation in text: [1] -
Friebe, A.; Volz, A.; Schmidt, J.; Voigt, B.; Adam, G.; Schnabl, H. Phytochemistry 1999, 52, 1607–1610. doi:10.1016/S0031-9422(99)00209-5
Return to citation in text: [1] -
Fried, J.; Edwards, J. A., Eds. Organic Reactions in Steroid Chemistry; Van Nostrand Reinhold Comp.: New York, 1972; Vol. 2, pp 2–10.
Return to citation in text: [1] [2] -
Lee, S. C.; Joo, S.-H.; Kim, S.-K. J. Plant Biol. 2011, 54, 10–14. doi:10.1007/s12374-010-9131-x
Return to citation in text: [1] -
Brosa, C. Steroids 1994, 59, 463–467. doi:10.1016/0039-128X(94)90058-2
Return to citation in text: [1] -
Yang, Y.-X.; Zheng, L.-T.; Shi, J.-J.; Gao, B.; Chen, Y.-K.; Yang, H.-C.; Chen, H.-L.; Li, Y.-C.; Zhen, X.-C. Bioorg. Med. Chem. Lett. 2014, 24, 1222–1227. doi:10.1016/j.bmcl.2013.12.055
Return to citation in text: [1] -
Richmond, V.; Santos, G. A. G.; Murray, A. P.; Maier, M. S. Steroids 2011, 76, 1160–1165. doi:10.1016/j.steroids.2011.05.005
Return to citation in text: [1] -
Voigt, B.; Porzel, A.; Adam, G.; Golsch, D.; Adam, W.; Wagner, C.; Merzweiler, K. Collect. Czech. Chem. Commun. 2002, 67, 91–102. doi:10.1135/cccc20020091
Return to citation in text: [1] [2] [3] -
Kim, S.-K. ACS Symp. Ser. 1991, 474, 26–35. doi:10.1021/bk-1991-0474.ch003
Return to citation in text: [1] -
Abe, H.; Honjo, C.; Kyokawa, Y.; Asakawa, S.; Natsume, M.; Narushima, M. Biosci., Biotechnol., Biochem. 1994, 58, 986–989. doi:10.1271/bbb.58.986
Return to citation in text: [1] -
Yokota, T.; Nakayama, M.; Wakisaka, T.; Schmidt, J.; Adam, G. Biosci., Biotechnol., Biochem. 1994, 58, 1183–1185. doi:10.1271/bbb.58.1183
Return to citation in text: [1] -
Griffiths, P. G.; Sasse, J. M.; Yokota, T.; Cameron, D. W. Biosci., Biotechnol., Biochem. 1995, 59, 956–959. doi:10.1271/bbb.59.956
Return to citation in text: [1] -
Bhardwaj, R.; Kaur, S.; Nagar, P. K.; Arora, H. K. Plant Growth Regul. 2007, 53, 1. doi:10.1007/s10725-007-9197-8
Return to citation in text: [1] -
Müller, R.; Rüedi, P. Helv. Chim. Acta 2003, 86, 439–456. doi:10.1002/hlca.200390044
Return to citation in text: [1] -
Li, W.-S.; Morrison, H. Org. Lett. 2000, 2, 15–18. doi:10.1021/ol990319s
Return to citation in text: [1] -
Hai, T.; Schneider, B.; Porzel, A.; Adam, G. Phytochemistry 1996, 41, 197–201. doi:10.1016/0031-9422(95)00585-4
Return to citation in text: [1] -
Paquette, L. A.; Hofferberth, J. E. Org. React. 2004, 62, 477–567. doi:10.1002/0471264180.or062.03
Return to citation in text: [1] -
Klinot, J.; Sejbal, J.; Vystrčil, A. Collect. Czech. Chem. Commun. 1989, 54, 400–412. doi:10.1135/cccc19890400
Return to citation in text: [1]
| 32. | Hai, T.; Schneider, B.; Porzel, A.; Adam, G. Phytochemistry 1996, 41, 197–201. doi:10.1016/0031-9422(95)00585-4 |
