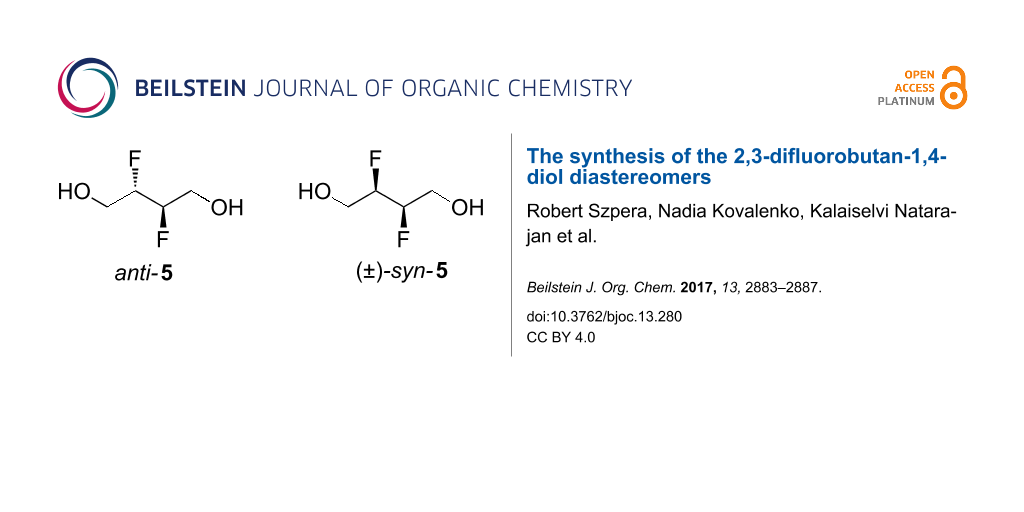
Abstract
The diastereoselective synthesis of fluorinated building blocks that contain chiral fluorine substituents is of interest. Here we describe optimisation efforts in the synthesis of anti-2,3-difluorobutane-1,4-diol, as well as the synthesis of the corresponding syn-diastereomer. Both targets were synthesised using an epoxide opening strategy.

Graphical Abstract
Introduction
The introduction of fluorine in organic compounds usually results in the modification of a range of chemical, physical and biological properties [1]. Fluorine incorporation is therefore a common strategy to optimise the properties of drugs/agrochemicals, as well as materials [2-6].
Many methods exist for the stereoselective introduction of the C–F group [7-11]. An alternative and often time-efficient approach is the use of fluorinated building blocks, where fluorine is introduced as part of a carbon containing fragment, sometimes also bearing other functionality [12,13]. The development of novel fluorinated building blocks is therefore of interest, particularly those that can be synthesised conveniently on a multigram scale. Interestingly, the majority of currently commercially available fluorinated building blocks do not contain stereogenic C–F bonds.
The vicinal-difluoride motif is known to exert conformational control through the fluorine gauche effect [14,15], and so building blocks containing this motif are of interest [16,17]. We have previously reported on the gram-scale synthesis of meso-2,3-difluorobutane-1,4-diol (anti-5) starting from commercially available cis-but-2-ene-1,4-diol (Scheme 1) [17]. The vicinal-difluoride group was introduced by a two-step sequence, with initial nucleophilic epoxide [18] opening by a fluoride source [19], followed by nucleophilic deoxyfluorination [9-11].
![[1860-5397-13-280-i1]](/bjoc/content/inline/1860-5397-13-280-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthesis of anti-2,3-difluorobutan-1,4-diol (anti-5) [17].
Scheme 1: The synthesis of anti-2,3-difluorobutan-1,4-diol (anti-5) [17].
In this contribution, we report on work directed at the further optimisation of the synthesis of anti-5, as well as on a gram-scale synthesis of its diastereomer (±)-syn-5, a novel compound.
Results and Discussion
Optimisation of the synthesis of anti-5
While the synthesis of anti-5 as described in Scheme 1 was high-yielding [17], two disadvantages were apparent. First, the epoxide opening takes 2.5 days at 115 °C and uses an expensive fluoride source (Landini’s reagent [18]: Bu4NH2F3). It was found that Bu4NH2F3 made in-house gave significantly reduced yields. Second, the use of the benzyl ether protecting group resulted in a significant increase in mass, and therefore, chromatographic purification of the protected intermediates upon scale-up was inconvenient.
