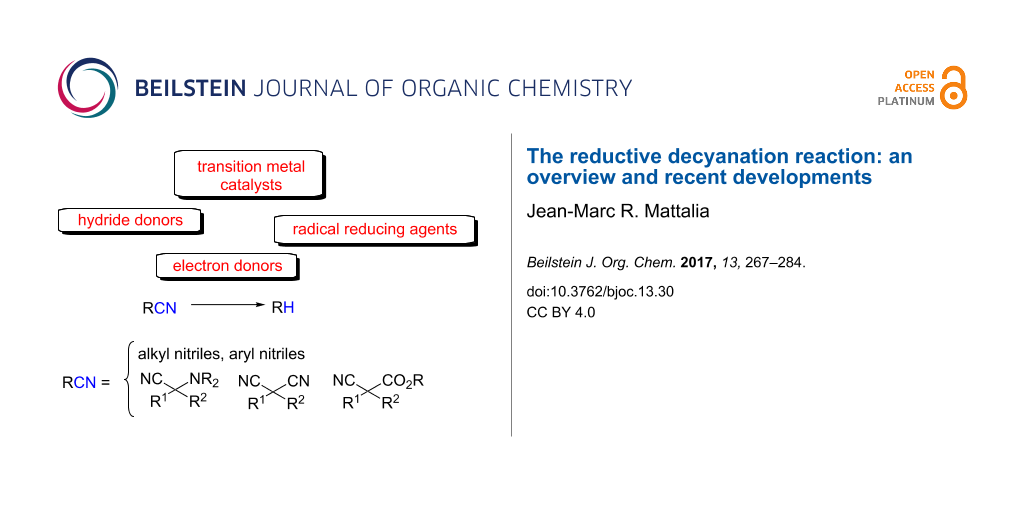
Abstract
This review presents an overview of the reductive decyanation reaction with a special interest for recent developments. This transformation allows synthetic chemists to take advantages of the nitrile functional group before its removal. Mechanistic details and applications to organic synthesis are provided.

Graphical Abstract
Introduction
Many strategies in organic synthesis involve the removal of a beneficial functional group. The electron-withdrawing properties of the nitrile functional group appear beneficial in a variety of reactions [1,2]. This group coordinates metal complexes and can be used as a directing group for C–H bond activation reactions catalyzed by transition metals [3-6]. The α-deprotonation of alkylnitriles generates active α-cyano carbanion nucleophiles. Recent investigations have resulted in different modes of alkylnitrile activations and in the development of new catalytic cyanoalkylation methodologies [7]. While Fleming and Zhang first focused on the removal of the cyano group from cyclic substrates [8], in 2006 we published a review reporting various methods allowing the reductive decyanation reaction that transforms organic nitriles into the parent alkane [9]. Even if chemical procedures previously described are still of relevance in organic synthesis, it is noteworthy that new methods have now emerged with the aim to develop mild reaction conditions that allow reduction of a wide scope of substrates and tolerate a variety of functional groups. This review attempts to be complementary to the paper published in 2006 and proposes an overview of the reductive decyanation reaction that focuses on more modern methods.
Review
Alkali-metal-promoted decyanation
Since the article by Arapakos in 1967 [10], decyanations using alkali metal dissolving conditions, typically Li or Na/NH3, are widely used in organic synthesis [11-15]. The mechanism proposed involves an electron transfer with formation of a radical anion intermediate, and then a cyanide anion is eliminated with concomitant formation of a radical. The latter is then reduced to a carbanion which can be in situ protonated by ammonia or, depending on the conditions used, another proton source (Scheme 1).
![[1860-5397-13-30-i1]](/bjoc/content/inline/1860-5397-13-30-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanism for the reduction under metal dissolving conditions.
Scheme 1: Mechanism for the reduction under metal dissolving conditions.
Using Na/NH3 or Li/EtNH2 solutions, Arapakos obtained the best yields for the decyanation of phenyl-substituted acetonitriles, tertiary alkyl, and aromatic nitriles. However, primary and secondary nitriles also led to the reduction to the amine [10,16]. This drawback can be overcome using K/HMPA/t-BuOH [17,18] or K/dicyclohexano-18-crown-6/toluene [19,20]. In the latter case, the toluene radical anion is believed to be the reactive species. LiDBB (lithium di-tert-butylbiphenylide) and Li naphthalenide are also common electron donors [15,21,22]. Because of the mechanism described in Scheme 1, the nature of the medium and the substrate strongly influence the course of the reaction. Then, in the absence of a proton source, the organolithium intermediate can cyclize or react with an electrophile giving the expected coupling products [23-27]. Metal dissolving conditions allow the reduction of various other functional groups [28]. Rychnovsky took advantage of this reactivity and achieved reductive decyanations with concomitant Birch reduction or benzyl ether cleavage [29-31]. An example related to the synthesis of polyene macrolides is described in Scheme 2.
![[1860-5397-13-30-i2]](/bjoc/content/inline/1860-5397-13-30-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Example of decyanation in metal dissolving conditions coupled with deprotection [30]. TBDMS = tert-butyldimethylsilyl.
Scheme 2: Example of decyanation in metal dissolving conditions coupled with deprotection [30]. TBDMS = tert-buty...
Rojas et al. proposed a convenient two-step pathway for the preparation of alkyl α,ω-dienes 3. These dienes are well-known precursors in ring-closing metathesis (RCM) and acyclic diene metathesis (ADMET) chemistry [32]. They first reported the quantitative α-alkylation of primary nitriles 1 [33]. In a second part of their work, they developed adequate conditions to carry out the decyanation reaction without olefin isomerization [18]. They explored several methods for the preparation of 12-butyltricosa-1,22-diene 3 (R = n-C4H9). The reaction was carried out in a slurry of K/Al2O3 in hexane, hexane/toluene (1:1) and toluene giving 20%, 63% and 75% of olefin isomerization (from NMR and GC), respectively, for each solvent [34]. This isomerization was attributed to the translocation of the tertiary radical intermediate to a more stable allyl radical leading to the double bond migration. This rearrangement was avoided using K/Ph3CH in hexane/ether (3 R = n-C4H9, 41% yield) or K/HMPA/t-BuOH in ether (3 R = n-C4H9, 99% yield). The latter optimized conditions allow the decyanation of alkylcyano α,ω-dienes 2 in quantitative yields with no detection of olefin isomerization (Scheme 3).
![[1860-5397-13-30-i3]](/bjoc/content/inline/1860-5397-13-30-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of α,ω-dienes [18,33].
Scheme 3: Preparation of α,ω-dienes [18,33].
Radical intermediates were trapped using the radical probe 4 (Scheme 4). Replacing t-BuOH with t-BuOD in Scheme 3 (R = CH3 case), yielded the decyanation product 3 with 92% deuterium incorporation. With respect to the mechanism described in Scheme 1, the authors suggest that t-BuOH (or Ph3CH) could act as H-atom donor that quenches the radical intermediate. However, this interpretation is opened to discussion because olefin functionalities also can undergo isomerization via anionic intermediates [35] and radicals usually abstract hydrogen atoms preferentially from the alkyl groups of t-BuOH [36-38]. Therefore t-BuOH could act as a proton donor and so prevent the olefin isomerization.
![[1860-5397-13-30-i4]](/bjoc/content/inline/1860-5397-13-30-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cyclization reaction using a radical probe [18].
Scheme 4: Cyclization reaction using a radical probe [18].
Alkali metals can also be used in suspension. As mentioned above, highly dispersed potassium over neutral alumina (K/Al2O3) in hexane is able to effect the reductive cleavage of alkylnitriles [18,34]. Zárraga et al. described an efficient synthesis of (±)-xanthorrhizol (8) [39]. The authors prepared the intermediate 7 by dialkylation of 6 and attempted to carry out a one-pot decyanation and demethylation [40] with a suspension of lithium in THF. The target compound 8 was obtained in 74% yield together with 24% of the byproduct 9 (Scheme 5). This compound seems to be formed by a cross-linked ether cleavage of the methoxy group induced by the anion intermediate resulting from the decyanation pathway (Scheme 1). The authors increased the yield to 99% by adding NH4Cl as proton source that immediately reacts with the anion before the ether cleavage.
![[1860-5397-13-30-i5]](/bjoc/content/inline/1860-5397-13-30-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of (±)-xanthorrhizol (8) [39].
Scheme 5: Synthesis of (±)-xanthorrhizol (8) [39].
Aluminium- and borohydrides and the use of sodium hydride
Reduction of α-aminonitriles
Bifunctional α-aminonitriles exhibit several modes of reactivity. Recent reviews demonstrated the richness of this chemistry and emphasized synthetic applications particularly in heterocyclic chemistry [41-44]. The reductive decyanation of α-aminonitriles under metal dissolving conditions is a common procedure that proceeds through a two-electron-transfer pathway (Scheme 1) [23,44]. In the ionic pathway, the loss of the cyanide ion yields an iminium cation that can be reduced by various hydride donors (Scheme 6). Alternatively, secondary amines could involve an elimination of HCN and reduction of the formed imine.
![[1860-5397-13-30-i6]](/bjoc/content/inline/1860-5397-13-30-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Mechanism for the reduction of α-aminonitriles by hydride donors.
Scheme 6: Mechanism for the reduction of α-aminonitriles by hydride donors.
NaBH4 [45-51] or NaBH3CN [47,52-59] are widely used hydride donors and, less frequently, BH3 [60,61], AgBF4/Zn(BH4)2 [62-64] or LiAlH4 [65,66]. These reactions usually require mild conditions but the course of the reaction depends on the ease of formation of the iminium ion [48,67-69]. With the highly reactive LiAlH4, diamines can also be formed by reduction of the nitrile moiety [70]. This competition with the decyanation reaction depends on the structure of the α-aminonitrile, stereoelectronic effects and internal strain of the molecule [68].
Chuang et al. prepared a set of α-aminoacrylonitriles 11 by a cyano-promoted aza-Diels–Alder cycloaddition [71]. The cyano groups were then removed in high yields by treatment with NaBH4 in 2-propanol by using both basic and nucleophilic properties of the hydride ion. The proposed mechanism involves a double-bond isomerization to the α-aminonitrile intermediate which is then reduced by the hydride ion in a classical way (Scheme 7). Interestingly, deuterium-labelling experiments indicate that one of the methylene hydrogens of the formed allylamine 12 is derived from the protic solvent and the other comes from the reducing agent. Finally, an oxidative aryl–aryl coupling promoted by vanadium oxytrifluoride (VOF3) afforded phenanthroindolizidines (13, n = 1) and phenanthroquinolizidines (13, n = 2). Anticancer activities of these 18 compounds were evaluated against three human cancer cell lines.
![[1860-5397-13-30-i7]](/bjoc/content/inline/1860-5397-13-30-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of phenanthroindolizidines and phenanthroquinolizidines [71].
Scheme 7: Synthesis of phenanthroindolizidines and phenanthroquinolizidines [71].
BH3·THF containing NaBH4 has been used for the reduction of diimines [72,73] and was studied in-depth by Zhang and co-workers in the reductive decyanation reaction. In their work, the cyano group activates the [3 + 2] cycloaddition of azomethine ylides and is then removed to yield 5-unsubstituted pyrrolidines [74]. These substructures appear in several biologically active natural products and drugs [75]. A range of decyanation conditions were screened such as NaBH4 in THF or MeOH, NaBH4/AgBF4 in THF and NaBH3CN in MeOH/AcOH. They also explored BH3 alone in THF and with varying amounts of NaBH4. They found that the addition of a catalytic amount of NaBH4 was very efficient for the reductive decyanation reaction. Scheme 8 shows the scope of this two-step transformation. This protocol is successfully used with various electron-deficient substituted phenyl groups (15b–e) and with heterocycles (15f–h). The double-bond of cyanopyrrolidine 14l is preserved from the hydroboration reaction (15l). Olefins bearing bulky ester, sulfone or amide groups afforded good to excellent yields (15i–k). A series of aliphatic α-iminonitriles also gave good results (15n,o). Notably the labile dimethylacetal group tolerates this two-step transformation (15m).
![[1860-5397-13-30-i8]](/bjoc/content/inline/1860-5397-13-30-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Two-step synthesis of 5-unsubstituted pyrrolidines (25 examples and 1 synthetic application, see below). The two yields refer successively to the cycloaddition (14) and decyanation (15) steps [74]. Boc = tert-butoxycarbonyl. EWG = electron-withdrawing group.
Scheme 8: Two-step synthesis of 5-unsubstituted pyrrolidines (25 examples and 1 synthetic application, see be...
Using this protocol, the authors described a total synthesis of (±)-isoretronecanol (19), a pyrrolizidine alkaloid (Scheme 9). In this case the two-step protocol is followed by a lactamization and the one-pot reduction of ester and lactam groups of 18.
