Abstract
Background: Due to the high reactivity towards various C-nucleophiles, trifluoromethylketimines are known to be useful reagents for the synthesis of α-trifluoromethylated amine derivatives. However, decarboxylative reactions with malonic acid and its mono(thio)esters have been poorly investigated so far despite the potential to become a convenient route to β-trifluoromethyl-β-amino acid derivatives and to their partially saturated heterocyclic analogues.
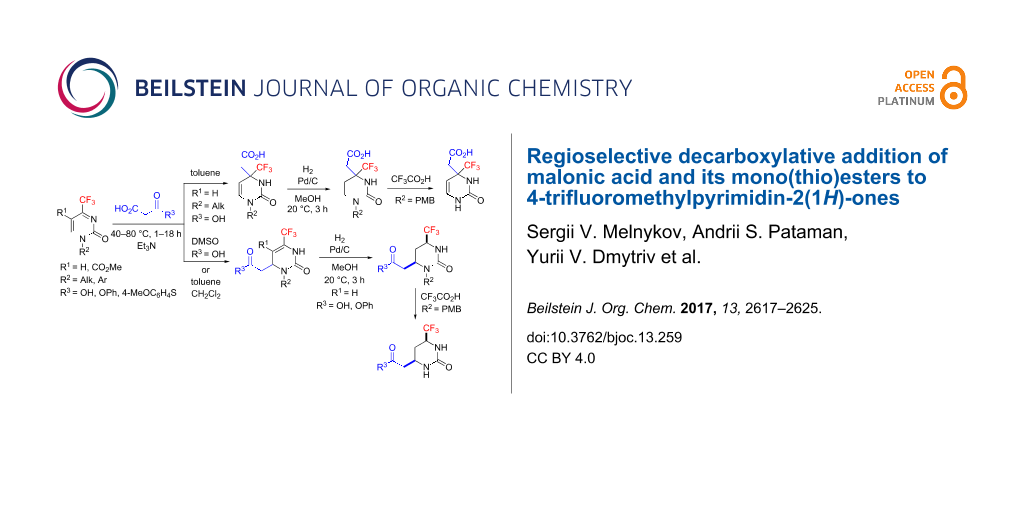
Results: In this paper we show that 4-trifluoromethylpyrimidin-2(1H)-ones, unique heterocyclic ketimines, react with malonic acid under organic base catalysis to regioselectively provide either Michael- or Mannich-type decarboxylative addition products depending on solvent polarity. Malonic mono(thio)esters give exclusively Michael-type products. The two regioisomeric products can be converted into saturated (2-oxohexahydropyrimidin-4-yl)acetic acid derivatives by mild hydrogenation of the endocyclic C=C double bond in the presence of Pd/C as catalyst. The cis-stereoisomers selectively formed upon reduction of the Michael-type products were structurally determined by X-ray diffraction. As a result of this study, a number of novel acetic acid derivatives containing trifluoromethylated, partially or fully saturated 2-oxopyrimidine cores were prepared and characterized as promising building blocks.
Conclusions: Regio- and stereoselective protocols have been developed for the synthesis of novel isomeric 4(6)-trifluoromethylated 1,2,3,4-tetrahydro- and perhydro-(2-oxopyrimidin-4-yl)acetic acid derivatives.

Graphical Abstract
Introduction
Organofluorine compounds now play an essential role in the development of new materials for solar cells [1-3], radiotracers for PET imaging [4], agrochemicals [5,6], sensitive chemical probes for 19F nuclear magnetic resonance investigation of biological experiments [7,8], and are most widely used in the modern drug discovery and development area [9,10]. As a result of intensive research efforts over the last decades, efficient fluorination and fluoroalkylation methods have emerged to prepare previously challenging molecules decorated with fluorine atoms or fluorinated groups which make them practically useful [11-14]. A building-block approach remains an alternative strategy to the synthesis of fluorine-containing compounds. This complementary method takes advantage of specific reagents featuring original fluorinated motives and/or functional groups which affords more complex derivatives via conventional functionalization or (hetero)cyclization [15-17]. Among these reagents, trifluoromethylketimines have drawn much research interest in recent years as key starting materials for the synthesis of trifluoromethyl-substituted amines [18,19], α-amino acids [20-23] as well as nitrogen-containing heterocyclic compounds [24-29]. It should be noted that the presence of a strong electron-withdrawing trifluoromethyl group is responsible for the sufficient reactivity of the electrophilic ketimine function with various carbon nucleophiles in these reactions.
Recently, the decarboxylative addition of malonic acid mono(thio)esters to aldehydes and imines has become an increasingly popular synthetic strategy [28,30-36]. However, the utility of trifluoromethylketimines as electrophilic substrates in this reaction remains underinvestigated. The only published work from the group of Ma described the development of a chiral thiourea-catalyzed enantioselective decarboxylative Mannich reaction of malonic acid monoesters with 4-trifluoromethylquinazolin-2(1H)-ones as heterocyclic trifluoromethylketimine substrates for the preparation of enantioenriched 3,4-dihydroquinazolin-2(1H)-ones and the anti-HIV drug DPC 083 [28]. No examples of any ketimines reacting directly with malonic acid have been reported so far.
