Abstract
The use of γ-Al2O3 as a heterogeneous catalyst in scCO2 has been successfully applied to the amination of alcohols for the synthesis of N-alkylated heterocycles. The optimal reaction conditions (temperature and substrate flow rate) were determined using an automated self-optimising reactor, resulting in moderate to high yields of the target products. Carrying out the reaction in scCO2 was shown to be beneficial, as higher yields were obtained in the presence of CO2 than in its absence. A surprising discovery is that, in addition to cyclic amines, cyclic ureas and urethanes could be synthesised by incorporation of CO2 from the supercritical solvent into the product.

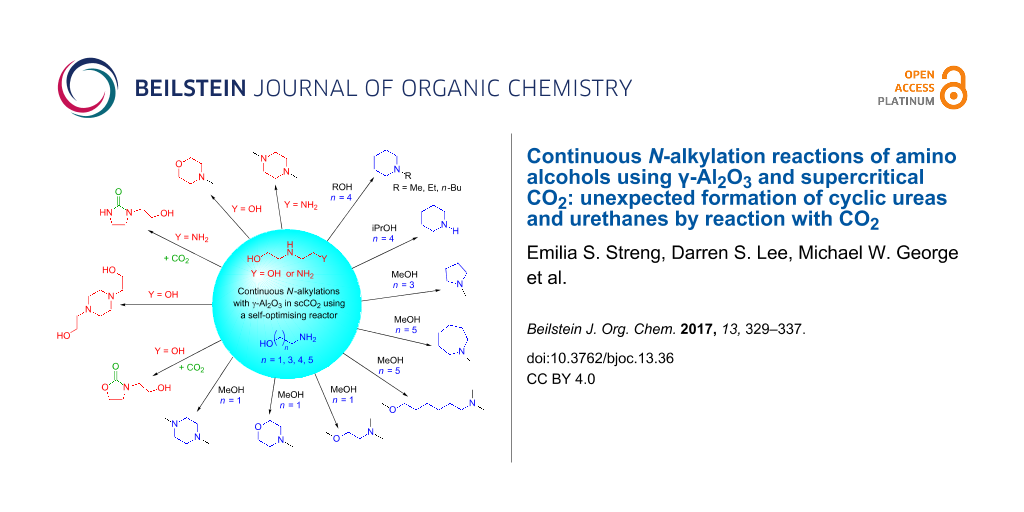
Graphical Abstract
Introduction
N-alkylated amines are an important motif present in a range of pharmaceutically and industrially useful chemicals; the alkylation of amines is a commonly used reaction in process R&D toward the synthesis of drug candidates [1-3]. Traditional methods to produce such compounds frequently employ toxic alkylating agents or harsh reagents that can generate stoichiometric quantities of waste, e.g., boron salts from reductive amination [4]. Hydrogenation offers a greener approach but is often only applicable to simple substrates due to chemoselectivity issues. An approach that has received much attention recently is the concept of hydrogen borrowing catalysis [5-19]. The coupling of alcohols and amines is made possible by the catalysts ability to take two H atoms from the alcohol, oxidising it to an aldehyde. The aldehyde then reacts with the amine affording an imine, which is subsequently reduced by transferring two H atoms back from the catalyst. In this case the only byproduct is water. Another approach to N-alkylation in which water is the only byproduct is the direct substitution of alcohols with amines. It is an attractive method; however, it requires significant activation of the alcohol or amine to proceed efficiently, and often a heterogeneous catalyst at elevated temperature and/or pressure is employed [20-28]. As these reactions are mostly carried out in high pressure systems, they are particularly suitable for the use of supercritical solvents. Supercritical solvents are highly compressed and/or heated gases that are beyond the critical point (e.g., the critical point for CO2 is 31.1 °C and 73.9 bar); in this phase the gas exhibits unique properties and behaves both like a liquid and gas. Using inert supercritical gases as reaction solvents is a greener alternative to using conventional flammable or toxic solvents; furthermore post-reaction separation is simplified as the gas/liquid phases separate upon cooling. The use of supercritical methanol (scMeOH) for N-alkylation reactions has been reported before [29,30].
Our own investigations with heterogeneous catalysis in supercritical carbon dioxide (scCO2) have mainly been focused on continuous flow systems and the etherification of alcohols, where alcohols are activated by heterogeneous catalysts [31-38]. We have usually employed γ-alumina as the catalyst, as this is a simple, readily available and environmentally benign catalyst that is often overlooked and it is used merely as a support for other catalysts [39-43]. The use of γ-alumina for the methylation of aniline with dimethyl carbonate has been reported [44]. In this paper, we chose to study the intramolecular and intermolecular alkylation of amino alcohols using γ-Al2O3 with scCO2 as the solvent and employed self-optimisation [45,46] to explore the defined parameter space to effectively identify the highest yielding and optimal conditions in a relatively short timeframe.