![[1860-5397-13-30-i9]](/bjoc/content/inline/1860-5397-13-30-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of (±)-isoretronecanol 19. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene [74].
Scheme 9: Synthesis of (±)-isoretronecanol 19. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene [74].
The authors proposed a reductive anionic chain mechanism described in Scheme 10. The exposure of 14a to BH3 generates a borane–amine complex 20 whose fragmentation could be promoted by NaBH4. The resulting imine 21 is reduced by BH3 with the help of the cyanoborohydride anion. The formed anion 22 abstracts a proton from complex 20 to produce 23 or 24 and regenerate 21 and BH3CN−. A set of experiments supports this proposal. Notably, borane is the major hydride source for the reduction and 22 (derived from the reaction of 23 and NaH) is efficient for this chain reaction. Stable intermediates 23 and 24 were fully characterized while the unstable complex 20 was only characterized with 1H NMR.
![[1860-5397-13-30-i10]](/bjoc/content/inline/1860-5397-13-30-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Proposed mechanism with 14a for the NaBH4 induced decyanation reaction (“BH3” = BH3·THF) [74].
Scheme 10: Proposed mechanism with 14a for the NaBH4 induced decyanation reaction (“BH3” = BH3·THF) [74].
Reduction of other substrates
The reductive decyanation promoted by aluminium- and borohydrides for substrates other than α-aminonitriles has been described for more specific cases and usually displays moderate yields [76-79]. Recently, the DIBAL-H-induced decyanation of gem-dicyanodihydroazulene derivatives was described but only poor yields were reported [80].
Chiba et al. accidently discovered the reductive decyanation of aryl substituted tertiary nitriles (Scheme 11) [81]. The protocol involves NaH in THF in the presence of LiI at 85 °C and appears suitable for the construction of the important 1,1-diarylalkane (26k–m) and triarylmethane (26n) derivatives. Strained cyclobutylarenes (26g,h) and heterocycles (26i,j) are prepared in this way and the electron-rich 4-methoxyphenyl group decreases the reaction rate (compare 26e,g with 26f,h). The decyanation of nitriles with the NaH–NaI system gives comparable yields but much longer reaction times are generally required.
![[1860-5397-13-30-i11]](/bjoc/content/inline/1860-5397-13-30-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Reductive decyanation by a sodium hydride–iodide composite (26 examples) [81].
Scheme 11: Reductive decyanation by a sodium hydride–iodide composite (26 examples) [81].
The reduction of radical probes (no rearranged products formed from 25d,l,m) and deuterium-labelling experiments (no deuterium incorporation using THF-d8 and quenching with D2O) discard the possibility of a single-electron transfer pathway. Other reductions suggest a hydride addition with formation of an iminyl anion intermediate. Particularly, when the reaction of 25f was quenched after 2.5 h, the corresponding aldehyde was formed in 42% yield together with 37% of the decyanated product 26f.
DFT calculations conducted on nitrile 25o support the hydride addition to the CN triple bond with formation of an iminyl anion intermediate 27. The latter easily isomerizes to its isomer 28 where a sodium cation–π-interaction occurs. The last step involves a C–C bond cleavage and proton transfer with elimination of NaCN (Scheme 12). This proton transfer occurs with retention of configuration as experimentally observed. Indeed, the kinetic profiles show that the decyanation reactions include an induction period (0.5 h and 2 h, respectively, for NaH–LiI and NaH–NaI systems) suggesting the formation of a new inorganic composite. These materials consist of metal iodide interspersed with activated NaH resulting in an unique hydride-donor reactivity [82]. An addition–elimination mechanism has been previously proposed for the LiAlH4 promoted decyanation of 2,2-diphenylpropionitrile and related nitriles. In such pathways, the phenyl groups probably favor the C–C bond cleavage by stabilizing the incipient negative charge on the carbon adjacent to the cyano group [76,77].
![[1860-5397-13-30-i12]](/bjoc/content/inline/1860-5397-13-30-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Proposed mechanism for the reduction by NaH [81].
Scheme 12: Proposed mechanism for the reduction by NaH [81].
Transition-metal-catalyzed reductive decyanation
Hydrogenation of α-aminonitriles
The decyanation of α-aminonitriles with hydrogen present in an excess of Raney nickel was described by Husson and co-workers on oxazolidine derivatives [83,84]. The authors logically proposed that the decyanation occurred via the reduction of an iminium ion intermediate. The hydrogenation of α-aminonitriles catalyzed by nickel nanoparticles results in reductive decyanation and yields 29–31. The colloid solution of nickel is prepared in situ via reduction of anhydrous NiCl2 with NaBH4 in t-BuOH, iPrOH or n-butanol, the reactions are then performed upon bubbling of hydrogen at atmospheric pressure through the reaction mixture (Scheme 13). The reductive decyanation of 2-hydroxyadamantane-2-carbonitrile is successful by this method and yields alcohol 32 [85].
![[1860-5397-13-30-i13]](/bjoc/content/inline/1860-5397-13-30-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Reductive decyanation catalyzed by nickel nanoparticles. Yields are given in weight % from GC–MS data [85].
Scheme 13: Reductive decyanation catalyzed by nickel nanoparticles. Yields are given in weight % from GC–MS da...
For less reactive nitriles, the C–CN bond cleavage usually requires hydrogen under pressure and high temperatures [86]. Tao et al. reported the decyanation of 2-cyanobenzo[b]thiophene using hydrogen and Pd, Pt/TiO2 or Pd/TiO2-Cu as catalyst at 200–300 °C with yields varying from 79 to 90% (Scheme 14) [87].
![[1860-5397-13-30-i14]](/bjoc/content/inline/1860-5397-13-30-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Decyanation of 2-cyanobenzo[b]thiophene [87].
Scheme 14: Decyanation of 2-cyanobenzo[b]thiophene [87].
Opatz et al. developed the enantioselective syntheses of various alkaloids using the rhodium catalyst developed by Noyori [88] for the asymmetric transfer hydrogenation of imines. Interestingly, imines are formed from unstable α-aminonitrile intermediates which spontaneously eliminate HCN [89-91].
Iron-catalyzed reductive decyanation
In 1982, Yamamoto et al. disclosed the C–CN bond cleavage promoted by an electron-rich cobalt complex [92]. Since that time, the activation of inert C–CN bonds by transition metals has been widely investigated. Improvements towards mild and green conditions with a large substrate scope have been achieved in many reactions including decyanation. Several reviews account for the richness of this chemistry [93-99]. Two major pathways for the C–CN bond activation have emerged (Scheme 15). One is the oxidative addition of the C–CN bond to a low-valent metal center (Ni case). The other pathway involves a silylmetal-assisted carbon cyano bond cleavage through an iminoacyl intermediate (Rh, Fe cases). In both pathways, the combination with a reducing agent gives a catalytic reductive system for the removal of the cyano group.
![[1860-5397-13-30-i15]](/bjoc/content/inline/1860-5397-13-30-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Simplified pathways involved in transition-metal-promoted reductive decyanations [93,95].
Scheme 15: Simplified pathways involved in transition-metal-promoted reductive decyanations [93,95].
The discussion below on transition-metal-catalyzed reactions focuses on selected examples that show the wide scope of reduced substrates including alkyl cyanides, challenging substrates due to their propensity to undergo β-hydride elimination from alkylmetal intermediates. Nakazawa et al. reported the photoinduced C–CN bond cleavage catalyzed by iron complexes of a few primary alkyl cyanides and aryl cyanides in the presence of Et3SiH [100,101]. Decyanated species were formed together with silyl cyanide. Both aliphatic and aromatic nitriles were successfully reduced (Scheme 16) but an electron-withdrawing, bulky or coordinating substituent on the C atom linked to the cyano group disfavors the reductive decyanation.
![[1860-5397-13-30-i16]](/bjoc/content/inline/1860-5397-13-30-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Fe-catalyzed reductive decyanation. Numbers in square brackets represent turnover numbers. The TONs were determined by GC and based on the amount of Et3SiCN produced (10 examples with TON > 4) [100]. Cp = cyclopentadienyl.
Scheme 16: Fe-catalyzed reductive decyanation. Numbers in square brackets represent turnover numbers. The TONs...
Rhodium-catalyzed reductive decyanation
Chatani and co-workers have investigated the rhodium-catalyzed carbon–cyano bond cleavage reactions using organosilicon reagents [94]. They reported a rhodium-catalyzed reductive decyanation with a hydrosilane as reducing agent. They selected [RhCl(cod)]2 (cod = 1,5-cyclooctadiene) as catalyst and found that the use of triisopropylsilane and P(OBu)3 or P(OiPr)3 as ligands led to the best results [102,103]. Aryl, heteroaryl (Scheme 17), benzyl and alkyl cyanides (Scheme 18) are applicable to this decyanation reaction.
![[1860-5397-13-30-i17]](/bjoc/content/inline/1860-5397-13-30-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Rh-catalyzed reductive decyanation of aryl nitriles (18 examples, 2 synthetic applications) [103].
Scheme 17: Rh-catalyzed reductive decyanation of aryl nitriles (18 examples, 2 synthetic applications) [103].
![[1860-5397-13-30-i18]](/bjoc/content/inline/1860-5397-13-30-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Rh-catalyzed reductive decyanation of aliphatic nitriles (15 examples, one synthetic application) [103].
Scheme 18: Rh-catalyzed reductive decyanation of aliphatic nitriles (15 examples, one synthetic application) [103].
The use of hydrosilane as a mild reducing agent allows a high functional group tolerance (34b–e, 35, 39b, 43) and sterically hindered cyano groups are successfully removed (36, 41, 43). Reactions of benzyl cyanides proceed smoothly even in the absence of the phosphite ligands (39b, 43). Interestingly, an ester group in the α-position increases the reactivity (43). The reaction works well with simple primary and secondary alkyl cyanides but requires a higher reaction temperature. Aliphatic nitriles containing β-hydrogen atoms are successfully reduced (40, 42a–c, 43, 44a). Even if alkenes are not formed, the authors show the occurrence of a β-hydride elimination/hydrometalation sequence. However, the reduction of the more hindered substrate 44b requires a larger excess of hydrosilane to prevent the formation of alkene. Finally, unactivated tertiary alkyl cyanides lead to a complex mixture.
Nickel-catalyzed reductive decyanation
Maiti et al. established that the Ni(acac)2 complex (acac = acetylacetonate) of PCy3 (Cy, cyclohexyl) in combination with TMDS (tetramethyldisiloxane) as hydride source can catalyze the reductive decyanation efficiently (method A, Scheme 19) [104]. The use of AlMe3 appeared beneficial by facilitating the oxidative addition of the C–CN bond to Ni. Soon after, the same team developed the use of cheaper, greener and milder hydrogen gas as the hydride source. They found that catalytic Ni(cod)2 in combination with PCy3 and AlMe3 under 1 bar pressure of H2 gas was the protocol of choice (method B) [105]. As well as the activation of the C–CN bond, AlMe3 helps to remove and consume the HCN gas produced under the reaction conditions. These protocols were successfully applied to a wide range of nitriles containing various other functional groups (Scheme 19). The ether bonds (33b, 49), ester (45), hydroxy (39c) and keto groups (46) are tolerated. With method B, aryl cyanides with amide (54), trifluoromethyl (55) and fluoro groups (47c) are reduced in attractive yields. For substrates bearing dicyano groups, monodecyanation is the major pathway (47b) and decyanations are efficient with sterically demanding ortho-substituents (54, 57) or with ortho-directing groups (45, 48, 50). Aliphatic nitriles underwent decyanation without β-hydride elimination (42a, 52b, 53).
![[1860-5397-13-30-i19]](/bjoc/content/inline/1860-5397-13-30-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Ni-catalyzed reductive decyanation (method A: 28 examples and 2 synthetic applications; method B: 31 examples and 5 synthetic applications) [104,105].
Scheme 19: Ni-catalyzed reductive decyanation (method A: 28 examples and 2 synthetic applications; method B: 3...