Here we present the results of the decarboxylative addition of malonic acid, malonic monoester 1a and thioester 1b to 4-trifluoromethylpyrimidin-2(1H)-ones 2 (Figure 1). These compounds are unique heterocyclic conjugated trifluoromethylketimines with two competing electrophilic centers which can enable either Michael- or Mannich-type nucleophilic additions. As found in our previous studies, organocatalytic addition of acetone [37], nitromethane [38] and trimethylsilyl cyanide [39] in most cases can be performed regioselectively after optimization of the reaction conditions (temperature, solvent, time and catalyst nature). In general, under kinetic reaction control, the Michael-type 1,4-adducts are the predominant products while under thermodynamic control, the regioisomeric Mannich-type 1,2-adducts are more likely to be formed. These observations allowed us to develop selective methods for the synthesis of functionalized partially saturated 4-trifluoromethyl-substituted pyrimidin-2(1H)-ones, in particular, 4,5-dihydroorotic acid analogues 3 [39]. Here we report the preparation of acid 3 homologues (with a methylene linker between the carboxylic group and the pyrimidine ring) and their isomers resulting from two alternative regioselective pathways for the decarboxylative nucleophilic addition of malonic acid and its mono(thio)esters.
![[1860-5397-13-259-1]](/bjoc/content/figures/1860-5397-13-259-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
We first screened organic base catalysts, solvents and temperature in the decarboxylative addition of malonic acid (nucleophilic component) to 1-methyl-4-trifluoromethylpyrimidin-2(1H)-one (2a, the simplest model substrate) aiming to find the optimal organocatalytic reaction conditions (Table 1). In the preliminary experiments, it was established that the reaction was quite slow; heating and a 5-fold excess of malonic acid were required to reach a reasonable conversion. Additionally, it was found that a stoichiometric amount of a model catalyst, triethylamine (TEA), was necessary for the reaction to proceed efficiently. Thus, heating the reaction mixture in toluene at 80 °C for 18 h in the presence of 1 equivalent of TEA resulted in a satisfactory 84% conversion and led to the Mannich-type product, (1-methyl-2-oxo-4-trifluoromethyl-1,2,3,4-tetrahydropyrimidin-4-yl)acetic acid (4a), along with a small amount of the Michael-type regioisomer 5a (Table 1, entry 1). The reaction course and the ratio of the regioisomers formed were conveniently monitored by 19F NMR spectroscopy. Acid 4a precipitated in pure form on evaporating toluene and treating the residue with diluted hydrochloric acid. Performing the reaction in a more polar solvent such as THF drastically shifted the regioselectivity to the Michael-type adduct formation (Table 1, entry 2). Using DMSO as solvent and heating the reaction at 80 °C provided exclusively (3-methyl-2-oxo-6-trifluoromethyl-1,2,3,4-tetrahydropyrimidin-4-yl)acetic acid (5a) in high isolated yield (Table 1, entry 3). Methanol proved to be an unsuitable solvent for this reaction in terms of both conversion and selectivity (Table 1, entry 4). Likewise, diisopropylethylamine (DIEA) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were found to be not superior to TEA as catalysts (Table 1, entries 5 and 6). Unfortunately, quinine and the chiral quinine-derived thiourea organocatalyst QT in toluene led to a mixture of racemic products along with 19% and 38% of unreacted starting material, respectively (Table 1, entries 7 and 8).
Table 1: Screening of the reaction conditions for organic base-catalyzed malonic acid addition to 1-methyl-4-trifluoromethylpyrimidin-2(1H)-one (2a).
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-259-i2.svg?max-width=637&scale=1.0)
|
||||||
| entry | base (1 equiv) | solvent | temp. (°C) | conv. (%) | 4a:5a ratio (%) | product (isolated yield, %) |
|---|---|---|---|---|---|---|
| 1 | TEA | toluene | 80 | 84 | 92:8 | 4a (68) |
| 2 | TEA | THF | 65 | 97 | 13:87 | 5a (63) |
| 3 | TEA | DMSO | 80 | 94 | 1:99 | 5a (85) |
| 4 | TEA | MeOH | 63 | 46 | 21:79 | – |
| 5 | DIEA | toluene | 80 | 83 | 88:12 | 4a (66) |
| 6 | DBU | toluene | 80 | 80 | 85:15 | – |
| 7 | quinine | toluene | 80 | 81 | 47:53a | – |
| 8 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-259-i3.svg?max-width=637&scale=1.0)
QT |
toluene | 80 | 62 | 34:66a | – |
aThe two regioisomers were racemic.
As seen from the screening results, the reaction regioselectivity is easily solvent controlled. Non-polar toluene is the preferential solvent for the Mannich-type decarboxylative addition to the C=N double bond while polar DMSO promotes the highly selective Michael-type addition to the C=C double bond. These observations are explained by the fact that the initially formed (kinetically controlled) Michael-type dicarboxylate adduct A is much more stable in a low-polar than in a high-polar solvent (Table 1). In the former case, the long-living intermediate A is gradually converted, via the reversible first reaction step, into the energetically advantageous (thermodynamically controlled) Mannich-type adduct B, followed by rapid irreversible decarboxylation of B into compound 4a. Contrastingly, in a high-polar solvent, the intermediate A is so labile that it undergoes decarboxylation to product 5a rather than rearrangement to B. The proposed reaction mechanism is supported by the known effect of solvent polarity on the decarboxylation rate of malonic acid derivatives which was claimed to be faster in polar media [40].