As previously reported [17], epoxide opening of cis-2 with Olah’s reagent (HF·py) led to an 80% yield of the fluorohydrin after just three hours, however, the product was obtained as a mixture of both the syn- and anti-diastereomers. Whilst no mechanistic studies were conducted, it is possible that competing SN1 and/or anchimeric assistance by the benzyloxy group occurred. Work by Schlosser has shown that 1,2-disubstituted epoxide opening with Et3N·3HF proceeds with excellent diastereoselectivity [19]. Et3N·3HF is less acidic than Olah’s reagent, disfavouring SN1 and rearrangement pathways [20,21]. Indeed, the use of this reagent for the epoxide opening of cis-2 led to (±)-syn-3 in excellent yield (Scheme 2), with no significant isomerisation (see Supporting Information File 1). Epoxide opening with the recently described TBAF/KHF2 [22] was also possible, but in lower yield (75%, not shown). Incidentally, it was also found that the subsequent deoxofluorination reaction was somewhat higher yielding when DAST was added at rt over just 5 min, immediately followed by the addition of pyridine and heating at 70 °C.
![[1860-5397-13-280-i2]](/bjoc/content/inline/1860-5397-13-280-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Improved epoxide opening and deoxofluorination conditions.
Scheme 2: Improved epoxide opening and deoxofluorination conditions.
It should be noted that DAST is known to undergo decomposition at temperatures above 90 °C [23]. Here we use DAST in solution. The initial mixing is at room temperature, and heating doesn’t exceed 70 °C, and therefore, the procedure is deemed to have low risk. Nonetheless, care must be taken and the reaction was run with the protection of a blast shield.
In order to reduce the relative contribution of the protecting group to the overall weight of the intermediates, the use of an acetonide was explored. Given the starting alkene was cis-configured, its introduction was possible from the start (Scheme 3).
![[1860-5397-13-280-i3]](/bjoc/content/inline/1860-5397-13-280-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Attempted synthesis of anti-5 via acetonide protection.
Scheme 3: Attempted synthesis of anti-5 via acetonide protection.
Hence, following literature procedures [24-26], the reaction of cis-1 with 2,2-dimethoxypropane and subsequent epoxidation led to 7. However, epoxide opening with Et3N·3HF was accompanied by acetonide rearrangement to afford fluorohydrin (±)-9, containing the thermodynamically favoured five-membered ring [24]. This is clearly indicated by the appearance of a doublet of doublets for the primary alcohol OH proton. DAST-mediated deoxofluorination then led to (±)-10, in which an alkyl fluoride signal at −232 ppm confirmed the presence of a primary fluoride.
Hence, non-acidic epoxide opening conditions were investigated to circumvent the rearrangement (Scheme 4). Both the use of Bu4NH2F3 [18] and of the TBAF/KHF2 reagent combination [22] were successful (56% and 64%, respectively). While subsequent fluorination using PyFluor only led to the formation of the 2-pyridinesulfonate intermediate (±)-12, the use of DAST at 60 °C proved successful. The difluoride meso-11 was not isolated due to its low boiling point, but was immediately subjected to acid hydrolysis to give anti-5. Unfortunately, the yield for this two-step process was only moderate (30%).
![[1860-5397-13-280-i4]](/bjoc/content/inline/1860-5397-13-280-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Completion of the synthesis of anti-5.
Scheme 4: Completion of the synthesis of anti-5.
Synthesis of (±)-syn-5
The synthesis of (±)-syn-5 (Scheme 5) was achieved starting from the trans-configured but-2-ene-1,4-diol (1), which is not commercially available in geometrically pure form.