Enthaler et al. reported the application of nickel complex 58 as precatalyst and tert-butylmagnesium chloride as reducing agent (method A) in the decyanation of alkyl and aryl cyanides [106]. In the same year, to easily generate nickel hydride complexes, the authors explored the possibility to apply more reactive hydride donors such as LiBH4 (method B) [107,108]. Finally, after investigating the reaction conditions and a precatalyst screening, the conditions described in Scheme 20 were selected. With method B, lower catalyst loading and shorter reaction times are necessary. With method A, benzonitrile yields 51% of benzene (34a) and an increase in yield is obtained with the methoxy and methyl groups (34c, 59, 34j para). For some substrates, the replacement of t-BuMgCl by n-Bu2Mg improves the yield (34b,c, 59). With method B, excellent yields are obtained for methoxy (34c, 59) and methyl substituted aryl cyanides 34j while with method A, changing the position of the methyl substituent from the para to the ortho or meta positions decreases the yield in toluene. Dimethylamino and thioether groups display a lower yield with method B (34b,g) contrary to the trifluoromethyl group (34i). With p-fluorobenzonitrile the decyanation is observed with both methods (34h) but, with the bromo counterpart, dehalogenation also occurs. 2-Cyanothiophene results in quantitative yield of thiophene 60 with both methods but cyanopyridines are not converted, probably due to the coordination abilities of the pyridine. Finally, decanenitrile is decyanated with method A (61) but aliphatic nitriles are not converted to the desired product with method B.
![[1860-5397-13-30-i20]](/bjoc/content/inline/1860-5397-13-30-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Reductive decyanation catalyzed by the nickel complex 58 (method A, 14 examples, yield ≥ 20% and 1 synthetic application; method B, 12 examples, yield ≥ 17% and 1 synthetic application). Yields determined by GC–MS and 1H NMR spectroscopy [106,107].
Scheme 20: Reductive decyanation catalyzed by the nickel complex 58 (method A, 14 examples, yield ≥ 20% and 1 ...
Trapping of radicals with method A led the authors to propose the electron transfer mechanism described in Scheme 21. The complex 62 reacts with the Grignard reagent to form the nickel-magnesium hydride intermediate 64 via a β-hydride elimination from 63. A single-electron transfer (SET) to the nitrile oxidizes the complex at the metal center into 65 and generates an aryl radical. The electron can also be located in the ligand (non-innocent ligand, not represented in Scheme 21). Then, elimination of MgXCN and radical recombination with the nickel species produce 66. Finally, an elimination of Ar-H closes the catalytic cycle. A similar mechanism was proposed with method B.
![[1860-5397-13-30-i21]](/bjoc/content/inline/1860-5397-13-30-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Proposed catalytic cycle for the nickel complex 58 catalyzed decyanation (method A). Only the cycle for 63 is shown [106].
Scheme 21: Proposed catalytic cycle for the nickel complex 58 catalyzed decyanation (method A). Only the cycle...
Radical reactions
This part is related to radical reactions not involving alkali metals and focuses on the reduction of malononitriles and cyanoacetates (α-cyanoesters). These compounds are particularly versatile reagents in organic synthesis including multicomponent reactions [109-113]. However, like decarboxylation for related malonic ester or acetoacetates, an easy access to the removal of the cyano group should encourage future developments using these compounds.
Bu3SnH and N-heterocyclic carbene boranes
The reductive decyanation of malononitriles to mononitriles using tributyltin hydride/AIBN in benzene was unexpectedly discovered by Curran and Seong [114]. Later, they made a full study and successfully reduced to mononitriles a variety of mono- and dialkylated malononitriles but under these conditions, the reduction of cyanoacetates failed [115]. Synthetic applications of this methodology were later described [116-118]. Chiba et al. have developed a concise and stereoselective methodology for the preparation of highly substituted carbocycles [119,120]. An example in described in Scheme 22. The 5-membered ring is formed via a K2CO3 mediated SN2 – conjugate addition sequence between malononitrile and 6-bromo-2-hexenoate derivatives 67a,b. Dicyanocyclopentanes 68a,b are treated with tributyltin hydride/AIBN giving the monodecyanated products 69a,b. Bicyclic lactones 70a,b are then obtained in 3 steps in 41% and 51% yields, respectively, from 69a,b.
![[1860-5397-13-30-i22]](/bjoc/content/inline/1860-5397-13-30-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of bicyclic lactones [119,120].
Scheme 22: Synthesis of bicyclic lactones [119,120].
Later Curran’s group discovered that NHC-boryl radicals, generated from NHC-boranes (N-heterocyclic carbene boranes), abstracted the cyano group from various organic nitriles and dinitriles and applied this reaction for the synthesis of new NHC-boryl nitrile and dinitrile compounds [121]. They observed that malononitrile was the most efficient donor to produce boryl nitriles and concluded that substituted malononitriles would be decyanated by NHC-boranes. For this transformation, malononitriles are reacting with a slight excess of NHC-borane 71, in refluxing t-BuOH with DTBP (di-tert-butylperoxide) as radical initiator. The yields are attractive while roughly comparable amounts of boryl nitriles 74 and 75 are formed (Scheme 23). Malononitriles 72d,e are successfully reduced to 73d,e while with photoactivated 78 such substrates afforded complex mixtures (see below). Contrary to the reductive decyanation with Bu3SnH [115], the NHC borane reduces the α-cyanoester 72g to 73g while the aryl chloride and bromide moieties are preserved (73h,i).
![[1860-5397-13-30-i23]](/bjoc/content/inline/1860-5397-13-30-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Reductive decyanation of malononitriles and cyanoacetates using NHC-boryl radicals (9 examples). For 74 and 75, isolated or NMR yields are given from 71 [121].
Scheme 23: Reductive decyanation of malononitriles and cyanoacetates using NHC-boryl radicals (9 examples). Fo...
The use of a 5-hexenyl radical probe led to the formation of 99% of cyclized products (relative yield). The authors proposed a radical chain mechanism similar to that proposed in the reaction with Bu3SnH. While the tin radical was proposed to add to nitrogen [115], here the evidence points more to the addition of the boryl radical 76 on the nitrile carbon to form the nitrogen centered radical 77. β-Fragmentation leads to NHC-boryl nitrile 74 and a carbon centered radical. A hydrogen atom transfer reaction between the electrophilic α-cyano radical and the nucleophile NHC-borane achieves the chain propagation (Scheme 24). The isolation of the NHC-boryl nitrile 74, EPR spectroscopy observations [122], and polar effects fit with this proposition.
![[1860-5397-13-30-i24]](/bjoc/content/inline/1860-5397-13-30-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Proposed mechanism for the reduction by NHC-boryl radicals. The other possible pathway (addition of 76 to the nitrogen) is not represented here [121].
Scheme 24: Proposed mechanism for the reduction by NHC-boryl radicals. The other possible pathway (addition of ...
Neutral organic electron donors
Powerful single-electron transfer reagents have been described. Kang et al. reported the decyanation of both malononitriles and α-cyanoesters using samarium(II) iodide/THF/HMPA at respectively 0 °C and room temperature [123]. Metallic samarium can also promote the decyanation [124].
Doni and Murphy have reported the reductive decyanation of malononitriles and α-cyanoesters by using the neutral organic electron donor 78 (Scheme 25) under photoactivation (method A, Scheme 26) [125]. The first observation was that surprisingly, the reaction of 2,2-dibenzylmalononitrile (72b) provided both debenzylated (2-benzylmalononitrile, 19%) and decyanation (73b, 75%) products. In contrast, the corresponding dibenzylcyanoacetate led exclusively to the debenzylation product [126]. Selected examples are presented in Scheme 26. Excellent yields are obtained even if decyanation of cyanoacetates requires higher amounts of 78 (6 equiv) with extended reaction times (72 h). In line with this work, the authors prepared the tetra(iminophosphorano)-substituted bispyridinylidene 80, a new highly efficient neutral organic electron donor (Scheme 25) [127]. This compound is able to reduce aryl halides and appears as the only reductant able to reduce dialkylarenesulfonamides as well as malononitriles without photoexcitation. Electron donor 80 can be isolated but is more conveniently generated in situ by deprotonation with KHMDS of its pyridinium ion precursor 79 in refluxing toluene (method B).
![[1860-5397-13-30-i25]](/bjoc/content/inline/1860-5397-13-30-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Structures of organic electron-donors. Only the major Z isomer of 80 is shown [125,127].
Scheme 25: Structures of organic electron-donors. Only the major Z isomer of 80 is shown [125,127].
![[1860-5397-13-30-i26]](/bjoc/content/inline/1860-5397-13-30-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Reductive decyanation of malononitriles and cyanoacetates using organic electron-donors (method A, 11 examples; method B, 3 examples). Yields in brackets refer to method B [125,127].
Scheme 26: Reductive decyanation of malononitriles and cyanoacetates using organic electron-donors (method A, ...
With method B, malononitriles are reduced to mononitriles with comparable yields to that achieved with photoactivated 78 (73k,m,n). No allylic bond cleavage is observed and alkene moieties are preserved in the decyanated product (73m,n,q). No radicals are trapped from the reduction of the 5-hexenyl radical probe 72m with both methods. Mononitrile 2,2-dimethylhexadecanenitrile appears inert with both methods, and organic bromides are not tolerated. The proposed mechanism is similar to Scheme 1. First, a SET from the electron donor to the nitrile forms a radical anion. This radical anion can fragment into a cyanide ion and a radical which is rapidly reduced into a stabilized carbanion before protonation. This carbanion appears as a key intermediate leading to complex mixtures when method A is applied to 72d or a substrate similar to 72e.
More recently, the reaction of dibenzylmalononitrile 72b with the hydroxyaryl-substituted benzimidazoline derivative 81 as photo-reductant in a basic medium has been described and led to 73b in fair yield (Scheme 27) [128].
![[1860-5397-13-30-i27]](/bjoc/content/inline/1860-5397-13-30-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Photoreaction of dibenzylmalononitrile with 81 [128].
Scheme 27: Photoreaction of dibenzylmalononitrile with 81 [128].
Acid, base or organometallic-induced reductive decyanation
Among the other procedures mentioned in our previous review [9], the base [129,130] or acid-induced [131-134] hydrolysis–decarboxylation sequence appears as a classical pathway (Scheme 28).
![[1860-5397-13-30-i28]](/bjoc/content/inline/1860-5397-13-30-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Examples of decyanation promoted in acid or basic media [129,131,134,135].
Scheme 28: Examples of decyanation promoted in acid or basic media [129,131,134,135].
In the particular case of diphenylacetonitriles, an addition–elimination mechanism is proposed (Scheme 29) [135,136]. Such a pathway could also apply for the reductive decyanation of diphenylacetonitriles induced by organolithiums or Grignard reagents [137,138]. This reaction, applied to nitriles substituted with suitable leaving groups appears as a cyanation method of organometallic reagents or other nucleophiles [139].
![[1860-5397-13-30-i29]](/bjoc/content/inline/1860-5397-13-30-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Mechanism proposed for the base-induced reductive decyanation of diphenylacetonitriles [136].
Scheme 29: Mechanism proposed for the base-induced reductive decyanation of diphenylacetonitriles [136].
Nambo et al described the preparation of triarylacetonitriles using sequential Pd-catalyzed arylations. Triarylacetonitriles obtained can be transformed into various species including triarylmethanes by treatment with MeMgCl (Scheme 30) [140]. In this case a SET mechanism could operate [141].
![[1860-5397-13-30-i30]](/bjoc/content/inline/1860-5397-13-30-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Reductive decyanation of triarylacetonitriles [140].
Scheme 30: Reductive decyanation of triarylacetonitriles [140].
Conclusion
The reductive decyanation reaction appears useful in organic synthesis because it offers the possibility to temporarily use the advantages from the nitrile functional group. While our previous review showed that several methods had a narrow substrate scope [9], new and convenient synthetic methods have now emerged. Classical metal dissolving conditions are still used but the method, while it works, shows that the course of the reaction may strongly depend on reaction conditions. The transition-metal-catalyzed defunctionalization reactions cover a wide range of nitriles including the most challenging alkanenitriles and allow a large functional group tolerance. The decyanation of α-aminonitriles by aluminium- and borohydrides has been widely investigated and offers synthetic applications in heterocyclic chemistry. Recent developments using N-heterocyclic carbene boranes and super electron donors provide new procedures for the reduction of malononitriles or α-cyanoesters. This opens the possibility to synthetic applications by using these intermediates in the future.