To study the substrate scope of the regioselective additions of malonic acid, we introduced substituted pyrimidones 2b–m in the reaction and performed it under optimal conditions using toluene or DMSO as solvent and TEA (1 equiv) as catalyst (Table 2). The alkyl substituent at the nitrogen atom of the substrate had no significant effect on the reaction course. In all cases, both regioisomers, 4b–i and 5b–i, were isolated in modest to high yields (Table 2, entries 1–16). The presence of the ester functionality at position 5 of the heterocycle led to product mixtures if toluene was used as solvent so that products 4j–m could not be obtained selectively and separated. In DMSO solution, the corresponding Michael-type adducts 5j–m were smoothly formed and obtained in 75–83% isolated yields (Table 2, entries 17–20). 4-Trifluoromethylpyrimidin-2(1H)-ones 2 lacking a substituent at position 1 (structures not shown) were found to be completely unreactive in the decarboxylative reaction under study.
With the aim of preparing the corresponding N1(3)-unsubstituted products 4j and 5n,o, we utilized N1(3)-(4-methoxybenzyl) derivatives 4i, 5i, 5k in trifluoroacetic acid (TFA); the resulting cleavage of the 4-methoxybenzyl (PMB) group afforded the target compounds in good yields (see Table 2).
Table 2: Regioselective decarboxylative addition of malonic acid to 4-trifluoromethylpyrimidin-2(1H)-ones 2b–m and preparation of N1(3)-unsubstituted compounds 4j and 5n,o.
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-259-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry | comp. 2 | R1 | R2 | isol. product | yield (%) |
|---|---|---|---|---|---|
| 1 | b | H | Et | 4b | 62 |
| 2 | b | H | Et | 5b | 58 |
| 3 | c | H | n-Bu | 4c | 67 |
| 4 | c | H | n-Bu | 5c | 55 |
| 5 | d | H | Me2CHCH2 | 4d | 57 |
| 6 | d | H | Me2CHCH2 | 5d | 51 |
| 7 | e | H | MeOCH2CH2 | 4e | 59 |
| 8 | e | H | MeOCH2CH2 | 5e | 64 |
| 9 | f | H | CH2=CHCH2 | 4f | 73 |
| 10 | f | H | CH2=CHCH2 | 5f | 82 |
| 11 | g | H | Bn | 4g | 68 |
| 12 | g | H | Bn | 5g | 89 |
| 13 | h | H | 4-FC6H4CH2 | 4h | 60 |
| 14 | h | H | 4-FC6H4CH2 | 5h | 80 |
| 15 | i | H | 4-MeOC6H4CH2 | 4i | 65 |
| 16 | i | H | 4-MeOC6H4CH2 | 5i | 82 |
| 17 | j | CO2Me | 4-FC6H4CH2 | 5j | 75 |
| 18 | k | CO2Me | 4-MeOC6H4CH2 | 5k | 83 |
| 19 | l | CO2Me | 4-ClC6H4 | 5l | 81 |
| 20 | m | CO2Me | 4-MeOC6H4 | 5m | 81 |
Next we studied the decarboxylative addition of reagent 1a to model substrate 2a (Table 3) to compare the reactivity of malonic acid and its monophenyl ester 1a. It was proved again that the reaction proceeded sufficiently fast in toluene only in the presence of a stoichiometric amount of TEA or DIEA (Table 3, entries 1 and 2). Under these conditions the reaction provided the Michael-type adduct, phenyl 2-(3-methyl-2-oxo-6-trifluoromethyl-1,2,3,4-tetrahydropyrimidin-4-yl)acetate (6a). The presence of DBU caused substantial decarboxylation of starting reagent 1a (Table 3, entry 3). This unwanted process necessitated using of up to 6 equivalents of 1a to reach a reasonable conversion with TEA as catalyst. Quinine and QT were again found to be ineffective to promote the enantioselective reaction (Table 3, entries 4 and 5). In contrast to the reaction with malonic acid under similar conditions, just trace amounts of regioisomeric Mannich-type adduct 7a were detected. Presumably, in this case, the kinetically controlled Michael-type intermediate C is even far more prone to decarboxylation than the dicarboxylate intermediate A (Table 1) and hence, the reaction is sufficiently regioselective irrespective of the solvent polarity (Table 3, entries 1, and 6–9). Performing the reaction in toluene in the presence of TEA (1 equiv) at 80 °C for 4 hours gave the best result in terms of regioselectivity and yield of 6a (Table 3, entry 1), virtually the only product formed in all the solvents used here (as evidenced by 19F NMR monitoring).
Table 3: Screening of the reaction conditions for organic base-catalyzed malonic acid monophenyl ester (1a) addition to 1-methyl-4-trifluoromethylpyrimidin-2(1H)-one (2a).
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-259-i5.svg?max-width=637&scale=1.0)
|
||||||
| entry | solvent | base | temp. (°C) | time (h) | conv. | yield 6a (%)a |
|---|---|---|---|---|---|---|
| 1 | toluene | TEA | 80 | 4 | 98 | 81 |
| 2 | toluene | DIEA | 80 | 4 | 96 | 77 |
| 3 | toluene | DBU | 80 | 4 | 10 | – |
| 4 | toluene | quinine | 80 | 4 | 92 | 68b |
| 5 | toluene | QT | 80 | 4 | 78 | 55b |
| 6 | CH2Cl2 | TEA | 40 | 8 | 94 | 75 |
| 7 | THF | TEA | 66 | 8 | 90 | 74 |
| 8 | dioxane | TEA | 80 | 4 | 91 | 80 |
| 9 | DMSO | TEA | 80 | 4 | 93 | 81 |
aThe regioisomeric product 7a was formed in a negligible amount in all cases; bRacemic product.