![[1860-5397-13-280-i5]](/bjoc/content/inline/1860-5397-13-280-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Hence, according to literature procedures, reduction of 1,4-butynediol (13) by LiAlH4 to give trans-1 [27] was followed by benzylation [28] and epoxidation with m-CPBA to give (±)-trans-2 [28]. When the reaction was performed on a small scale, excess m-CPBA and the byproduct 3-chlorobenzoic acid were removed by extraction with a saturated Na2S2O3 solution. However, on scale-up this proved inconvenient due to the large volumes of solvent required, and so these impurities were precipitated out the reaction mixture by cooling to 0 °C and collected by filtration through Celite. After work-up, the obtained epoxide was of high purity and no additional chromatographic purification was required, which was convenient on scale. The reaction of (±)-trans-2 with neat Et3N·3HF at 120 °C for 16 h led, after aqueous work-up, to (±)-anti-3 in high diastereomeric purity (see Supporting Information File 1). The 19F shift of −195.3 ppm is different compared to that of (±)-syn-3 (−204.4 ppm) [17]. Upon scale-up of the reaction to 10 g of (±)-trans-2, a similarly high yield of 90% (crude) was obtained, which again could be used directly in the next step without purification. Conversion of fluorohydrin (±)-anti-3 to difluoride (±)-syn-4 under the same conditions as shown in Scheme 2 resulted in 56% yield after column chromatography. 19F NMR analysis of the crude product showed a dr of 98:2 in favour of (±)-syn-4. However, given a diastereomerically pure starting material was used, this indicates that SN1 or neighbouring group participation pathways may have occurred, although only to a very small extent. Separation of the diastereomers proved not possible. Finally, deprotection of (±)-syn-4 by palladium catalysed hydrogenolysis led to (±)-syn-5. Recrystallization to remove the minor diastereomer was not successful.
Conclusion
A gram-scale synthesis of both syn- and anti-2,3-difluorobutan-1,4-diol diastereomers is described. The key steps involve epoxide opening and subsequent deoxyfluorination. For the first step, Et3N·3HF was found to be the best reagent, giving an excellent yield with no formation of diastereomeric byproducts. Unfortunately it was found that the subsequent DAST-mediated deoxyfluorination gives rise to a small amount of the undesired diastereomer. The primary alcohol groups require protection, for which the benzyl group has been employed. While this group is effective for this purpose, there is a significant mass increase upon its introduction (roughly three fold increase). An investigation to use the much smaller acetonide protecting group, which can be used for the cis-1,4-butenediol starting material, was carried out. It was found that the use of Et3N·3HF for the epoxide opening step also lead to acetal rearrangement, leading to a more stable 1,3-dioxolane ring. While the use of Bu4NH2F3/KHF2 and TBAF/KHF2 achieves epoxide opening without acetonide rearrangement, the subsequent deoxyfluorination/deprotection sequence is low yielding (30%). Overall, the protocols provided will be of use for the large-scale synthesis of both syn- and anti-2,3-difluorobutan-1,4-diol building blocks.
Supporting Information
| Supporting Information File 1: Experimental part and NMR spectra. | ||
| Format: PDF | Size: 3.3 MB | Download |
References
-
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432. doi:10.1021/cr4002879
Return to citation in text: [1] -
Fujiwara, T.; O'Hagan, D. J. Fluorine Chem. 2014, 167, 16. doi:10.1016/j.jfluchem.2014.06.014
Return to citation in text: [1] -
O'Hagan, D. J. Fluorine Chem. 2010, 131, 1071. doi:10.1016/j.jfluchem.2010.03.003
Return to citation in text: [1] -
Kirsch, P.; Bremer, M. ChemPhysChem 2010, 11, 357. doi:10.1002/cphc.200900745
Return to citation in text: [1] -
Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073. doi:10.1021/cr500706a
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398. doi:10.1039/C6OB00764C
Return to citation in text: [1] [2] -
Al-Maharik, N.; O'Hagan, D. Aldrichimica Acta 2011, 44, 65.
Return to citation in text: [1] [2] -
Hu, W.-L.; Hu, X.-G.; Hunter, L. Synthesis 2017, 49, 4917. doi:10.1055/s-0036-1590881
Return to citation in text: [1] [2] -
Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R. D., Ed.; Topics in Current Chemistry, Vol. 193; Springer: Berlin, Heidelberg, 1997; pp 131–196.