References
-
Zhao, L.; Wen, M.; Wang, Z.-X. Eur. J. Org. Chem. 2012, 3587–3597. doi:10.1002/ejoc.201200121
Return to citation in text: [1] -
Huang, Q.; Tran, G.; Pardo, D. G.; Tsuchiya, T.; Hillebrand, S.; Vors, J.-P.; Cossy, J. Tetrahedron 2015, 71, 7250–7259. doi:10.1016/j.tet.2015.03.099
Return to citation in text: [1] -
Reddy, M. C.; Jeganmohan, M. Chem. Commun. 2015, 51, 10738–10741. doi:10.1039/c5cc03112e
Return to citation in text: [1] -
Bera, M.; Modak, A.; Patra, T.; Maji, A.; Maiti, D. Org. Lett. 2014, 16, 5760–5763. doi:10.1021/ol502823c
Return to citation in text: [1] -
Bera, M.; Maji, A.; Sahoo, S. K.; Maiti, D. Angew. Chem., Int. Ed. 2015, 54, 8515–8519. doi:10.1002/anie.201503112
Return to citation in text: [1] -
Du, B.; Jiang, X.; Sun, P. J. Org. Chem. 2013, 78, 2786–2791. doi:10.1021/jo302765g
Return to citation in text: [1] -
López, R.; Palomo, C. Angew. Chem., Int. Ed. 2015, 54, 13170–13184. doi:10.1002/anie.201502493
Return to citation in text: [1] -
Fleming, F. F.; Zhang, Z. Tetrahedron 2005, 61, 747–789. doi:10.1016/j.tet.2004.11.012
Return to citation in text: [1] -
Mattalia, J.-M.; Marchi-Delapierre, C.; Hazimeh, H.; Chanon, M. ARKIVOC 2006, iv, 90–118. doi:10.3998/ark.5550190.0007.408
Return to citation in text: [1] [2] [3] -
Arapakos, P. G. J. Am. Chem. Soc. 1967, 89, 6794–6796. doi:10.1021/ja01001a090
Return to citation in text: [1] [2] -
Rychnovsky, S. D.; Rogers, B. N.; Richardson, T. I. Acc. Chem. Res. 1998, 31, 9–17. doi:10.1021/ar960223n
Return to citation in text: [1] -
Schleicher, K. D.; Jamison, T. F. Beilstein J. Org. Chem. 2013, 9, 1533–1550. doi:10.3762/bjoc.9.175
Return to citation in text: [1] -
McLachlan, M. M. W.; O'Connor, P. D.; Fairweather, K. A.; Willis, A. C.; Mander, L. N. Aust. J. Chem. 2010, 63, 742–760. doi:10.1071/CH10056
Return to citation in text: [1] -
Sinz, C. J.; Rychnovsky, S. D. Top. Curr. Chem. 2001, 216, 51–92. doi:10.1007/3-540-44726-1_2
Return to citation in text: [1] -
Rychnovsky, S. D. Chem. Rev. 1995, 95, 2021–2040. doi:10.1021/cr00038a011
Return to citation in text: [1] [2] -
Arapakos, P. G.; Scott, M. K.; Huber, F. E., Jr. J. Am. Chem. Soc. 1969, 91, 2059–2062. doi:10.1021/ja01036a033
Return to citation in text: [1] -
Larchevêque, M.; Cuvigny, T. Tetrahedron Lett. 1975, 16, 3851–3854. doi:10.1016/S0040-4039(00)91294-2
Return to citation in text: [1] -
Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962–4970. doi:10.1021/jo800640j
Return to citation in text: [1] [2] [3] [4] [5] -
Ohsawa, T.; Kobayashi, T.; Mizuguchi, Y.; Saitoh, T.; Oishi, T. Tetrahedron Lett. 1985, 26, 6103–6106. doi:10.1016/S0040-4039(00)95137-2
Return to citation in text: [1] -
Baek, D. J.; Bittman, R. Chem. Phys. Lipids 2013, 175–176, 99–104. doi:10.1016/j.chemphyslip.2013.08.003
Return to citation in text: [1] -
Amancha, P. K.; Lai, Y.-C.; Chen, I.-C.; Liu, H.-J.; Zhu, J.-L. Tetrahedron 2010, 66, 871–877. doi:10.1016/j.tet.2009.11.105
Return to citation in text: [1] -
Amancha, P. K.; Liu, H.-J.; Ly, T. W.; Shia, K.-S. Eur. J. Org. Chem. 2010, 3473–3480. doi:10.1002/ejoc.201000318
Return to citation in text: [1] -
Perry, M. A.; Rychnovsky, S. D. Nat. Prod. Rep. 2015, 32, 517–533. doi:10.1039/c4np00125g
Return to citation in text: [1] [2] -
Zeller, E.; Sajus, H.; Grierson, D. S. Synlett 1991, 44–46. doi:10.1055/s-1991-20623
Return to citation in text: [1] -
La Cruz, T. E.; Rychnovsky, S. D. Org. Lett. 2005, 7, 1873–1875. doi:10.1021/ol050589c
Return to citation in text: [1] -
Tsao, J.-P.; Tsai, T.-Y.; Chen, I.-C.; Liu, H.-J.; Zhu, J.-L.; Tsao, S.-W. Synthesis 2010, 4242–4250. doi:10.1055/s-0030-1258301
Return to citation in text: [1] -
Cacciarini, M.; Jevric, M.; Elm, J.; Petersen, A. U.; Mikkelsen, K. V.; Nielsen, M. B. RSC Adv. 2016, 6, 49003–49010. doi:10.1039/c6ra06045e
Return to citation in text: [1] -
Yus, M.; Foubelo, F. Dissolving Metals. In Modern Reduction Methods; Andersson, P. G.; Munslow, I. J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp 419–445. doi:10.1002/9783527622115.ch17
Return to citation in text: [1] -
Mander, L. N.; McLachlan, M. M. J. Am. Chem. Soc. 2003, 125, 2400–2401. doi:10.1021/ja029725o
Return to citation in text: [1] -
Richardson, T. I.; Rychnovsky, S. D. Tetrahedron 1999, 55, 8977–8996. doi:10.1016/s0040-4020(99)00457-3
Return to citation in text: [1] [2] -
Sinz, C. J.; Rychnovsky, S. D. Angew. Chem., Int. Ed. 2001, 40, 3224–3227. doi:10.1002/1521-3773(20010903)40:17<3224::aid-anie3224>3.0.co;2-d
Return to citation in text: [1] -
da Silva, L. C.; Wagener, K. B. Macromol. Chem. Phys. 2016, 217, 850–855. doi:10.1002/macp.201500508
Return to citation in text: [1] -
Rojas, G.; Baughman, T. W.; Wagener, K. B. Synth. Commun. 2007, 37, 3923–3931. doi:10.1080/00397910701572456
Return to citation in text: [1] [2] -
Savoia, D.; Tagliavini, E.; Trombini, C.; Umani-Ronchi, A. J. Org. Chem. 1980, 45, 3227–3229. doi:10.1021/jo01304a016
Return to citation in text: [1] [2] -
Bailey, W. F.; Punzalan, E. R. J. Am. Chem. Soc. 1994, 116, 6577–6580. doi:10.1021/ja00094a012
Return to citation in text: [1] -
Sánchez-Sánchez, C.; Pérez-Inestrosa, E.; García-Segura, R.; Suau, R. Tetrahedron 2002, 58, 7267–7274. doi:10.1016/s0040-4020(02)00756-1
Return to citation in text: [1] -
Mertens, R.; von Sonntag, C. J. Photochem. Photobiol., A: Chem. 1995, 85, 1–9. doi:10.1016/1010-6030(94)03903-8
Return to citation in text: [1] -
Ashworth, B.; Gilbert, B. C.; Norman, R. O. C. J. Chem. Res., Synop. 1977, 94–95.
Return to citation in text: [1] -
Luján-Montelongo, J. A.; Covarrubias-Zúñiga, A.; Romero-Ortega, M.; Avila-Zárraga, J. G. Lett. Org. Chem. 2008, 5, 470–472. doi:10.2174/157017808785740525
Return to citation in text: [1] [2] -
Maercker, A. Angew. Chem., Int. Ed. Engl. 1987, 26, 972–989. doi:10.1002/anie.198709721
Return to citation in text: [1] -
Lahm, G.; Orejarena Pacheco, J. C.; Opatz, T. Synthesis 2014, 46, 2413–2421. doi:10.1055/s-0034-1378393
Return to citation in text: [1] -
Opatz, T. Synthesis 2009, 1941–1959. doi:10.1055/s-0029-1216839
Return to citation in text: [1] -
Otto, N.; Opatz, T. Chem. – Eur. J. 2014, 20, 13064–13077. doi:10.1002/chem.201403956
Return to citation in text: [1] -
Husson, H.-P.; Royer, J. Chem. Soc. Rev. 1999, 28, 383–394. doi:10.1039/a900153k
Return to citation in text: [1] [2] -
Girard, N.; Pouchain, L.; Hurvois, J.-P.; Moinet, C. Synlett 2006, 1679–1682. doi:10.1055/s-2006-944214
Return to citation in text: [1] -
Louafi, F.; Hurvois, J.-P.; Chibani, A.; Roisnel, T. J. Org. Chem. 2010, 75, 5721–5724. doi:10.1021/jo100714y
Return to citation in text: [1] -
Orejarena Pacheco, J. C.; Lahm, G.; Opatz, T. J. Org. Chem. 2013, 78, 4985–4992. doi:10.1021/jo400659n
Return to citation in text: [1] [2] -
Shahane, S.; Louafi, F.; Moreau, J.; Hurvois, J.-P.; Renaud, J.-L.; van de Weghe, P.; Roisnel, T. Eur. J. Org. Chem. 2008, 4622–4631. doi:10.1002/ejoc.200800512
Return to citation in text: [1] [2] -
Vu, V. H.; Louafi, F.; Girard, N.; Marion, R.; Roisnel, T.; Dorcet, V.; Hurvois, J.-P. J. Org. Chem. 2014, 79, 3358–3373. doi:10.1021/jo500104c
Return to citation in text: [1] -
Louafi, F.; Moreau, J.; Shahane, S.; Golhen, S.; Roisnel, T.; Sinbandhit, S.; Hurvois, J.-P. J. Org. Chem. 2011, 76, 9720–9732. doi:10.1021/jo2017982
Return to citation in text: [1] -
Benmekhbi, L.; Louafi, F.; Roisnel, T.; Hurvois, J.-P. J. Org. Chem. 2016, 81, 6721–6739. doi:10.1021/acs.joc.6b01419
Return to citation in text: [1] -
Amat, M.; Gómez-Esqué, A.; Escolano, C.; Santos, M. M. M.; Molins, E.; Bosch, J. J. Org. Chem. 2009, 74, 1205–1211. doi:10.1021/jo802387c
Return to citation in text: [1] -
Lahm, G.; Stoye, A.; Opatz, T. J. Org. Chem. 2012, 77, 6620–6623. doi:10.1021/jo3011045
Return to citation in text: [1] -
Bergner, I.; Wiebe, C.; Meyer, N.; Opatz, T. J. Org. Chem. 2009, 74, 8243–8253. doi:10.1021/jo901759u
Return to citation in text: [1] -
Bergner, I.; Opatz, T. Synthesis 2007, 918–928. doi:10.1055/s-2007-965937
Return to citation in text: [1] -
Maloney, K. M.; Danheiser, R. L. Org. Lett. 2005, 7, 3115–3118. doi:10.1021/ol051185n
Return to citation in text: [1] -
Meyer, N.; Opatz, T. Synlett 2003, 1427–1430. doi:10.1055/s-2003-40842
Return to citation in text: [1] -
Meyer, N.; Opatz, T. Synlett 2004, 787–790. doi:10.1055/s-2004-817774
Return to citation in text: [1] -
Orejarena Pacheco, J. C.; Lipp, A.; Nauth, A. M.; Acke, F.; Dietz, J.-P.; Opatz, T. Chem. – Eur. J. 2016, 22, 5409–5415. doi:10.1002/chem.201504845
Return to citation in text: [1] -
Kison, C.; Opatz, T. Eur. J. Org. Chem. 2008, 2740–2745. doi:10.1002/ejoc.200701205
Return to citation in text: [1] -
Ogura, K.; Shimamura, Y.; Fujita, M. J. Org. Chem. 1991, 56, 2920–2922. doi:10.1021/jo00008a062
Return to citation in text: [1] -
Guerrier, L.; Royer, J.; Grierson, D. S.; Husson, H.-P. J. Am. Chem. Soc. 1983, 105, 7754–7755. doi:10.1021/ja00364a053
Return to citation in text: [1] -
Royer, J.; Husson, H.-P. J. Org. Chem. 1985, 50, 670–673. doi:10.1021/jo00205a023
Return to citation in text: [1] -
Grierson, D. S.; Royer, J.; Guerrier, L.; Husson, H.-P. J. Org. Chem. 1986, 51, 4475–4477. doi:10.1021/jo00373a027
Return to citation in text: [1] -
Orejarena Pacheco, J. C.; Opatz, T. J. Org. Chem. 2014, 79, 5182–5192. doi:10.1021/jo500749x
Return to citation in text: [1] -
Tankabekyan, N. A.; Mokhov, V. M.; Popov, Yu. V. Russ. J. Org. Chem. 2014, 50, 1056–1057. doi:10.1134/s1070428014070215
Return to citation in text: [1] -
Yamada, S.; Tomioka, K.; Koga, K. Tetrahedron Lett. 1976, 17, 61–64. doi:10.1016/s0040-4039(00)71323-2
Return to citation in text: [1] -
Zhu, J.; Quirion, J.-C.; Husson, H.-P. J. Org. Chem. 1993, 58, 6451–6456. doi:10.1021/jo00075a048