Results and Discussion
To investigate our hypothesis that γ-Al2O3 with scCO2 could be successfully applied to the amination of alcohols, we chose to employ a self-optimising reactor (Figure 1, see Supporting Information File 1 for details) to streamline the optimisation process using 5-amino-1-pentanol (1) as the model substrate and methanol as the alkylating agent (Scheme 1). For this reaction, self-optimisation is important as multiple products were identified that could form in parallel; from 1 the possible products we expected to see were a mixture of piperidine (2a), N-methylpiperidine (2b), N- and O-methylated 1, as well as oligomers. We chose to target 2b only for self-optimisation.
![[1860-5397-13-36-i1]](/bjoc/content/inline/1860-5397-13-36-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Target reaction – intramolecular cyclisation of 1 followed by N-methylation with methanol to yield 2b.
Scheme 1: Target reaction – intramolecular cyclisation of 1 followed by N-methylation with methanol to yield ...
We targeted N-methylpiperidine (2b) using the self-optimisation approach with SNOBFIT as the optimising algorithm [47] and GC analysis as the analytical tool providing the responses for the self-optimisation. This methodology allows high yielding conditions to be found, minimising the formation of byproducts. The temperature and the flow rate of the reaction were optimised in both the presence and absence of scCO2 (Figure 1).
![[1860-5397-13-36-1]](/bjoc/content/figures/1860-5397-13-36-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Simplified schematic demonstrating a self-optimising reactor [34,35,37,44]. The reagents are pumped into the system where they are mixed and then flowed through a reactor filled with catalyst. The output of the reactor is analysed by an on-line GC. The response (e.g., yield) of this analysis is then sent to an optimising search algorithm (e.g., SNOBFIT), which then changes the conditions (e.g., flow rates and temperature) in order to maximise the response of the analysis.
Figure 1: Simplified schematic demonstrating a self-optimising reactor [34,35,37,44]. The reagents are pumped into the sys...
The results of the optimisations are shown in Figure 2, and the conditions with the highest yields of 2b are shown in Table 1. During these experiments the parameter space was extensively studied and high yields were achieved at several different conditions. This provides confidence that our optimal yield was the global optimum within the studied limits of the reaction. It can be seen from Figure 2 that, when the reaction was carried out in scCO2, high yields (up to 96%) for 2b were achieved (Figure 2a, Table 1, entries 1–3). In the absence of scCO2 the percentage yield was good but the highest yields were ca. 8–11% less (Figure 2b, Table 1, entries 4–6) compared to when scCO2 was present. Clearly scCO2 is beneficial as a solvent in the formation of 2b.
![[1860-5397-13-36-2]](/bjoc/content/figures/1860-5397-13-36-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Result of the SNOBFIT optimisation for N-methylpiperidine (2b) with and without CO2 showing yields ≥70%. Figure a (left) shows the yields for the experiment carried out in scCO2 at different temperatures and flow rates; Figure b (right) shows the results without CO2. Conditions: Temperature 250–350 °C, substrate flow (0.5 M solution in MeOH) 0.1–0.5 mL min−1, 100 bar, when applicable 0.5 mL min−1 CO2.
Figure 2: Result of the SNOBFIT optimisation for N-methylpiperidine (2b) with and without CO2 showing yields ...
Table 1: The highest yields of 2b found by the optimisations carried out with CO2 (entries 1–3) and without CO2 (entries 4–6).a
| Entry | T (°C) | Flow rate (mL min−1) | Yield 2b (%)b |
|---|---|---|---|
| 1c | 340 | 0.3 | 94 |
| 2c | 310 | 0.1 | 94 |
| 3c | 330 | 0.15 | 96 |
| 4d | 350 | 0.4 | 86 |
| 5d | 350 | 0.3 | 85 |
| 6d | 350 | 0.5 | 83 |
a0.5 M solution of 1 in MeOH, 100 bar system pressure. bYields based on GC analysis. cWith 0.5 mL min−1 CO2. dNo CO2 used.