The addition of malonic acid monophenyl ester (1a) to substituted pyrimidones 2b–e carried out in the presence of TEA in toluene for 8 h has shown that a substituent at position 1 of the pyrimidine ring can significantly influence the progress of the reaction (Table 4). Thus, N1-alkyl-substituted compounds 2b–e exhibited a lower reactivity compared to 2a and the corresponding products 6b–e were not isolated due to low conversion and regioselectivity (Table 4, entries 1–4). These are likely caused by the enhanced steric hindrance around the neighboring electrophilic position 6 and also the lowered electrophilicity of the reaction center. Consequently, the nucleophilic attack on the C=N double bond becomes equally probable thus leading to the loss of regioselectivity. Fortunately, allyl and various benzyl or phenyl substituents in derivatives 2f–m allowed the regioselective synthesis of products 6f–m in high yields (Table 4, entries 5–12). We found that the ester group at position 5 significantly increases the electrophilicity of the endocyclic C=C double bond giving rise to faster addition of 1a and higher regioselectivity of products 6j–m (Table 4, entries 9–12). Like N3-unsubstituted compounds 5n,o, their phenyl ester analogues 6n,o were obtained by the cleavage of the N3-PMB substituent on short heating in TFA (see Table 4). It has been shown that acids 4a,f–m can be synthesized alternatively by alkaline hydrolysis of esters 6a,f–m (see Supporting Information File 1 for full experimental data). The ester group at position 5 remained intact during the hydrolysis.
Table 4: Regioselective decarboxylative addition of malonic acid monophenyl ester (1a) to 4-trifluoromethylpyrimidin-2(1H)-ones 2b–m and preparation of N3-unsubstituted compounds 6n,o.
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-259-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | comp. 2, 6 | R1 | R2 | time (h) | conv. | yield 6 (%) |
|---|---|---|---|---|---|---|
| 1 | b | H | Et | 8 | 50 | – |
| 2 | c | H | n-Bu | 8 | 55 | – |
| 3 | d | H | Me2CHCH2 | 8 | 49 | – |
| 4 | e | H | MeOCH2CH2 | 8 | 44 | – |
| 5 | f | H | CH2=CHCH2 | 4 | 97 | 75 |
| 6 | g | H | Bn | 4 | 99 | 70 |
| 7 | h | H | 4-FC6H4CH2 | 4 | 99 | 74 |
| 8 | i | H | 4-MeOC6H4CH2 | 4 | 98 | 69 |
| 9 | j | CO2Me | 4-FC6H4CH2 | 2 | 99 | 80 |
| 10 | k | CO2Me | 4-MeOC6H4CH2 | 2 | 97 | 71 |
| 11 | l | CO2Me | 4-ClC6H4 | 2 | 98 | 73 |
| 12 | m | CO2Me | 4-MeOC6H4 | 2 | 99 | 75 |
Malonic acid monothioesters are known to be more reactive C-nucleophiles than the corresponding esters [41]. Therefore, we studied the decarboxylative addition of compound 1b as representative example to substrates 2a–m (Table 5). They were found to furnish Michael-type addition products 8a–m on heating in CH2Cl2 at 40 °C in excellent yields. Moreover, 3 equivalents excess of 1b was sufficient for the reaction to be completed within 1–3 hours.
Table 5: Regioselective decarboxylative addition of malonic acid mono-4-methoxyphenyl thioester (1b) to 4-trifluoromethylpyrimidin-2(1H)-ones 2a–m.
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-259-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | comp. 2, 8 | R1 | R2 | time (h) | yield 8 (%) |
|---|---|---|---|---|---|
| 1 | a | H | Me | 3 | 83 |
| 2 | b | H | Et | 3 | 71 |
| 3 | c | H | n-Bu | 3 | 77 |
| 4 | d | H | Me2CHCH2 | 3 | 77 |
| 5 | e | H | MeOCH2CH2 | 3 | 75 |
| 6 | f | H | CH2=CHCH2 | 3 | 73 |
| 7 | g | H | Bn | 3 | 74 |
| 8 | h | H | 4-FC6H4CH2 | 3 | 71 |
| 9 | i | H | 4-MeOC6H4CH2 | 3 | 70 |
| 10 | j | CO2Me | 4-FC6H4CH2 | 1 | 77 |
| 11 | k | CO2Me | 4-MeOC6H4CH2 | 1 | 75 |
| 12 | l | CO2Me | 4-ClC6H4 | 1 | 81 |
| 13 | m | CO2Me | 4-MeOC6H4 | 1 | 72 |
Satisfactory conversion and regioselectivity were achieved even with substrates 2b–e bearing ethyl, n-butyl, isobutyl and 2-methoxyethyl substituents which demonstrated low reactivity in the addition reaction with ester analogue 1a. It can thus be inferred that the substituents R1 and R2 have almost no impact on the outcome of the decarboxylative addition provided a highly reactive nucleophilic component such as malonic acid monothioester 1b is used.