Return to citation in text: [1] [2] -
Meyer, N.; Werner, F.; Opatz, T. Synthesis 2005, 945–956. doi:10.1055/s-2005-861838
Return to citation in text: [1] -
Badru, R.; Singh, B. RSC Adv. 2014, 4, 38978–38985. doi:10.1039/c4ra08048c
Return to citation in text: [1] -
Chang, C.-F.; Li, C.-F.; Tsai, C.-C.; Chuang, T.-H. Org. Lett. 2016, 18, 638–641. doi:10.1021/acs.orglett.5b03395
Return to citation in text: [1] [2] -
Kison, C.; Opatz, T. Synthesis 2006, 3727–3738. doi:10.1055/s-2006-950237
Return to citation in text: [1] -
Kison, C.; Meyer, N.; Opatz, T. Angew. Chem., Int. Ed. 2005, 44, 5662–5664. doi:10.1002/anie.200501705
Return to citation in text: [1] -
Li, J.; Zhao, H.; Jiang, X.; Wang, X.; Hu, H.; Yu, L.; Zhang, Y. Angew. Chem., Int. Ed. 2015, 54, 6306–6310. doi:10.1002/anie.201500961
Return to citation in text: [1] [2] [3] [4] -
Li, J.; Zhao, H.; Zhang, Y. Synlett 2015, 2745–2750. doi:10.1055/s-0035-1560178
Return to citation in text: [1] -
Black, D. S. C.; Doyle, J. E. Aust. J. Chem. 1978, 31, 2323–2326. doi:10.1071/ch9782323
Return to citation in text: [1] [2] -
Mattalia, J.-M.; Bodineau, N.; Negrel, J.-C.; Chanon, M. J. Phys. Org. Chem. 2000, 13, 233–236. doi:10.1002/1099-1395(200005)13:5<233::aid-poc235>3.0.co;2-o
Return to citation in text: [1] [2] -
Mattalia, J.-M.; Samat, A.; Chanon, M. J. Chem. Soc., Perkin Trans. 1 1991, 1769–1770. doi:10.1039/P19910001769
Return to citation in text: [1] -
Németh, G.; Poszávácz, L.; Bózsing, D.; Simig, G. J. Fluorine Chem. 1996, 78, 87–89. doi:10.1016/0022-1139(96)03433-1
Return to citation in text: [1] -
Cacciarini, M.; Skov, A. B.; Jevric, M.; Hansen, A. S.; Elm, J.; Kjaergaard, H. G.; Mikkelsen, K. V.; Nielsen, M. B. Chem. – Eur. J. 2015, 21, 7454–7461. doi:10.1002/chem.201500100
Return to citation in text: [1] -
Too, P. C.; Chan, G. H.; Tnay, Y. L.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2016, 55, 3719–3723. doi:10.1002/anie.201600305
Return to citation in text: [1] [2] [3] -
Hong, Z.; Ong, D. Y.; Muduli, S. K.; Too, P. C.; Chan, G. H.; Tnay, Y. L.; Chiba, S.; Nishiyama, Y.; Hirao, H.; Soo, H. S. Chem. – Eur. J. 2016, 22, 7108–7114. doi:10.1002/chem.201600340
Return to citation in text: [1] -
François, D.; Poupon, E.; Kunesch, N.; Husson, H.-P. Eur. J. Org. Chem. 2004, 4823–4829. doi:10.1002/ejoc.200400404
Return to citation in text: [1] -
François, D.; Poupon, E.; Lallemand, M.-C.; Kunesch, N.; Husson, H.-P. J. Org. Chem. 2000, 65, 3209–3212. doi:10.1021/jo990706f
Return to citation in text: [1] -
Mokhov, V. M.; Popov, Yu. V.; Shcherbakova, K. V. Russ. J. Gen. Chem. 2016, 86, 273–280. doi:10.1134/s1070363216020110
Return to citation in text: [1] [2] -
Weigert, F. J.; Moguel, M. J. Mol. Catal. 1992, 75, 209–218. doi:10.1016/0304-5102(92)80123-x
Return to citation in text: [1] -
Liao, Z.; Lv, X.; Tao, M. Res. Chem. Intermed. 2013, 39, 4021–4024. doi:10.1007/s11164-012-0918-x
Return to citation in text: [1] [2] -
Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996, 118, 4916–4917. doi:10.1021/ja960364k
Return to citation in text: [1] -
Werner, F.; Blank, N.; Opatz, T. Eur. J. Org. Chem. 2007, 3911–3915. doi:10.1002/ejoc.200700261
Return to citation in text: [1] -
Blank, N.; Opatz, T. J. Org. Chem. 2011, 76, 9777–9784. doi:10.1021/jo201871c
Return to citation in text: [1] -
Geffe, M.; Opatz, T. Org. Lett. 2014, 16, 5282–5285. doi:10.1021/ol5023849
Return to citation in text: [1] -
Ozawa, F.; Iri, K.; Yamamoto, A. Chem. Lett. 1982, 11, 1707–1710. doi:10.1246/cl.1982.1707
Return to citation in text: [1] -
Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613–8661. doi:10.1021/cr400628s
Return to citation in text: [1] [2] -
Kita, Y.; Tobisu, M.; Chatani, N. J. Synth. Org. Chem., Jpn. 2010, 68, 1112–1122. doi:10.5059/yukigoseikyokaishi.68.1112
Return to citation in text: [1] [2] -
Modak, A.; Maiti, D. Org. Biomol. Chem. 2016, 14, 21–35. doi:10.1039/c5ob01949d
Return to citation in text: [1] [2] -
Nakao, Y. Top. Curr. Chem. 2014, 346, 33–58. doi:10.1007/128_2013_494
Return to citation in text: [1] -
Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410–9464. doi:10.1021/acs.chemrev.5b00138
Return to citation in text: [1] -
Wen, Q.; Lu, P.; Wang, Y. RSC Adv. 2014, 4, 47806–47826. doi:10.1039/c4ra08675a
Return to citation in text: [1] -
Wang, R.; Falck, J. R. Catal. Rev.: Sci. Eng. 2014, 56, 288–331. doi:10.1080/01614940.2014.920178
Return to citation in text: [1] -
Nakazawa, H.; Itazaki, M.; Mamata, K.; Ueda, K. Chem. – Asian J. 2007, 2, 882–888. doi:10.1002/asia.200700076
Return to citation in text: [1] [2] -
Nakazawa, H.; Kamata, K.; Itazaki, M. Chem. Commun. 2005, 4004–4006. doi:10.1039/b504131g
Return to citation in text: [1] -
Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 3174–3175. doi:10.1021/ja810142v
Return to citation in text: [1] -
Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. Bull. Korean Chem. Soc. 2010, 31, 582–587. doi:10.5012/bkcs.2010.31.03.582
Return to citation in text: [1] [2] [3] -
Patra, T.; Agasti, S.; Maiti, A.; Maiti, D. Chem. Commun. 2013, 49, 69–71. doi:10.1039/c2cc36883h
Return to citation in text: [1] [2] -
Patra, T.; Agasti, S.; Modak, A.; Maiti, D. Chem. Commun. 2013, 49, 8362–8364. doi:10.1039/c3cc44562c
Return to citation in text: [1] [2] -
Weidauer, M.; Someya, C. I.; Irran, E.; Enthaler, S. Asian J. Org. Chem. 2013, 2, 150–156. doi:10.1002/ajoc.201200185
Return to citation in text: [1] [2] [3] -
Enthaler, S.; Weidauer, M.; Irran, E.; Epping, J. D.; Kretschmer, R.; Someya, C. I. J. Organomet. Chem. 2013, 745–746, 262–274. doi:10.1016/j.jorganchem.2013.07.068
Return to citation in text: [1] [2] -
Someya, C. I.; Inoue, S.; Irran, E.; Enthaler, S. Inorg. Chem. Commun. 2014, 44, 114–118. doi:10.1016/j.inoche.2014.03.014
Return to citation in text: [1] -
Dandia, A.; Singh, R.; Maheshwari, S. Curr. Org. Chem. 2014, 18, 2513–2529. doi:10.2174/138527281819141028114524
Return to citation in text: [1] -
Diaz-de-Villegas, M. D.; Gálvez, J. A.; Badorrey, R.; López-Ram-de-Viu, P. Adv. Synth. Catal. 2014, 356, 3261–3288. doi:10.1002/adsc.201400404
Return to citation in text: [1] -
Inokuma, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2006, 128, 9413–9419. doi:10.1021/ja061364f
Return to citation in text: [1] -
Pal, S.; Khan, M. N.; Karamthulla, S.; Choudhury, L. H. Tetrahedron Lett. 2015, 56, 359–364. doi:10.1016/j.tetlet.2014.11.095
Return to citation in text: [1] -
Kang, S. R.; Lee, Y. R. Synthesis 2013, 45, 2593–2599. doi:10.1055/s-0033-1338506
Return to citation in text: [1] -
Curran, D. P.; Seong, C. M. J. Am. Chem. Soc. 1990, 112, 9401–9403. doi:10.1021/ja00181a057
Return to citation in text: [1] -
Curran, D. P.; Seong, C. M. Synlett 1991, 107–108. doi:10.1055/s-1991-20644
Return to citation in text: [1] [2] [3] -
Deme, R.; Schlich, M.; Mucsi, Z.; Karvaly, G.; Tóth, G.; Mátyus, P. ARKIVOC 2016, v, 164–196. doi:10.3998/ark.5550190.p009.692
Return to citation in text: [1] -
Gerlach, U. Tetrahedron Lett. 1995, 36, 5159–5162. doi:10.1016/0040-4039(95)01006-4
Return to citation in text: [1] -
Hattori, K.; Grossman, R. B. J. Org. Chem. 2003, 68, 1409–1417. doi:10.1021/jo026643+
Return to citation in text: [1] -
Tong, B. M. K.; Chen, H.; Chong, S. Y.; Heng, Y. L.; Chiba, S. Org. Lett. 2012, 14, 2826–2829. doi:10.1021/ol301044e
Return to citation in text: [1] [2] -
Tong, B. M. K.; Chen, H.; Chong, S. Y.; Heng, Y. L.; Chiba, S. Org. Lett. 2013, 15, 6111–6112. doi:10.1021/ol4032842
Return to citation in text: [1] [2] -
Kawamoto, T.; Geib, S. J.; Curran, D. P. J. Am. Chem. Soc. 2015, 137, 8617–8622. doi:10.1021/jacs.5b04677
Return to citation in text: [1] [2] [3] -
Giles, J. R. M.; Roberts, B. P. J. Chem. Soc., Perkin Trans. 2 1983, 743–755. doi:10.1039/P29830000743
Return to citation in text: [1] -
Kang, H.-Y.; Hong, W. S.; Cho, Y. S.; Koh, H. Y. Tetrahedron Lett. 1995, 36, 7661–7664. doi:10.1016/0040-4039(95)01606-i
Return to citation in text: [1] -
Liu, Y.; Zhang, F.; Qi, Y.; Zhang, Y.; Zhang, S. Eur. J. Org. Chem. 2008, 5470–5476. doi:10.1002/ejoc.200800258
Return to citation in text: [1] -
Doni, E.; Murphy, J. A. Org. Chem. Front. 2014, 1, 1072–1076. doi:10.1039/c4qo00202d
Return to citation in text: [1] [2] [3] -
Doni, E.; Mondal, B.; O'Sullivan, S.; Tuttle, T.; Murphy, J. A. J. Am. Chem. Soc. 2013, 135, 10934–10937. doi:10.1021/ja4050168
Return to citation in text: [1] -
Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378
Return to citation in text: [1] [2] [3] -
Hasegawa, E.; Izumiya, N.; Fukuda, T.; Nemoto, K.; Iwamoto, H.; Takizawa, S.-y.; Murata, S. Tetrahedron 2016, 72, 7805–7812. doi:10.1016/j.tet.2016.05.078
Return to citation in text: [1] [2] -
Krasodomski, W.; Łuczyński, M. K.; Wilamowski, J.; Sepioł, J. J. Tetrahedron 2003, 59, 5677–5683. doi:10.1016/S0040-4020(03)00884-6
Return to citation in text: [1] [2] -
Yalçin, E.; Kutlu, Y. C.; Korkmaz, V.; Şahin, E.; Seferoğlu, Z. ARKIVOC 2015, v, 202–218. doi:10.3998/ark.5550190.p009.102
Return to citation in text: [1] -
Hédou, D.; Guillon, R.; Lecointe, C.; Logé, C.; Chosson, E.; Besson, T. Tetrahedron 2013, 69, 3182–3191. doi:10.1016/j.tet.2013.02.066
Return to citation in text: [1] [2] -
Nandi, S.; Panda, K.; Suresh, J. R.; Ila, H.; Junjappa, H. Tetrahedron 2004, 60, 3663–3673. doi:10.1016/j.tet.2004.02.053
Return to citation in text: [1] -
Panda, K.; Suresh, J. R.; Ila, H.; Junjappa, H. J. Org. Chem. 2003, 68, 3498–3506. doi:10.1021/jo026786w
Return to citation in text: [1] -
Peruncheralathan, S.; Khan, T. A.; Ila, H.; Junjappa, H. Tetrahedron 2004, 60, 3457–3464. doi:10.1016/j.tet.2004.02.029
Return to citation in text: [1] [2] -
Bendale, P. M.; Chowdhury, B. R.; Khadilkar, B. M. Indian J. Chem. 2001, 40B, 433–435.