The optimal region for synthesising 2b turned out to be quite broad, as high yields were obtained at a variety of conditions. At lower flow rates (0.1 mL min–1) and hence longer residence times, yields of 94% were observed at 310 °C (Table 1, entry 2). Increasing the temperature by 30 °C led to an increase in the rate of cyclisation and methylation which then allowed for faster flow rates to be used under this operating temperature whilst still maintaining the same yield of 2b (Table 1, entry 1). Hence, three times the amount of material could be processed in the same time using this elevated temperature, i.e., higher productivity.
After optimisation with the model substrate 1 in methanol, the application of these reaction conditions to a small range of different alcohols was studied. Initially we repeated the model reaction to demonstrate that the approach is repeatable and that the conditions found during the optimisation were indeed the optimum (N.B. We chose the conditions that afforded the highest yield). Pleasingly, full conversion of 1 was obtained and an identical yield of 2b was observed (Table 2, entry 1). After showing that the conditions were repeatable, we applied them to several different alcohols by flowing a starting mixture of 1 with the alcohol as the solvent (Table 2, entries 2–4). As might be expected, the cyclisation to N-alkylated piperidines was observed for the primary alcohols. The yield of the corresponding N-alkylated piperidine falls as the longer chain alcohols are reacted. When the secondary alcohol isopropanol was used as the solvent, no N-alkylation was observed and piperidine 2a was found as the major product. As this catalyst system has been used previously for the etherification of alcohols [31-38], it is possible that ethers of the alcohols could be formed. In the case of 2d, dibutyl ether was the major byproduct, but in most other cases only small amounts of the corresponding ethers were observed. When the reaction with isopropanol was repeated without scCO2 the same selectivity was observed. However, when primary alcohols were run in the absence of scCO2 the yields of the corresponding N-alkylated products were lower and more piperidine 2a was observed. These results suggest that the rate of intermolecular alkylation is faster in scCO2, while the rate of intramolecular cyclisation is not significantly affected by the presence of scCO2 and thus proceeds faster than the intermolecular reaction.
Table 2: Cyclisation and N-alkylation of 1 with different alcohols.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-36-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | R = | Yield (%)b,c |
|---|---|---|
| 1 | Me 2b | 94% |
| 2 | Et 2c | 82% |
| 3 | n-Bu 2d | 73% |
| 4 | iPr 2e | 0% (2a 80%) |
aConditions: 1 (0.5 M in ROH), 340 °C, substrate flow: 0.3 mL min−1, CO2 flow: 0.5 mL min−1, 100 bar.; bDetermined by GC analysis of the reaction mixture. cThe remaining materials are unidentified side products.
We also explored the cyclisation and N-alkylation of different amino alcohol substrates. Initially we investigated the effect of simply changing the alkane chain length. Starting with 4-amino-1-butanol (3) under the model conditions afforded the desired N-methylpyrrolidine (4) in 95% yield. Extending the alkyl chain using 6-amino-1-hexanol (5), however, favoured methylation over intramolecular cyclisation as only 20% of the cyclised product 6 was observed. The major product was 6-(dimethylamino)-1-methoxyhexane (7, Scheme 2), which was formed by both O- and N-methylation of the starting material. Self-optimisation of the reaction of this substrate was performed in order to try and locate the optimal conditions for the highest yield of 6. Within the parameters explored, it was found that higher reaction temperatures increased the selectivity and yield of 6 up to 55%. This relatively modest yield could not be optimised further.
![[1860-5397-13-36-i2]](/bjoc/content/inline/1860-5397-13-36-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cyclisation and N-alkylation of 1,4- and 1,6-amino alcohols.
Scheme 2: Cyclisation and N-alkylation of 1,4- and 1,6-amino alcohols.
Ethanolamine 8 was used to explore the potential competition between the intra- and intermolecular etherification and amination. In this case we observed no azridine or N-methylaziridine, which would be expected from the intramolecular closure of 8, consistent with the results observed with bromoalkylamines [48], and suggesting the rate of closure for three-membered rings is slower than that of five- and six-membered rings. We cannot rule out the formation of aziridine as an intermediate in the formation of the dimeric products that were observed. The reaction with ethanolamine yielded three products (Table 3), N-methylmorpholine (9), 1,4-dimethylpiperazine (10) and the fully N- and O-methylated ethanolamine 11. Under the standard conditions, 11 was the major product, and as the temperature was increased, the amount of 10 increased. When the parameter space was explored using the self-optimisation approach the selectivity to 10 was increased to 63%. The etherification/deamination pathway forming 9 could not be optimised above 11% as the dehydration or methylated products were present as the major products in all cases. These results prompted us to explore the use of more functionalised amino alcohols in an attempt to access these heterocycles more cleanly and to allow us to further examine the deamination reactivity that produces 9.