| 24. | Voigt, B.; Porzel, A.; Adam, G.; Golsch, D.; Adam, W.; Wagner, C.; Merzweiler, K. Collect. Czech. Chem. Commun. 2002, 67, 91–102. doi:10.1135/cccc20020091 |
| 33. | Paquette, L. A.; Hofferberth, J. E. Org. React. 2004, 62, 477–567. doi:10.1002/0471264180.or062.03 |
| 34. | Klinot, J.; Sejbal, J.; Vystrčil, A. Collect. Czech. Chem. Commun. 1989, 54, 400–412. doi:10.1135/cccc19890400 |
| 1. | Bajguz, A. In Brassinosteroids: A Class of Plant Hormone; Hayat, S.; Ahmad, A., Eds.; Springer: Dordrecht, 2011; pp 1–27. doi:10.1007/978-94-007-0189-2_1 |
| 8. | Khripach, V. A.; Zhabinskii, V. N.; Khripach, N. B. New Practical Aspects of Brassinosteroids and Results of Their Ten-Year Agricultural Use in Russia and Belarus. In Brassinosteroids; Hayat, S.; Ahmad, A., Eds.; Springer: Dordrecht, 2003; pp 189–230. doi:10.1007/978-94-017-0948-4_9 |
| 19. | Fried, J.; Edwards, J. A., Eds. Organic Reactions in Steroid Chemistry; Van Nostrand Reinhold Comp.: New York, 1972; Vol. 2, pp 2–10. |
| 4. | Alshakova, I. D.; Ermolovich, Y. V.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2015, 97, 72–77. doi:10.1016/j.steroids.2014.08.020 |
| 5. | Baradzenka, A. G.; Barysau, B. M.; Hurski, A. L.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2015, 101, 90–95. doi:10.1016/j.steroids.2015.06.004 |
| 6. | Hurski, A. L.; Ermolovich, Y. V.; Zhabinskii, V. N.; Khripach, V. A. Org. Biomol. Chem. 2015, 13, 1446–1452. doi:10.1039/c4ob02197e |
| 7. | Zhabinskii, V. N.; Gulyakevich, O. V.; Kurman, P. V.; Shabunya, P. S.; Fatykhava, S. A.; Khripach, V. A. J. Labelled Compd. Radiopharm. 2015, 58, 469–472. doi:10.1002/jlcr.3353 |
| 20. | Lee, S. C.; Joo, S.-H.; Kim, S.-K. J. Plant Biol. 2011, 54, 10–14. doi:10.1007/s12374-010-9131-x |
| 3. | Khripach, V. A.; Zhabinskii, V. N.; de Groot, A. Brassinosteroids. A New Class of Plant Hormones; Academic Press: San Diego, 1999. |
| 10. | Antonchick, A. P.; Schneider, B.; Zhabinskii, V. N.; Konstantinova, O. V.; Khripach, V. A. Phytochemistry 2003, 63, 771–776. doi:10.1016/S0031-9422(03)00354-6 |
| 17. | Schmidt, J.; Spengler, B.; Yokota, T.; Nakayama, M.; Takatsuto, S.; Voigt, B.; Adam, G. Phytochemistry 1995, 38, 1095–1097. doi:10.1016/0031-9422(94)00797-W |
| 2. | Fujioka, S.; Yokota, T. Annu. Rev. Plant Biol. 2003, 54, 137–164. doi:10.1146/annurev.arplant.54.031902.134921 |
| 18. | Friebe, A.; Volz, A.; Schmidt, J.; Voigt, B.; Adam, G.; Schnabl, H. Phytochemistry 1999, 52, 1607–1610. doi:10.1016/S0031-9422(99)00209-5 |
| 14. | Zhylitskaya, H. A.; Chashchina, N. M.; Litvinovskaya, R. P.; Zavadskaya, M. I.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2017, 117, 2–10. doi:10.1016/j.steroids.2016.06.006 |
| 16. | Tipson, R. S.; Cohen, A. Carbohydr. Res. 1965, 1, 338–340. doi:10.1016/S0008-6215(00)81770-X |