Return to citation in text: [1] [2] -
Berkoff, C. E.; Rivard, D. E.; Kirkpatrick, D.; Ives, J. L. Synth. Commun. 1980, 10, 939–945. doi:10.1080/00397918008061855
Return to citation in text: [1] [2] -
Blicke, F. F.; Tsao, E.-P. J. Am. Chem. Soc. 1953, 75, 5587–5590. doi:10.1021/ja01118a032
Return to citation in text: [1] -
Kulp, S. S.; Romanelli, A. Org. Prep. Proced. Int. 1992, 24, 7–12. doi:10.1080/00304949209356689
Return to citation in text: [1] -
Reeves, J. T.; Malapit, C. A.; Buono, F. G.; Sidhu, K. P.; Marsini, M. A.; Sader, C. A.; Fandrick, K. R.; Busacca, C. A.; Senanayake, C. H. J. Am. Chem. Soc. 2015, 137, 9481–9488. doi:10.1021/jacs.5b06136
Return to citation in text: [1] -
Nambo, M.; Yar, M.; Smith, J. D.; Crudden, C. M. Org. Lett. 2015, 17, 50–53. doi:10.1021/ol503213z
Return to citation in text: [1] [2] -
Polivin, Y. N.; Karakhanov, R. A.; Sheveleva, T. S.; Ageev, E. A.; Remizov, A. S. Izv. Akad. Nauk SSSR, Ser. Khim. 1991, 2886–2889.
Return to citation in text: [1]
| 106. | Weidauer, M.; Someya, C. I.; Irran, E.; Enthaler, S. Asian J. Org. Chem. 2013, 2, 150–156. doi:10.1002/ajoc.201200185 |
| 107. | Enthaler, S.; Weidauer, M.; Irran, E.; Epping, J. D.; Kretschmer, R.; Someya, C. I. J. Organomet. Chem. 2013, 745–746, 262–274. doi:10.1016/j.jorganchem.2013.07.068 |
| 108. | Someya, C. I.; Inoue, S.; Irran, E.; Enthaler, S. Inorg. Chem. Commun. 2014, 44, 114–118. doi:10.1016/j.inoche.2014.03.014 |
| 105. | Patra, T.; Agasti, S.; Modak, A.; Maiti, D. Chem. Commun. 2013, 49, 8362–8364. doi:10.1039/c3cc44562c |
| 104. | Patra, T.; Agasti, S.; Maiti, A.; Maiti, D. Chem. Commun. 2013, 49, 69–71. doi:10.1039/c2cc36883h |
| 105. | Patra, T.; Agasti, S.; Modak, A.; Maiti, D. Chem. Commun. 2013, 49, 8362–8364. doi:10.1039/c3cc44562c |
| 104. | Patra, T.; Agasti, S.; Maiti, A.; Maiti, D. Chem. Commun. 2013, 49, 69–71. doi:10.1039/c2cc36883h |
| 115. | Curran, D. P.; Seong, C. M. Synlett 1991, 107–108. doi:10.1055/s-1991-20644 |
| 109. | Dandia, A.; Singh, R.; Maheshwari, S. Curr. Org. Chem. 2014, 18, 2513–2529. doi:10.2174/138527281819141028114524 |
| 110. | Diaz-de-Villegas, M. D.; Gálvez, J. A.; Badorrey, R.; López-Ram-de-Viu, P. Adv. Synth. Catal. 2014, 356, 3261–3288. doi:10.1002/adsc.201400404 |
| 111. | Inokuma, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2006, 128, 9413–9419. doi:10.1021/ja061364f |
| 112. | Pal, S.; Khan, M. N.; Karamthulla, S.; Choudhury, L. H. Tetrahedron Lett. 2015, 56, 359–364. doi:10.1016/j.tetlet.2014.11.095 |
| 113. | Kang, S. R.; Lee, Y. R. Synthesis 2013, 45, 2593–2599. doi:10.1055/s-0033-1338506 |
| 114. | Curran, D. P.; Seong, C. M. J. Am. Chem. Soc. 1990, 112, 9401–9403. doi:10.1021/ja00181a057 |
| 106. | Weidauer, M.; Someya, C. I.; Irran, E.; Enthaler, S. Asian J. Org. Chem. 2013, 2, 150–156. doi:10.1002/ajoc.201200185 |
| 107. | Enthaler, S.; Weidauer, M.; Irran, E.; Epping, J. D.; Kretschmer, R.; Someya, C. I. J. Organomet. Chem. 2013, 745–746, 262–274. doi:10.1016/j.jorganchem.2013.07.068 |
| 106. | Weidauer, M.; Someya, C. I.; Irran, E.; Enthaler, S. Asian J. Org. Chem. 2013, 2, 150–156. doi:10.1002/ajoc.201200185 |
| 115. | Curran, D. P.; Seong, C. M. Synlett 1991, 107–108. doi:10.1055/s-1991-20644 |
| 140. | Nambo, M.; Yar, M.; Smith, J. D.; Crudden, C. M. Org. Lett. 2015, 17, 50–53. doi:10.1021/ol503213z |
| 121. | Kawamoto, T.; Geib, S. J.; Curran, D. P. J. Am. Chem. Soc. 2015, 137, 8617–8622. doi:10.1021/jacs.5b04677 |
| 136. | Berkoff, C. E.; Rivard, D. E.; Kirkpatrick, D.; Ives, J. L. Synth. Commun. 1980, 10, 939–945. doi:10.1080/00397918008061855 |
| 119. | Tong, B. M. K.; Chen, H.; Chong, S. Y.; Heng, Y. L.; Chiba, S. Org. Lett. 2012, 14, 2826–2829. doi:10.1021/ol301044e |
| 120. | Tong, B. M. K.; Chen, H.; Chong, S. Y.; Heng, Y. L.; Chiba, S. Org. Lett. 2013, 15, 6111–6112. doi:10.1021/ol4032842 |
| 140. | Nambo, M.; Yar, M.; Smith, J. D.; Crudden, C. M. Org. Lett. 2015, 17, 50–53. doi:10.1021/ol503213z |
| 121. | Kawamoto, T.; Geib, S. J.; Curran, D. P. J. Am. Chem. Soc. 2015, 137, 8617–8622. doi:10.1021/jacs.5b04677 |
| 141. | Polivin, Y. N.; Karakhanov, R. A.; Sheveleva, T. S.; Ageev, E. A.; Remizov, A. S. Izv. Akad. Nauk SSSR, Ser. Khim. 1991, 2886–2889. |
| 116. | Deme, R.; Schlich, M.; Mucsi, Z.; Karvaly, G.; Tóth, G.; Mátyus, P. ARKIVOC 2016, v, 164–196. doi:10.3998/ark.5550190.p009.692 |
| 117. | Gerlach, U. Tetrahedron Lett. 1995, 36, 5159–5162. doi:10.1016/0040-4039(95)01006-4 |
| 118. | Hattori, K.; Grossman, R. B. J. Org. Chem. 2003, 68, 1409–1417. doi:10.1021/jo026643+ |
| 135. | Bendale, P. M.; Chowdhury, B. R.; Khadilkar, B. M. Indian J. Chem. 2001, 40B, 433–435. |
| 136. | Berkoff, C. E.; Rivard, D. E.; Kirkpatrick, D.; Ives, J. L. Synth. Commun. 1980, 10, 939–945. doi:10.1080/00397918008061855 |
| 119. | Tong, B. M. K.; Chen, H.; Chong, S. Y.; Heng, Y. L.; Chiba, S. Org. Lett. 2012, 14, 2826–2829. doi:10.1021/ol301044e |
| 120. | Tong, B. M. K.; Chen, H.; Chong, S. Y.; Heng, Y. L.; Chiba, S. Org. Lett. 2013, 15, 6111–6112. doi:10.1021/ol4032842 |
| 129. | Krasodomski, W.; Łuczyński, M. K.; Wilamowski, J.; Sepioł, J. J. Tetrahedron 2003, 59, 5677–5683. doi:10.1016/S0040-4020(03)00884-6 |
| 131. | Hédou, D.; Guillon, R.; Lecointe, C.; Logé, C.; Chosson, E.; Besson, T. Tetrahedron 2013, 69, 3182–3191. doi:10.1016/j.tet.2013.02.066 |
| 134. | Peruncheralathan, S.; Khan, T. A.; Ila, H.; Junjappa, H. Tetrahedron 2004, 60, 3457–3464. doi:10.1016/j.tet.2004.02.029 |
| 135. | Bendale, P. M.; Chowdhury, B. R.; Khadilkar, B. M. Indian J. Chem. 2001, 40B, 433–435. |
| 139. | Reeves, J. T.; Malapit, C. A.; Buono, F. G.; Sidhu, K. P.; Marsini, M. A.; Sader, C. A.; Fandrick, K. R.; Busacca, C. A.; Senanayake, C. H. J. Am. Chem. Soc. 2015, 137, 9481–9488. doi:10.1021/jacs.5b06136 |
| 137. | Blicke, F. F.; Tsao, E.-P. J. Am. Chem. Soc. 1953, 75, 5587–5590. doi:10.1021/ja01118a032 |
| 138. | Kulp, S. S.; Romanelli, A. Org. Prep. Proced. Int. 1992, 24, 7–12. doi:10.1080/00304949209356689 |
| 131. | Hédou, D.; Guillon, R.; Lecointe, C.; Logé, C.; Chosson, E.; Besson, T. Tetrahedron 2013, 69, 3182–3191. doi:10.1016/j.tet.2013.02.066 |
| 132. | Nandi, S.; Panda, K.; Suresh, J. R.; Ila, H.; Junjappa, H. Tetrahedron 2004, 60, 3663–3673. doi:10.1016/j.tet.2004.02.053 |
| 133. | Panda, K.; Suresh, J. R.; Ila, H.; Junjappa, H. J. Org. Chem. 2003, 68, 3498–3506. doi:10.1021/jo026786w |
| 134. | Peruncheralathan, S.; Khan, T. A.; Ila, H.; Junjappa, H. Tetrahedron 2004, 60, 3457–3464. doi:10.1016/j.tet.2004.02.029 |
| 121. | Kawamoto, T.; Geib, S. J.; Curran, D. P. J. Am. Chem. Soc. 2015, 137, 8617–8622. doi:10.1021/jacs.5b04677 |
| 123. | Kang, H.-Y.; Hong, W. S.; Cho, Y. S.; Koh, H. Y. Tetrahedron Lett. 1995, 36, 7661–7664. doi:10.1016/0040-4039(95)01606-i |
| 115. | Curran, D. P.; Seong, C. M. Synlett 1991, 107–108. doi:10.1055/s-1991-20644 |
| 122. | Giles, J. R. M.; Roberts, B. P. J. Chem. Soc., Perkin Trans. 2 1983, 743–755. doi:10.1039/P29830000743 |
| 9. | Mattalia, J.-M.; Marchi-Delapierre, C.; Hazimeh, H.; Chanon, M. ARKIVOC 2006, iv, 90–118. doi:10.3998/ark.5550190.0007.408 |
| 76. | Black, D. S. C.; Doyle, J. E. Aust. J. Chem. 1978, 31, 2323–2326. doi:10.1071/ch9782323 |
| 77. | Mattalia, J.-M.; Bodineau, N.; Negrel, J.-C.; Chanon, M. J. Phys. Org. Chem. 2000, 13, 233–236. doi:10.1002/1099-1395(200005)13:5<233::aid-poc235>3.0.co;2-o |
| 81. | Too, P. C.; Chan, G. H.; Tnay, Y. L.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2016, 55, 3719–3723. doi:10.1002/anie.201600305 |
| 82. | Hong, Z.; Ong, D. Y.; Muduli, S. K.; Too, P. C.; Chan, G. H.; Tnay, Y. L.; Chiba, S.; Nishiyama, Y.; Hirao, H.; Soo, H. S. Chem. – Eur. J. 2016, 22, 7108–7114. doi:10.1002/chem.201600340 |
| 1. | Zhao, L.; Wen, M.; Wang, Z.-X. Eur. J. Org. Chem. 2012, 3587–3597. doi:10.1002/ejoc.201200121 |
| 2. | Huang, Q.; Tran, G.; Pardo, D. G.; Tsuchiya, T.; Hillebrand, S.; Vors, J.-P.; Cossy, J. Tetrahedron 2015, 71, 7250–7259. doi:10.1016/j.tet.2015.03.099 |
| 9. | Mattalia, J.-M.; Marchi-Delapierre, C.; Hazimeh, H.; Chanon, M. ARKIVOC 2006, iv, 90–118. doi:10.3998/ark.5550190.0007.408 |