Importantly, representative compounds 6a,j and 8f readily reacted with benzylamine thus showing the possibility of esters 6 and thioesters 8 to be convenient amine acylating agents [32] and, hence, building blocks for direct preparation of the amide derivatives (see Supporting Information File 1 for examples of the corresponding amide syntheses).
In a next set of experiments, the endocyclic C=C double bond of the decarboxylative adducts 4–6 were hydrogenated to prepare compounds with a saturated 3,4,5,6-tetrahydropyrimidin-2(1H)-one ring functionalized with an acetic acid moiety and a trifluoromethyl group. Thus, the acids 4a,g,i quantitatively yielded reduced products 9a–c under mild catalytic conditions (when reacted with hydrogen at atmospheric pressure and room temperature for 3 hours in the presence of 10% Pd/C catalyst) as shown in Scheme 1. The simplest acetic acid derivative 9d was synthesized from 9c in a good yield by using the general procedure for N1-PMB cleavage. Likewise, regioisomeric acids 5a,g,i and their phenyl esters 6a,g,i were reduced to the respective saturated compounds 10a–c and 11a–c. In this case a high hydrogenation cis-stereoselectivity is provided when the Pd/C catalyst loading is smaller than 20 weight % (otherwise the reaction proceeds too fast leading to diastereomeric mixtures with a cis- to trans-ratio of up to 3:1).
![[1860-5397-13-259-i1]](/bjoc/content/inline/1860-5397-13-259-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Hydrogenation of compounds 4–6 and preparation of N1(3)-unsubstituted compounds 9–11d.
Scheme 1: Hydrogenation of compounds 4–6 and preparation of N1(3)-unsubstituted compounds 9–11d.
The relative cis-configuration of the CF3 and CH2COOPh substituents in the prepared phenyl (2-oxo-6-trifluoromethylhexahydropyrimidin-4-yl)acetates 11a–c was unambiguously corroborated by a single-crystal X-ray diffraction study of compound 11b (Figure 2, see Supporting Information File 1 for full structure description and experimental data). The configuration-preserving conversion of ester 11b into acid 10b by simple alkaline hydrolysis has also confirmed the cis-geometry for acids 10a–c obtained by direct hydrogenation of compounds 5a,g,i (see Supporting Information File 1). N3-Unsubstituted compounds 10d and 11d with the preserved cis-configuration of the substituents were readily prepared from the corresponding N3-PMB derivatives 10c and 11c by using the general procedure for N1(3)-PMB cleavage (see Scheme 1).
![[1860-5397-13-259-2]](/bjoc/content/figures/1860-5397-13-259-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of compound 11b. Two enantiomers form a heterochiral dimer in the crystal state. Intermolecular hydrogen bonds in the dimer are shown as dashed lines. Thermal ellipsoids are defined at 50% probability.
Figure 2: Molecular structure of compound 11b. Two enantiomers form a heterochiral dimer in the crystal state...
Conclusion
In conclusion, it has been demonstrated that the efficient and highly regioselective organocatalytic decarboxylative addition of malonic acid or its derivatives to 4-trifluoromethylpyrimidin-2(1H)-ones 2 is perfectly feasible with a precise control of the reaction conditions. A remarkable solvent effect has been observed which governs the ratio of the resulting regioisomeric decarboxylated adducts 4 and 5 and allows their preparative selective isolation. This effect may well be attributed to a two-step mechanism of the decarboxylative nucleophilic addition which is characterized by a faster decarboxylation of kinetically-controlled Michael-type intermediates in high-polar solvents.
Though malonic monoester 1a appears to be similar to malonic acid in reactivity towards compounds 2, it produces exclusively Michael-type adducts 6 regardless of the reaction conditions used. Likewise, the more reactive malonic mono thioester 1b, when reacted with a broader scope of substrates 2 under milder conditions, gives rise only to analogous Michael-type products 8. In general, the reactivity of substrates 2 can be increased by introduction of the ester functionality at position 5 as well as allyl, various benzyl or phenyl substituents at position 1 of the pyrimidine core. Notably, esters 6 and thioesters 8 are remarkable for their potential application as smooth amine acylating agents.
It has been shown that the 3,4-dihydropyrimidin-2(1H)-one ring in both Mannich- and Michael-type products 4 and 5, 6 can be readily hydrogenated under mild catalytic conditions to furnish saturated compounds 9 and 10, 11, respectively. Products 10 and 11 featuring two stereogenic centers were obtained only as cis-isomers.
N1(3)-Unsubstituted products 4j, 5n,o, 6n,o, and 9–11d, unavailable by direct decarboxylative addition, are readily accessible by TFA-mediated cleavage of the corresponding N1(3)-PMB substituted precursors.