Return to citation in text: [1] -
Harsanyi, A.; Sandford, G. Org. Process Res. Dev. 2014, 18, 981. doi:10.1021/op500141c
Return to citation in text: [1] -
Thiehoff, C.; Rey, Y. P.; Gilmour, R. Isr. J. Chem. 2017, 57, 92. doi:10.1002/ijch.201600038
Return to citation in text: [1] -
Hunter, L. Beilstein J. Org. Chem. 2010, 6, No. 38. doi:10.3762/bjoc.6.38
Return to citation in text: [1] -
O'Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19
Return to citation in text: [1] -
Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Landini, D.; Penso, M. Tetrahedron Lett. 1990, 31, 7209. doi:10.1016/S0040-4039(00)97281-2
Return to citation in text: [1] [2] [3] -
Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875. doi:10.1016/S0040-4020(88)90023-3
Return to citation in text: [1] [2] -
Haufe, G. J. Fluorine Chem. 2004, 125, 875. doi:10.1016/j.jfluchem.2004.01.023
Return to citation in text: [1] -
Wölker, D.; Haufe, G. J. Org. Chem. 2002, 67, 3015. doi:10.1021/jo016331r
Return to citation in text: [1] -
Yan, N.; Lei, Z.-W.; Su, J.-K.; Liao, W.-L.; Hu, X.-G. Chin. Chem. Lett. 2016, 28, 467. doi:10.1016/j.cclet.2016.10.006
Return to citation in text: [1] [2] -
Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048. doi:10.1021/jo990566+
Return to citation in text: [1] -
Elliott, W. J.; Fried, J. J. Org. Chem. 1976, 41, 2469. doi:10.1021/jo00876a027
Return to citation in text: [1] [2] -
Al-Dulayymi, A.; Li, X.; Neuenschwander, M. Helv. Chim. Acta 2000, 83, 1633. doi:10.1002/1522-2675(20000705)83:7<1633::AID-HLCA1633>3.0.CO;2-5
Return to citation in text: [1] -
Mischitz, M.; Kroutil, W.; Wandel, U.; Faber, K. Tetrahedron: Asymmetry 1995, 6, 1261. doi:10.1016/0957-4166(95)00158-L
Return to citation in text: [1] -
Organ, M. G.; Cooper, J. T.; Rogers, L. R.; Soleymanzadeh, F.; Paul, T. J. Org. Chem. 2000, 65, 7959. doi:10.1021/jo001045l
Return to citation in text: [1] -
Hachiya, I.; Matsumoto, T.; Inagaki, T.; Takahashi, A.; Shimizu, M. Heterocycles 2010, 82, 449. doi:10.3987/COM-10-S(E)16
Return to citation in text: [1] [2]
| 1. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315. doi:10.1021/acs.jmedchem.5b00258 |
| 14. | Thiehoff, C.; Rey, Y. P.; Gilmour, R. Isr. J. Chem. 2017, 57, 92. doi:10.1002/ijch.201600038 |
| 15. | Hunter, L. Beilstein J. Org. Chem. 2010, 6, No. 38. doi:10.3762/bjoc.6.38 |
| 19. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875. doi:10.1016/S0040-4020(88)90023-3 |
| 12. | Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R. D., Ed.; Topics in Current Chemistry, Vol. 193; Springer: Berlin, Heidelberg, 1997; pp 131–196. |
| 13. | Harsanyi, A.; Sandford, G. Org. Process Res. Dev. 2014, 18, 981. doi:10.1021/op500141c |
| 20. | Haufe, G. J. Fluorine Chem. 2004, 125, 875. doi:10.1016/j.jfluchem.2004.01.023 |
| 21. | Wölker, D.; Haufe, G. J. Org. Chem. 2002, 67, 3015. doi:10.1021/jo016331r |
| 7. | Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073. doi:10.1021/cr500706a |
| 8. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214. doi:10.1002/anie.201206566 |
| 9. | Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398. doi:10.1039/C6OB00764C |
| 10. | Al-Maharik, N.; O'Hagan, D. Aldrichimica Acta 2011, 44, 65. |