| 88. | Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996, 118, 4916–4917. doi:10.1021/ja960364k |
| 8. | Fleming, F. F.; Zhang, Z. Tetrahedron 2005, 61, 747–789. doi:10.1016/j.tet.2004.11.012 |
| 7. | López, R.; Palomo, C. Angew. Chem., Int. Ed. 2015, 54, 13170–13184. doi:10.1002/anie.201502493 |
| 87. | Liao, Z.; Lv, X.; Tao, M. Res. Chem. Intermed. 2013, 39, 4021–4024. doi:10.1007/s11164-012-0918-x |
| 3. | Reddy, M. C.; Jeganmohan, M. Chem. Commun. 2015, 51, 10738–10741. doi:10.1039/c5cc03112e |
| 4. | Bera, M.; Modak, A.; Patra, T.; Maji, A.; Maiti, D. Org. Lett. 2014, 16, 5760–5763. doi:10.1021/ol502823c |
| 5. | Bera, M.; Maji, A.; Sahoo, S. K.; Maiti, D. Angew. Chem., Int. Ed. 2015, 54, 8515–8519. doi:10.1002/anie.201503112 |
| 6. | Du, B.; Jiang, X.; Sun, P. J. Org. Chem. 2013, 78, 2786–2791. doi:10.1021/jo302765g |
| 87. | Liao, Z.; Lv, X.; Tao, M. Res. Chem. Intermed. 2013, 39, 4021–4024. doi:10.1007/s11164-012-0918-x |
| 17. | Larchevêque, M.; Cuvigny, T. Tetrahedron Lett. 1975, 16, 3851–3854. doi:10.1016/S0040-4039(00)91294-2 |
| 18. | Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962–4970. doi:10.1021/jo800640j |
| 85. | Mokhov, V. M.; Popov, Yu. V.; Shcherbakova, K. V. Russ. J. Gen. Chem. 2016, 86, 273–280. doi:10.1134/s1070363216020110 |
| 10. | Arapakos, P. G. J. Am. Chem. Soc. 1967, 89, 6794–6796. doi:10.1021/ja01001a090 |
| 16. | Arapakos, P. G.; Scott, M. K.; Huber, F. E., Jr. J. Am. Chem. Soc. 1969, 91, 2059–2062. doi:10.1021/ja01036a033 |
| 86. | Weigert, F. J.; Moguel, M. J. Mol. Catal. 1992, 75, 209–218. doi:10.1016/0304-5102(92)80123-x |
| 11. | Rychnovsky, S. D.; Rogers, B. N.; Richardson, T. I. Acc. Chem. Res. 1998, 31, 9–17. doi:10.1021/ar960223n |
| 12. | Schleicher, K. D.; Jamison, T. F. Beilstein J. Org. Chem. 2013, 9, 1533–1550. doi:10.3762/bjoc.9.175 |
| 13. | McLachlan, M. M. W.; O'Connor, P. D.; Fairweather, K. A.; Willis, A. C.; Mander, L. N. Aust. J. Chem. 2010, 63, 742–760. doi:10.1071/CH10056 |
| 14. | Sinz, C. J.; Rychnovsky, S. D. Top. Curr. Chem. 2001, 216, 51–92. doi:10.1007/3-540-44726-1_2 |
| 15. | Rychnovsky, S. D. Chem. Rev. 1995, 95, 2021–2040. doi:10.1021/cr00038a011 |
| 83. | François, D.; Poupon, E.; Kunesch, N.; Husson, H.-P. Eur. J. Org. Chem. 2004, 4823–4829. doi:10.1002/ejoc.200400404 |
| 84. | François, D.; Poupon, E.; Lallemand, M.-C.; Kunesch, N.; Husson, H.-P. J. Org. Chem. 2000, 65, 3209–3212. doi:10.1021/jo990706f |
| 10. | Arapakos, P. G. J. Am. Chem. Soc. 1967, 89, 6794–6796. doi:10.1021/ja01001a090 |
| 85. | Mokhov, V. M.; Popov, Yu. V.; Shcherbakova, K. V. Russ. J. Gen. Chem. 2016, 86, 273–280. doi:10.1134/s1070363216020110 |
| 93. | Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613–8661. doi:10.1021/cr400628s |
| 94. | Kita, Y.; Tobisu, M.; Chatani, N. J. Synth. Org. Chem., Jpn. 2010, 68, 1112–1122. doi:10.5059/yukigoseikyokaishi.68.1112 |
| 95. | Modak, A.; Maiti, D. Org. Biomol. Chem. 2016, 14, 21–35. doi:10.1039/c5ob01949d |
| 96. | Nakao, Y. Top. Curr. Chem. 2014, 346, 33–58. doi:10.1007/128_2013_494 |
| 97. | Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410–9464. doi:10.1021/acs.chemrev.5b00138 |
| 98. | Wen, Q.; Lu, P.; Wang, Y. RSC Adv. 2014, 4, 47806–47826. doi:10.1039/c4ra08675a |
| 99. | Wang, R.; Falck, J. R. Catal. Rev.: Sci. Eng. 2014, 56, 288–331. doi:10.1080/01614940.2014.920178 |
| 93. | Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613–8661. doi:10.1021/cr400628s |
| 95. | Modak, A.; Maiti, D. Org. Biomol. Chem. 2016, 14, 21–35. doi:10.1039/c5ob01949d |
| 89. | Werner, F.; Blank, N.; Opatz, T. Eur. J. Org. Chem. 2007, 3911–3915. doi:10.1002/ejoc.200700261 |
| 90. | Blank, N.; Opatz, T. J. Org. Chem. 2011, 76, 9777–9784. doi:10.1021/jo201871c |
| 91. | Geffe, M.; Opatz, T. Org. Lett. 2014, 16, 5282–5285. doi:10.1021/ol5023849 |
| 92. | Ozawa, F.; Iri, K.; Yamamoto, A. Chem. Lett. 1982, 11, 1707–1710. doi:10.1246/cl.1982.1707 |
| 103. | Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. Bull. Korean Chem. Soc. 2010, 31, 582–587. doi:10.5012/bkcs.2010.31.03.582 |
| 103. | Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. Bull. Korean Chem. Soc. 2010, 31, 582–587. doi:10.5012/bkcs.2010.31.03.582 |
| 94. | Kita, Y.; Tobisu, M.; Chatani, N. J. Synth. Org. Chem., Jpn. 2010, 68, 1112–1122. doi:10.5059/yukigoseikyokaishi.68.1112 |
| 102. | Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 3174–3175. doi:10.1021/ja810142v |
| 103. | Tobisu, M.; Nakamura, R.; Kita, Y.; Chatani, N. Bull. Korean Chem. Soc. 2010, 31, 582–587. doi:10.5012/bkcs.2010.31.03.582 |
| 100. | Nakazawa, H.; Itazaki, M.; Mamata, K.; Ueda, K. Chem. – Asian J. 2007, 2, 882–888. doi:10.1002/asia.200700076 |
| 101. | Nakazawa, H.; Kamata, K.; Itazaki, M. Chem. Commun. 2005, 4004–4006. doi:10.1039/b504131g |
| 100. | Nakazawa, H.; Itazaki, M.; Mamata, K.; Ueda, K. Chem. – Asian J. 2007, 2, 882–888. doi:10.1002/asia.200700076 |
| 41. | Lahm, G.; Orejarena Pacheco, J. C.; Opatz, T. Synthesis 2014, 46, 2413–2421. doi:10.1055/s-0034-1378393 |
| 42. | Opatz, T. Synthesis 2009, 1941–1959. doi:10.1055/s-0029-1216839 |
| 43. | Otto, N.; Opatz, T. Chem. – Eur. J. 2014, 20, 13064–13077. doi:10.1002/chem.201403956 |
| 44. | Husson, H.-P.; Royer, J. Chem. Soc. Rev. 1999, 28, 383–394. doi:10.1039/a900153k |
| 23. | Perry, M. A.; Rychnovsky, S. D. Nat. Prod. Rep. 2015, 32, 517–533. doi:10.1039/c4np00125g |
| 44. | Husson, H.-P.; Royer, J. Chem. Soc. Rev. 1999, 28, 383–394. doi:10.1039/a900153k |
| 45. | Girard, N.; Pouchain, L.; Hurvois, J.-P.; Moinet, C. Synlett 2006, 1679–1682. doi:10.1055/s-2006-944214 |
| 46. | Louafi, F.; Hurvois, J.-P.; Chibani, A.; Roisnel, T. J. Org. Chem. 2010, 75, 5721–5724. doi:10.1021/jo100714y |
| 47. | Orejarena Pacheco, J. C.; Lahm, G.; Opatz, T. J. Org. Chem. 2013, 78, 4985–4992. doi:10.1021/jo400659n |
| 48. | Shahane, S.; Louafi, F.; Moreau, J.; Hurvois, J.-P.; Renaud, J.-L.; van de Weghe, P.; Roisnel, T. Eur. J. Org. Chem. 2008, 4622–4631. doi:10.1002/ejoc.200800512 |
| 49. | Vu, V. H.; Louafi, F.; Girard, N.; Marion, R.; Roisnel, T.; Dorcet, V.; Hurvois, J.-P. J. Org. Chem. 2014, 79, 3358–3373. doi:10.1021/jo500104c |
| 50. | Louafi, F.; Moreau, J.; Shahane, S.; Golhen, S.; Roisnel, T.; Sinbandhit, S.; Hurvois, J.-P. J. Org. Chem. 2011, 76, 9720–9732. doi:10.1021/jo2017982 |
| 51. | Benmekhbi, L.; Louafi, F.; Roisnel, T.; Hurvois, J.-P. J. Org. Chem. 2016, 81, 6721–6739. doi:10.1021/acs.joc.6b01419 |
| 68. | Zhu, J.; Quirion, J.-C.; Husson, H.-P. J. Org. Chem. 1993, 58, 6451–6456. doi:10.1021/jo00075a048 |
| 71. | Chang, C.-F.; Li, C.-F.; Tsai, C.-C.; Chuang, T.-H. Org. Lett. 2016, 18, 638–641. doi:10.1021/acs.orglett.5b03395 |
| 48. | Shahane, S.; Louafi, F.; Moreau, J.; Hurvois, J.-P.; Renaud, J.-L.; van de Weghe, P.; Roisnel, T. Eur. J. Org. Chem. 2008, 4622–4631. doi:10.1002/ejoc.200800512 |
| 67. | Yamada, S.; Tomioka, K.; Koga, K. Tetrahedron Lett. 1976, 17, 61–64. doi:10.1016/s0040-4039(00)71323-2 |
| 68. | Zhu, J.; Quirion, J.-C.; Husson, H.-P. J. Org. Chem. 1993, 58, 6451–6456. doi:10.1021/jo00075a048 |
| 69. | Meyer, N.; Werner, F.; Opatz, T. Synthesis 2005, 945–956. doi:10.1055/s-2005-861838 |
| 62. | Guerrier, L.; Royer, J.; Grierson, D. S.; Husson, H.-P. J. Am. Chem. Soc. 1983, 105, 7754–7755. doi:10.1021/ja00364a053 |
| 63. | Royer, J.; Husson, H.-P. J. Org. Chem. 1985, 50, 670–673. doi:10.1021/jo00205a023 |
| 64. | Grierson, D. S.; Royer, J.; Guerrier, L.; Husson, H.-P. J. Org. Chem. 1986, 51, 4475–4477. doi:10.1021/jo00373a027 |
| 65. | Orejarena Pacheco, J. C.; Opatz, T. J. Org. Chem. 2014, 79, 5182–5192. doi:10.1021/jo500749x |
| 66. | Tankabekyan, N. A.; Mokhov, V. M.; Popov, Yu. V. Russ. J. Org. Chem. 2014, 50, 1056–1057. doi:10.1134/s1070428014070215 |
| 47. | Orejarena Pacheco, J. C.; Lahm, G.; Opatz, T. J. Org. Chem. 2013, 78, 4985–4992. doi:10.1021/jo400659n |
| 52. | Amat, M.; Gómez-Esqué, A.; Escolano, C.; Santos, M. M. M.; Molins, E.; Bosch, J. J. Org. Chem. 2009, 74, 1205–1211. doi:10.1021/jo802387c |
| 53. | Lahm, G.; Stoye, A.; Opatz, T. J. Org. Chem. 2012, 77, 6620–6623. doi:10.1021/jo3011045 |