References
-
Liang, Y.; Feng, D.; Wu, Y.; Tsai, S.-T.; Li, G.; Ray, C.; Yu, L. J. Am. Chem. Soc. 2009, 131, 7792–7799. doi:10.1021/ja901545q
Return to citation in text: [1] -
Son, H. J.; Wang, W.; Xu, T.; Liang, Y.; Wu, Y.; Li, G.; Yu, L. J. Am. Chem. Soc. 2011, 133, 1885–1894. doi:10.1021/ja108601g
Return to citation in text: [1] -
Stuart, A. C.; Tumbleston, J. R.; Zhou, H.; Li, W.; Liu, S.; Ade, H.; You, W. J. Am. Chem. Soc. 2013, 135, 1806–1815. doi:10.1021/ja309289u
Return to citation in text: [1] -
Brooks, A. F.; Topczewski, J. J.; Ichiishi, N.; Sanford, M. S.; Scott, P. J. H. Chem. Sci. 2014, 5, 4545–4553. doi:10.1039/C4SC02099E
Return to citation in text: [1] -
Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833
Return to citation in text: [1] -
Jeschke, P. Pest Manage. Sci. 2010, 66, 10–27. doi:10.1002/ps.1829
Return to citation in text: [1] -
Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. Chem. Soc. Rev. 2013, 42, 7971–7982. doi:10.1039/c3cs60129c
Return to citation in text: [1] -
Marsh, E. N. G.; Suzuki, Y. ACS Chem. Biol. 2014, 9, 1242–1250. doi:10.1021/cb500111u
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398–8427. doi:10.1039/c6ob00764c
Return to citation in text: [1] -
Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b
Return to citation in text: [1] -
Bizet, V.; Besset, T.; Ma, J.-A.; Cahard, D. Curr. Top. Med. Chem. 2014, 14, 901–940. doi:10.2174/1568026614666140202205531
Return to citation in text: [1] -
Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry, Techniques and Synthons; Chambers, R. D., Ed.; Topics in Current Chemistry, Vol. 193; Springer: Berlin, 1998; pp 131–195. doi:10.1007/3-540-69197-9_4
Return to citation in text: [1] -
Ren, X.; Wan, W.; Jiang, H.; Hao, J. Mini-Rev. Org. Chem. 2007, 4, 330–337. doi:10.2174/157019307782411662
Return to citation in text: [1] -
Zhu, S. Z.; Wang, Y. L.; Peng, W. M.; Song, L. P.; Jin, G. F. Curr. Org. Chem. 2002, 6, 1057–1096. doi:10.2174/1385272023373635
Return to citation in text: [1] -
Wu, Y.; Hu, L.; Li, Z.; Deng, L. Nature 2015, 523, 445–450. doi:10.1038/nature14617
Return to citation in text: [1] -
Kutovaya, I. V.; Shmatova, O. I.; Tkachuk, V. M.; Melnichenko, N. V.; Vovk, M. V.; Nenajdenko, V. G. Eur. J. Org. Chem. 2015, 6749–6761. doi:10.1002/ejoc.201500898
Return to citation in text: [1] -
Morisaki, K.; Sawa, M.; Yonesaki, R.; Morimoto, H.; Mashima, K.; Ohshima, T. J. Am. Chem. Soc. 2016, 138, 6194–6203. doi:10.1021/jacs.6b01590
Return to citation in text: [1] -
Sanz-Vidal, Á.; Miró, J.; Sánchez-Roselló, M.; del Pozo, C.; Fustero, S. J. Org. Chem. 2016, 81, 6515–6524. doi:10.1021/acs.joc.6b01139
Return to citation in text: [1] -
Miró, J.; Sánchez-Roselló, M.; González, J.; del Pozo, C.; Fustero, S. Chem. – Eur. J. 2015, 21, 5459–5466. doi:10.1002/chem.201406224
Return to citation in text: [1] -
Han, X.; Wu, H.; Wang, W.; Dong, C.; Tien, P.; Wu, S.; Zhou, H.-B. Org. Biomol. Chem. 2014, 12, 8308–8317. doi:10.1039/C4OB01333F
Return to citation in text: [1] -
Lou, H.; Wang, Y.; Jin, E.; Lin, X. J. Org. Chem. 2016, 81, 2019–2026. doi:10.1021/acs.joc.5b02848
Return to citation in text: [1] -
Zhou, B.; Jiang, C.; Rao Gandi, V.; Lu, Y.; Hayashi, T. Chem. – Eur. J. 2016, 22, 13068–13071. doi:10.1002/chem.201603105
Return to citation in text: [1] -
Zhou, D.; Huang, Z.; Yu, X.; Wang, Y.; Li, J.; Wang, W.; Xie, H. Org. Lett. 2015, 17, 5554–5557. doi:10.1021/acs.orglett.5b02668
Return to citation in text: [1] -
Yang, L.-J.; Li, S.; Wang, S.; Nie, J.; Ma, J.-A. J. Org. Chem. 2014, 79, 3547–3557. doi:10.1021/jo500356t
Return to citation in text: [1] -
Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307
Return to citation in text: [1] [2] [3] -
Shmatova, O. I.; Shevchenko, N. E.; Balenkova, E. S.; Röschenthaler, G.-V.; Nenajdenko, V. G. Eur. J. Org. Chem. 2013, 15, 3049–3058. doi:10.1002/ejoc.201201725