| 11. | Hu, W.-L.; Hu, X.-G.; Hunter, L. Synthesis 2017, 49, 4917. doi:10.1055/s-0036-1590881 |
| 18. | Landini, D.; Penso, M. Tetrahedron Lett. 1990, 31, 7209. doi:10.1016/S0040-4039(00)97281-2 |
| 2. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422. doi:10.1021/acs.chemrev.5b00392 |
| 3. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432. doi:10.1021/cr4002879 |
| 4. | Fujiwara, T.; O'Hagan, D. J. Fluorine Chem. 2014, 167, 16. doi:10.1016/j.jfluchem.2014.06.014 |
| 5. | O'Hagan, D. J. Fluorine Chem. 2010, 131, 1071. doi:10.1016/j.jfluchem.2010.03.003 |
| 6. | Kirsch, P.; Bremer, M. ChemPhysChem 2010, 11, 357. doi:10.1002/cphc.200900745 |
| 17. | Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62 |
| 19. | Hamatani, T.; Matsubara, S.; Matsuda, H.; Schlosser, M. Tetrahedron 1988, 44, 2875. doi:10.1016/S0040-4020(88)90023-3 |
| 17. | Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62 |
| 18. | Landini, D.; Penso, M. Tetrahedron Lett. 1990, 31, 7209. doi:10.1016/S0040-4039(00)97281-2 |
| 17. | Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62 |
| 17. | Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62 |
| 16. | O'Hagan, D.; Rzepa, H. S.; Schüler, M.; Slawin, A. M. Z. Beilstein J. Org. Chem. 2006, 2, No. 19. doi:10.1186/1860-5397-2-19 |
| 17. | Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62 |
| 9. | Yerien, D. E.; Bonesi, S.; Postigo, A. Org. Biomol. Chem. 2016, 14, 8398. doi:10.1039/C6OB00764C |
| 10. | Al-Maharik, N.; O'Hagan, D. Aldrichimica Acta 2011, 44, 65. |
| 11. | Hu, W.-L.; Hu, X.-G.; Hunter, L. Synthesis 2017, 49, 4917. doi:10.1055/s-0036-1590881 |
| 24. | Elliott, W. J.; Fried, J. J. Org. Chem. 1976, 41, 2469. doi:10.1021/jo00876a027 |
| 25. | Al-Dulayymi, A.; Li, X.; Neuenschwander, M. Helv. Chim. Acta 2000, 83, 1633. doi:10.1002/1522-2675(20000705)83:7<1633::AID-HLCA1633>3.0.CO;2-5 |
| 26. | Mischitz, M.; Kroutil, W.; Wandel, U.; Faber, K. Tetrahedron: Asymmetry 1995, 6, 1261. doi:10.1016/0957-4166(95)00158-L |
| 22. | Yan, N.; Lei, Z.-W.; Su, J.-K.; Liao, W.-L.; Hu, X.-G. Chin. Chem. Lett. 2016, 28, 467. doi:10.1016/j.cclet.2016.10.006 |
| 23. | Lal, G. S.; Pez, G. P.; Pesaresi, R. J.; Prozonic, F. M.; Cheng, H. J. Org. Chem. 1999, 64, 7048. doi:10.1021/jo990566+ |
| 17. | Linclau, B.; Leung, L.; Nonnenmacher, J.; Tizzard, G. Beilstein J. Org. Chem. 2010, 6, No. 62. doi:10.3762/bjoc.6.62 |
| 28. | Hachiya, I.; Matsumoto, T.; Inagaki, T.; Takahashi, A.; Shimizu, M. Heterocycles 2010, 82, 449. doi:10.3987/COM-10-S(E)16 |
| 28. | Hachiya, I.; Matsumoto, T.; Inagaki, T.; Takahashi, A.; Shimizu, M. Heterocycles 2010, 82, 449. doi:10.3987/COM-10-S(E)16 |
| 22. | Yan, N.; Lei, Z.-W.; Su, J.-K.; Liao, W.-L.; Hu, X.-G. Chin. Chem. Lett. 2016, 28, 467. doi:10.1016/j.cclet.2016.10.006 |
| 27. | Organ, M. G.; Cooper, J. T.; Rogers, L. R.; Soleymanzadeh, F.; Paul, T. J. Org. Chem. 2000, 65, 7959. doi:10.1021/jo001045l |
| 24. | Elliott, W. J.; Fried, J. J. Org. Chem. 1976, 41, 2469. doi:10.1021/jo00876a027 |
| 18. | Landini, D.; Penso, M. Tetrahedron Lett. 1990, 31, 7209. doi:10.1016/S0040-4039(00)97281-2 |
© 2017 Szpera et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)