| 54. | Bergner, I.; Wiebe, C.; Meyer, N.; Opatz, T. J. Org. Chem. 2009, 74, 8243–8253. doi:10.1021/jo901759u |
| 55. | Bergner, I.; Opatz, T. Synthesis 2007, 918–928. doi:10.1055/s-2007-965937 |
| 56. | Maloney, K. M.; Danheiser, R. L. Org. Lett. 2005, 7, 3115–3118. doi:10.1021/ol051185n |
| 57. | Meyer, N.; Opatz, T. Synlett 2003, 1427–1430. doi:10.1055/s-2003-40842 |
| 58. | Meyer, N.; Opatz, T. Synlett 2004, 787–790. doi:10.1055/s-2004-817774 |
| 59. | Orejarena Pacheco, J. C.; Lipp, A.; Nauth, A. M.; Acke, F.; Dietz, J.-P.; Opatz, T. Chem. – Eur. J. 2016, 22, 5409–5415. doi:10.1002/chem.201504845 |
| 60. | Kison, C.; Opatz, T. Eur. J. Org. Chem. 2008, 2740–2745. doi:10.1002/ejoc.200701205 |
| 61. | Ogura, K.; Shimamura, Y.; Fujita, M. J. Org. Chem. 1991, 56, 2920–2922. doi:10.1021/jo00008a062 |
| 71. | Chang, C.-F.; Li, C.-F.; Tsai, C.-C.; Chuang, T.-H. Org. Lett. 2016, 18, 638–641. doi:10.1021/acs.orglett.5b03395 |
| 72. | Kison, C.; Opatz, T. Synthesis 2006, 3727–3738. doi:10.1055/s-2006-950237 |
| 73. | Kison, C.; Meyer, N.; Opatz, T. Angew. Chem., Int. Ed. 2005, 44, 5662–5664. doi:10.1002/anie.200501705 |
| 74. | Li, J.; Zhao, H.; Jiang, X.; Wang, X.; Hu, H.; Yu, L.; Zhang, Y. Angew. Chem., Int. Ed. 2015, 54, 6306–6310. doi:10.1002/anie.201500961 |
| 81. | Too, P. C.; Chan, G. H.; Tnay, Y. L.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2016, 55, 3719–3723. doi:10.1002/anie.201600305 |
| 81. | Too, P. C.; Chan, G. H.; Tnay, Y. L.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2016, 55, 3719–3723. doi:10.1002/anie.201600305 |
| 76. | Black, D. S. C.; Doyle, J. E. Aust. J. Chem. 1978, 31, 2323–2326. doi:10.1071/ch9782323 |
| 77. | Mattalia, J.-M.; Bodineau, N.; Negrel, J.-C.; Chanon, M. J. Phys. Org. Chem. 2000, 13, 233–236. doi:10.1002/1099-1395(200005)13:5<233::aid-poc235>3.0.co;2-o |
| 78. | Mattalia, J.-M.; Samat, A.; Chanon, M. J. Chem. Soc., Perkin Trans. 1 1991, 1769–1770. doi:10.1039/P19910001769 |
| 79. | Németh, G.; Poszávácz, L.; Bózsing, D.; Simig, G. J. Fluorine Chem. 1996, 78, 87–89. doi:10.1016/0022-1139(96)03433-1 |
| 80. | Cacciarini, M.; Skov, A. B.; Jevric, M.; Hansen, A. S.; Elm, J.; Kjaergaard, H. G.; Mikkelsen, K. V.; Nielsen, M. B. Chem. – Eur. J. 2015, 21, 7454–7461. doi:10.1002/chem.201500100 |
| 74. | Li, J.; Zhao, H.; Jiang, X.; Wang, X.; Hu, H.; Yu, L.; Zhang, Y. Angew. Chem., Int. Ed. 2015, 54, 6306–6310. doi:10.1002/anie.201500961 |
| 74. | Li, J.; Zhao, H.; Jiang, X.; Wang, X.; Hu, H.; Yu, L.; Zhang, Y. Angew. Chem., Int. Ed. 2015, 54, 6306–6310. doi:10.1002/anie.201500961 |
| 75. | Li, J.; Zhao, H.; Zhang, Y. Synlett 2015, 2745–2750. doi:10.1055/s-0035-1560178 |
| 74. | Li, J.; Zhao, H.; Jiang, X.; Wang, X.; Hu, H.; Yu, L.; Zhang, Y. Angew. Chem., Int. Ed. 2015, 54, 6306–6310. doi:10.1002/anie.201500961 |
| 125. | Doni, E.; Murphy, J. A. Org. Chem. Front. 2014, 1, 1072–1076. doi:10.1039/c4qo00202d |
| 127. | Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378 |
| 128. | Hasegawa, E.; Izumiya, N.; Fukuda, T.; Nemoto, K.; Iwamoto, H.; Takizawa, S.-y.; Murata, S. Tetrahedron 2016, 72, 7805–7812. doi:10.1016/j.tet.2016.05.078 |
| 127. | Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378 |
| 125. | Doni, E.; Murphy, J. A. Org. Chem. Front. 2014, 1, 1072–1076. doi:10.1039/c4qo00202d |
| 127. | Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378 |
| 125. | Doni, E.; Murphy, J. A. Org. Chem. Front. 2014, 1, 1072–1076. doi:10.1039/c4qo00202d |
| 126. | Doni, E.; Mondal, B.; O'Sullivan, S.; Tuttle, T.; Murphy, J. A. J. Am. Chem. Soc. 2013, 135, 10934–10937. doi:10.1021/ja4050168 |
| 124. | Liu, Y.; Zhang, F.; Qi, Y.; Zhang, Y.; Zhang, S. Eur. J. Org. Chem. 2008, 5470–5476. doi:10.1002/ejoc.200800258 |
| 30. | Richardson, T. I.; Rychnovsky, S. D. Tetrahedron 1999, 55, 8977–8996. doi:10.1016/s0040-4020(99)00457-3 |
| 32. | da Silva, L. C.; Wagener, K. B. Macromol. Chem. Phys. 2016, 217, 850–855. doi:10.1002/macp.201500508 |
| 28. | Yus, M.; Foubelo, F. Dissolving Metals. In Modern Reduction Methods; Andersson, P. G.; Munslow, I. J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp 419–445. doi:10.1002/9783527622115.ch17 |
| 29. | Mander, L. N.; McLachlan, M. M. J. Am. Chem. Soc. 2003, 125, 2400–2401. doi:10.1021/ja029725o |
| 30. | Richardson, T. I.; Rychnovsky, S. D. Tetrahedron 1999, 55, 8977–8996. doi:10.1016/s0040-4020(99)00457-3 |
| 31. | Sinz, C. J.; Rychnovsky, S. D. Angew. Chem., Int. Ed. 2001, 40, 3224–3227. doi:10.1002/1521-3773(20010903)40:17<3224::aid-anie3224>3.0.co;2-d |
| 15. | Rychnovsky, S. D. Chem. Rev. 1995, 95, 2021–2040. doi:10.1021/cr00038a011 |
| 21. | Amancha, P. K.; Lai, Y.-C.; Chen, I.-C.; Liu, H.-J.; Zhu, J.-L. Tetrahedron 2010, 66, 871–877. doi:10.1016/j.tet.2009.11.105 |
| 22. | Amancha, P. K.; Liu, H.-J.; Ly, T. W.; Shia, K.-S. Eur. J. Org. Chem. 2010, 3473–3480. doi:10.1002/ejoc.201000318 |
| 129. | Krasodomski, W.; Łuczyński, M. K.; Wilamowski, J.; Sepioł, J. J. Tetrahedron 2003, 59, 5677–5683. doi:10.1016/S0040-4020(03)00884-6 |
| 130. | Yalçin, E.; Kutlu, Y. C.; Korkmaz, V.; Şahin, E.; Seferoğlu, Z. ARKIVOC 2015, v, 202–218. doi:10.3998/ark.5550190.p009.102 |
| 23. | Perry, M. A.; Rychnovsky, S. D. Nat. Prod. Rep. 2015, 32, 517–533. doi:10.1039/c4np00125g |
| 24. | Zeller, E.; Sajus, H.; Grierson, D. S. Synlett 1991, 44–46. doi:10.1055/s-1991-20623 |
| 25. | La Cruz, T. E.; Rychnovsky, S. D. Org. Lett. 2005, 7, 1873–1875. doi:10.1021/ol050589c |
| 26. | Tsao, J.-P.; Tsai, T.-Y.; Chen, I.-C.; Liu, H.-J.; Zhu, J.-L.; Tsao, S.-W. Synthesis 2010, 4242–4250. doi:10.1055/s-0030-1258301 |
| 27. | Cacciarini, M.; Jevric, M.; Elm, J.; Petersen, A. U.; Mikkelsen, K. V.; Nielsen, M. B. RSC Adv. 2016, 6, 49003–49010. doi:10.1039/c6ra06045e |
| 128. | Hasegawa, E.; Izumiya, N.; Fukuda, T.; Nemoto, K.; Iwamoto, H.; Takizawa, S.-y.; Murata, S. Tetrahedron 2016, 72, 7805–7812. doi:10.1016/j.tet.2016.05.078 |
| 19. | Ohsawa, T.; Kobayashi, T.; Mizuguchi, Y.; Saitoh, T.; Oishi, T. Tetrahedron Lett. 1985, 26, 6103–6106. doi:10.1016/S0040-4039(00)95137-2 |
| 20. | Baek, D. J.; Bittman, R. Chem. Phys. Lipids 2013, 175–176, 99–104. doi:10.1016/j.chemphyslip.2013.08.003 |
| 9. | Mattalia, J.-M.; Marchi-Delapierre, C.; Hazimeh, H.; Chanon, M. ARKIVOC 2006, iv, 90–118. doi:10.3998/ark.5550190.0007.408 |
| 34. | Savoia, D.; Tagliavini, E.; Trombini, C.; Umani-Ronchi, A. J. Org. Chem. 1980, 45, 3227–3229. doi:10.1021/jo01304a016 |
| 33. | Rojas, G.; Baughman, T. W.; Wagener, K. B. Synth. Commun. 2007, 37, 3923–3931. doi:10.1080/00397910701572456 |
| 18. | Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962–4970. doi:10.1021/jo800640j |
| 40. | Maercker, A. Angew. Chem., Int. Ed. Engl. 1987, 26, 972–989. doi:10.1002/anie.198709721 |
| 39. | Luján-Montelongo, J. A.; Covarrubias-Zúñiga, A.; Romero-Ortega, M.; Avila-Zárraga, J. G. Lett. Org. Chem. 2008, 5, 470–472. doi:10.2174/157017808785740525 |
| 18. | Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962–4970. doi:10.1021/jo800640j |
| 34. | Savoia, D.; Tagliavini, E.; Trombini, C.; Umani-Ronchi, A. J. Org. Chem. 1980, 45, 3227–3229. doi:10.1021/jo01304a016 |
| 39. | Luján-Montelongo, J. A.; Covarrubias-Zúñiga, A.; Romero-Ortega, M.; Avila-Zárraga, J. G. Lett. Org. Chem. 2008, 5, 470–472. doi:10.2174/157017808785740525 |
| 36. | Sánchez-Sánchez, C.; Pérez-Inestrosa, E.; García-Segura, R.; Suau, R. Tetrahedron 2002, 58, 7267–7274. doi:10.1016/s0040-4020(02)00756-1 |
| 37. | Mertens, R.; von Sonntag, C. J. Photochem. Photobiol., A: Chem. 1995, 85, 1–9. doi:10.1016/1010-6030(94)03903-8 |
| 38. | Ashworth, B.; Gilbert, B. C.; Norman, R. O. C. J. Chem. Res., Synop. 1977, 94–95. |
| 18. | Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962–4970. doi:10.1021/jo800640j |
| 18. | Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962–4970. doi:10.1021/jo800640j |
| 33. | Rojas, G.; Baughman, T. W.; Wagener, K. B. Synth. Commun. 2007, 37, 3923–3931. doi:10.1080/00397910701572456 |
| 35. | Bailey, W. F.; Punzalan, E. R. J. Am. Chem. Soc. 1994, 116, 6577–6580. doi:10.1021/ja00094a012 |
© 2017 Mattalia; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)