| 11. | Misharin, A. Y.; Mehtiev, A. R.; Zhabinskii, V. N.; Khripach, V. A.; Timofeev, V. P.; Tkachev, Y. V. Steroids 2010, 75, 287–294. doi:10.1016/j.steroids.2010.01.006 |
| 12. | Khripach, V. A.; Zhabinskii, V. N.; Gulyakevich, O. V.; Konstantinova, O. V.; Misharin, A. Y.; Mekhtiev, A. R.; Timofeev, V. P.; Tkachev, Y. V. Russ. J. Bioorg. Chem. 2010, 36, 746–754. doi:10.1134/S1068162010060117 |
| 13. | Khripach, V. A.; Zhabinskii, V. N.; Ermolovich, Y. V.; Gulyakevich, O. V.; Mekhtiev, A. R.; Karalkin, P. A. Russ. J. Bioorg. Chem. 2012, 38, 438–446. doi:10.1134/S1068162012040097 |
| 10. | Antonchick, A. P.; Schneider, B.; Zhabinskii, V. N.; Konstantinova, O. V.; Khripach, V. A. Phytochemistry 2003, 63, 771–776. doi:10.1016/S0031-9422(03)00354-6 |
| 10. | Antonchick, A. P.; Schneider, B.; Zhabinskii, V. N.; Konstantinova, O. V.; Khripach, V. A. Phytochemistry 2003, 63, 771–776. doi:10.1016/S0031-9422(03)00354-6 |
| 9. | Wada, K.; Marumo, S. Agric. Biol. Chem. 1981, 45, 2579–2585. doi:10.1080/00021369.1981.10864908 |
| 15. | Corey, E. J.; Winter, R. A. E. J. Am. Chem. Soc. 1963, 85, 2677–2678. doi:10.1021/ja00900a043 |
| 24. | Voigt, B.; Porzel, A.; Adam, G.; Golsch, D.; Adam, W.; Wagner, C.; Merzweiler, K. Collect. Czech. Chem. Commun. 2002, 67, 91–102. doi:10.1135/cccc20020091 |
| 22. | Yang, Y.-X.; Zheng, L.-T.; Shi, J.-J.; Gao, B.; Chen, Y.-K.; Yang, H.-C.; Chen, H.-L.; Li, Y.-C.; Zhen, X.-C. Bioorg. Med. Chem. Lett. 2014, 24, 1222–1227. doi:10.1016/j.bmcl.2013.12.055 |
| 23. | Richmond, V.; Santos, G. A. G.; Murray, A. P.; Maier, M. S. Steroids 2011, 76, 1160–1165. doi:10.1016/j.steroids.2011.05.005 |
| 24. | Voigt, B.; Porzel, A.; Adam, G.; Golsch, D.; Adam, W.; Wagner, C.; Merzweiler, K. Collect. Czech. Chem. Commun. 2002, 67, 91–102. doi:10.1135/cccc20020091 |
| 19. | Fried, J.; Edwards, J. A., Eds. Organic Reactions in Steroid Chemistry; Van Nostrand Reinhold Comp.: New York, 1972; Vol. 2, pp 2–10. |
| 29. | Bhardwaj, R.; Kaur, S.; Nagar, P. K.; Arora, H. K. Plant Growth Regul. 2007, 53, 1. doi:10.1007/s10725-007-9197-8 |
| 30. | Müller, R.; Rüedi, P. Helv. Chim. Acta 2003, 86, 439–456. doi:10.1002/hlca.200390044 |
| 26. | Abe, H.; Honjo, C.; Kyokawa, Y.; Asakawa, S.; Natsume, M.; Narushima, M. Biosci., Biotechnol., Biochem. 1994, 58, 986–989. doi:10.1271/bbb.58.986 |
| 27. | Yokota, T.; Nakayama, M.; Wakisaka, T.; Schmidt, J.; Adam, G. Biosci., Biotechnol., Biochem. 1994, 58, 1183–1185. doi:10.1271/bbb.58.1183 |
| 28. | Griffiths, P. G.; Sasse, J. M.; Yokota, T.; Cameron, D. W. Biosci., Biotechnol., Biochem. 1995, 59, 956–959. doi:10.1271/bbb.59.956 |
| 14. | Zhylitskaya, H. A.; Chashchina, N. M.; Litvinovskaya, R. P.; Zavadskaya, M. I.; Zhabinskii, V. N.; Khripach, V. A. Steroids 2017, 117, 2–10. doi:10.1016/j.steroids.2016.06.006 |
© 2017 Zhabinskii et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)