Return to citation in text: [1] -
Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536
Return to citation in text: [1] -
Li, X.-J.; Xiong, H.-Y.; Hua, M.-Q.; Nie, J.; Zheng, Y.; Ma, J.-A. Tetrahedron Lett. 2012, 53, 2117–2120. doi:10.1016/j.tetlet.2012.02.053
Return to citation in text: [1] -
Bae, H. Y.; Sim, J. H.; Lee, J.-W.; List, B.; Song, C. E. Angew. Chem., Int. Ed. 2013, 52, 12143–12147. doi:10.1002/anie.201306297
Return to citation in text: [1] [2] -
Jia, C.-M.; Zhang, H.-X.; Nie, J.; Ma, J.-A. J. Org. Chem. 2016, 81, 8561–8569. doi:10.1021/acs.joc.6b01750
Return to citation in text: [1] -
Saadi, J.; Wennemers, H. Nat. Chem. 2016, 8, 276–280. doi:10.1038/nchem.2437
Return to citation in text: [1] -
Bernardi, L.; Fochi, M.; Franchini, M. C.; Ricci, A. Org. Biomol. Chem. 2012, 10, 2911–2922. doi:10.1039/C2OB07037E
Return to citation in text: [1] -
Nakamura, S. Org. Biomol. Chem. 2014, 12, 394–405. doi:10.1039/C3OB42161A
Return to citation in text: [1] -
Sukach, V. A.; Tkachuk, V. M.; Shoba, V. M.; Pirozhenko, V. V.; Rusanov, E. B.; Chekotilo, A. A.; Röschenthaler, G.-V.; Vovk, M. V. Eur. J. Org. Chem. 2014, 1452–1460. doi:10.1002/ejoc.201301542
Return to citation in text: [1] -
Tkachuk, V. M.; Sukach, V. A.; Kovalchuk, K. V.; Vovk, M. V.; Nenajdenko, V. G. Org. Biomol. Chem. 2015, 13, 1420–1428. doi:10.1039/C4OB02233E
Return to citation in text: [1] -
Sukach, V. A.; Resetnic, A. A.; Tkachuk, V. M.; Lin, Z.; Kortz, U.; Vovk, M. V.; Röschenthaler, G.-V. Eur. J. Org. Chem. 2015, 1290–1301. doi:10.1002/ejoc.201403495
Return to citation in text: [1] [2] -
Midyana, G. G.; Makitra, R. G.; Pal’chikova, E. Y. Russ. J. Gen. Chem. 2010, 80, 944–947. doi:10.1134/S1070363210050142
Return to citation in text: [1] -
Bew, S. P.; Stephenson, G. R.; Rouden, J.; Godemert, J.; Seylani, H.; Martinez-Lozano, L. A. Chem. – Eur. J. 2017, 23, 4557–4569. doi:10.1002/chem.201605148
Return to citation in text: [1]
| 32. | Bae, H. Y.; Sim, J. H.; Lee, J.-W.; List, B.; Song, C. E. Angew. Chem., Int. Ed. 2013, 52, 12143–12147. doi:10.1002/anie.201306297 |
| 40. | Midyana, G. G.; Makitra, R. G.; Pal’chikova, E. Y. Russ. J. Gen. Chem. 2010, 80, 944–947. doi:10.1134/S1070363210050142 |
| 41. | Bew, S. P.; Stephenson, G. R.; Rouden, J.; Godemert, J.; Seylani, H.; Martinez-Lozano, L. A. Chem. – Eur. J. 2017, 23, 4557–4569. doi:10.1002/chem.201605148 |
| 1. | Liang, Y.; Feng, D.; Wu, Y.; Tsai, S.-T.; Li, G.; Ray, C.; Yu, L. J. Am. Chem. Soc. 2009, 131, 7792–7799. doi:10.1021/ja901545q |
| 2. | Son, H. J.; Wang, W.; Xu, T.; Liang, Y.; Wu, Y.; Li, G.; Yu, L. J. Am. Chem. Soc. 2011, 133, 1885–1894. doi:10.1021/ja108601g |
| 3. | Stuart, A. C.; Tumbleston, J. R.; Zhou, H.; Li, W.; Liu, S.; Ade, H.; You, W. J. Am. Chem. Soc. 2013, 135, 1806–1815. doi:10.1021/ja309289u |
| 9. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 10. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 39. | Sukach, V. A.; Resetnic, A. A.; Tkachuk, V. M.; Lin, Z.; Kortz, U.; Vovk, M. V.; Röschenthaler, G.-V. Eur. J. Org. Chem. 2015, 1290–1301. doi:10.1002/ejoc.201403495 |
| 7. | Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. Chem. Soc. Rev. 2013, 42, 7971–7982. doi:10.1039/c3cs60129c |
| 8. | Marsh, E. N. G.; Suzuki, Y. ACS Chem. Biol. 2014, 9, 1242–1250. doi:10.1021/cb500111u |
| 39. | Sukach, V. A.; Resetnic, A. A.; Tkachuk, V. M.; Lin, Z.; Kortz, U.; Vovk, M. V.; Röschenthaler, G.-V. Eur. J. Org. Chem. 2015, 1290–1301. doi:10.1002/ejoc.201403495 |
| 5. | Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833 |
| 6. | Jeschke, P. Pest Manage. Sci. 2010, 66, 10–27. doi:10.1002/ps.1829 |
| 37. | Sukach, V. A.; Tkachuk, V. M.; Shoba, V. M.; Pirozhenko, V. V.; Rusanov, E. B.; Chekotilo, A. A.; Röschenthaler, G.-V.; Vovk, M. V. Eur. J. Org. Chem. 2014, 1452–1460. doi:10.1002/ejoc.201301542 |
| 4. | Brooks, A. F.; Topczewski, J. J.; Ichiishi, N.; Sanford, M. S.; Scott, P. J. H. Chem. Sci. 2014, 5, 4545–4553. doi:10.1039/C4SC02099E |
| 38. | Tkachuk, V. M.; Sukach, V. A.; Kovalchuk, K. V.; Vovk, M. V.; Nenajdenko, V. G. Org. Biomol. Chem. 2015, 13, 1420–1428. doi:10.1039/C4OB02233E |
| 20. | Morisaki, K.; Sawa, M.; Yonesaki, R.; Morimoto, H.; Mashima, K.; Ohshima, T. J. Am. Chem. Soc. 2016, 138, 6194–6203. doi:10.1021/jacs.6b01590 |
| 21. | Sanz-Vidal, Á.; Miró, J.; Sánchez-Roselló, M.; del Pozo, C.; Fustero, S. J. Org. Chem. 2016, 81, 6515–6524. doi:10.1021/acs.joc.6b01139 |
| 22. | Miró, J.; Sánchez-Roselló, M.; González, J.; del Pozo, C.; Fustero, S. Chem. – Eur. J. 2015, 21, 5459–5466. doi:10.1002/chem.201406224 |
| 23. | Han, X.; Wu, H.; Wang, W.; Dong, C.; Tien, P.; Wu, S.; Zhou, H.-B. Org. Biomol. Chem. 2014, 12, 8308–8317. doi:10.1039/C4OB01333F |
| 28. | Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307 |
| 30. | Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536 |
| 31. | Li, X.-J.; Xiong, H.-Y.; Hua, M.-Q.; Nie, J.; Zheng, Y.; Ma, J.-A. Tetrahedron Lett. 2012, 53, 2117–2120. doi:10.1016/j.tetlet.2012.02.053 |
| 32. | Bae, H. Y.; Sim, J. H.; Lee, J.-W.; List, B.; Song, C. E. Angew. Chem., Int. Ed. 2013, 52, 12143–12147. doi:10.1002/anie.201306297 |
| 33. | Jia, C.-M.; Zhang, H.-X.; Nie, J.; Ma, J.-A. J. Org. Chem. 2016, 81, 8561–8569. doi:10.1021/acs.joc.6b01750 |
| 34. | Saadi, J.; Wennemers, H. Nat. Chem. 2016, 8, 276–280. doi:10.1038/nchem.2437 |
| 35. | Bernardi, L.; Fochi, M.; Franchini, M. C.; Ricci, A. Org. Biomol. Chem. 2012, 10, 2911–2922. doi:10.1039/C2OB07037E |
| 36. | Nakamura, S. Org. Biomol. Chem. 2014, 12, 394–405. doi:10.1039/C3OB42161A |
| 18. | Wu, Y.; Hu, L.; Li, Z.; Deng, L. Nature 2015, 523, 445–450. doi:10.1038/nature14617 |
| 19. | Kutovaya, I. V.; Shmatova, O. I.; Tkachuk, V. M.; Melnichenko, N. V.; Vovk, M. V.; Nenajdenko, V. G. Eur. J. Org. Chem. 2015, 6749–6761. doi:10.1002/ejoc.201500898 |
| 28. | Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307 |
| 15. | Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry, Techniques and Synthons; Chambers, R. D., Ed.; Topics in Current Chemistry, Vol. 193; Springer: Berlin, 1998; pp 131–195. doi:10.1007/3-540-69197-9_4 |
| 16. | Ren, X.; Wan, W.; Jiang, H.; Hao, J. Mini-Rev. Org. Chem. 2007, 4, 330–337. doi:10.2174/157019307782411662 |
| 17. | Zhu, S. Z.; Wang, Y. L.; Peng, W. M.; Song, L. P.; Jin, G. F. Curr. Org. Chem. 2002, 6, 1057–1096. doi:10.2174/1385272023373635 |
| 11. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 12. | Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398–8427. doi:10.1039/c6ob00764c |
| 13. | Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b |
| 14. | Bizet, V.; Besset, T.; Ma, J.-A.; Cahard, D. Curr. Top. Med. Chem. 2014, 14, 901–940. doi:10.2174/1568026614666140202205531 |
| 24. | Lou, H.; Wang, Y.; Jin, E.; Lin, X. J. Org. Chem. 2016, 81, 2019–2026. doi:10.1021/acs.joc.5b02848 |
| 25. | Zhou, B.; Jiang, C.; Rao Gandi, V.; Lu, Y.; Hayashi, T. Chem. – Eur. J. 2016, 22, 13068–13071. doi:10.1002/chem.201603105 |
| 26. | Zhou, D.; Huang, Z.; Yu, X.; Wang, Y.; Li, J.; Wang, W.; Xie, H. Org. Lett. 2015, 17, 5554–5557. doi:10.1021/acs.orglett.5b02668 |
| 27. | Yang, L.-J.; Li, S.; Wang, S.; Nie, J.; Ma, J.-A. J. Org. Chem. 2014, 79, 3547–3557. doi:10.1021/jo500356t |
| 28. | Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307 |
| 29. | Shmatova, O. I.; Shevchenko, N. E.; Balenkova, E. S.; Röschenthaler, G.-V.; Nenajdenko, V. G. Eur. J. Org. Chem. 2013, 15, 3049–3058. doi:10.1002/ejoc.201201725 |
© 2017 Melnykov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)