Table 3: Reactions of ethanolamine.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-36-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Flow rate (mL min−1) | Temperature (°C) | Conversion (%) | Selectivity (%)b | ||
|---|---|---|---|---|---|---|
| 9 | 10 | 11 | ||||
| 1a | 0.3 | 340 | 100 | <1 | 13 | 72 |
| 2c | 0.1 | 370 | 100 | 11 | 48 | 0 |
| 3c,d | 0.1 | 360 | 100 | 5 | 63 | 3 |
aConditions: 8 0.5 M (or 1.0 M) solution in MeOH, 0.5 mL min−1 CO2, 100 bar; bBased on GC analysis of the reaction mixture, remaining material is a mixture of unidentified side products; cSubstrate 1.0 M solution in MeOH; dAfter self-optimisation had been run targeting high yield of 10.
Diethanolamine 12 is expected to produce a cleaner cyclisation pathway to N-methylmorpholine (9) via intramolecular etherification. When diethanolamine 12 in methanol was reacted using the standard conditions (Table 1, entry 1), N-methylmorpholine (9) was obtained but only in 24% yield; however, when the conditions were changed in an attempt to optimise the yield, it became apparent that the reactivity of 12 was more complicated. Running the reaction at 380 °C and 0.3 mL min−1 resulted in 46% of 9 being obtained but, at lower temperatures, different products were obtained. For example, when the reaction was run at 250 °C (Table 4, entry 1), oxazolidinone 13 was observed as the major product (52%) together with 14, a dimer of the starting material 12 as the main byproduct (42%).
Table 4: Showing the effect of conditions on the reaction of diethanolamine 12 to form carbamate 13 and piperazine 14.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-36-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Conc. (M) | T (°C) | P (bar) | Flow rate (mL min−1) | Conv. (%)b | Selectivity (%)b | |
|---|---|---|---|---|---|---|---|
| 13 | 14 | ||||||
| 1 | 0.5 | 250 | 100 | 0.3 | 53 | 52 | 42 |
| 2 | 0.5 | 250 | 100 | 0.2 | 98 | 20 | 65 |
| 3 | 0.5 | 250 | 100 | 0.1 | 100 | 0 | 61c |
| 4 | 0.5 | 240 | 100 | 0.3 | 48 | 69 | 26 |
| 5 | 0.2 | 250 | 100 | 0.3 | 80 | 42 | 38 |
| 6 | 0.2 | 250 | 150 | 0.3 | 73 | 65 | 19 |
| 7 | 1.0 | 250 | 150 | 0.2 | 56 | 73 | 22 |
| 8 | 1.0 | 275 | 100 | 0.2 | 100 | 8 | 63d |
a12 in ethanol, 0.5 mL min−1 CO2. bBased on GC analysis of the reaction mixture. c12% of mono-O-ethylated 14. dTrace of mono- and bis-ethylated 14.
Formation of 13 involves incorporation of the CO2 solvent into the product. Despite the very large number of reactions studied in scCO2, there are relatively few examples of incorporation of CO2 into the product. In this case, incorporation presumably occurs via the formation of a carbamate intermediate. This surprising formation of 13 suggests the incorporation of CO2 into 12 with the dimer formation as a competing reaction. In fact, when further conditions were studied, it became apparent that the dimer 14 could be formed from oxazolidinone 13 as increasing the residence time led to an increase in selectivity of 14 over 13 (Table 4, entry 2). Indeed, when 13 was used as the starting material, the major product that was isolated was 14; and this reactivity of 13 has been reported previously in batch reactions [49]. Increasing the residence time further (Table 4, entry 3) resulted in the oxazolidinone 13 not being detected and 14 was the major product together with a small quantity of mono O-ethylated 14. Reducing the temperature gave a better selectivity to the oxazolidinone 13 (Table 4, entry 4) and lowering the concentration, increased the conversion but gave a poor selectivity (Table 4, entry 5). Increasing the pressure to 150 bar had a positive effect on the selectivity toward 13 (Table 4, entry 6) and increasing the concentration of 12 to 1 M gave the highest selectivity for 13 (Table 4, entry 7). Further increasing the temperature to 275 °C only served to increase the selectivity towards 14 (Table 4, entry 8). From these conditions, it appears that the incorporation of CO2 is fast but the rate of conversion to 14 is dependent on the pressure of the system, the temperature of the reactor, the residence time and to some extent the concentration of the amino alcohol in the alcohol. A higher pressure of CO2 appears to slow the rate of conversion of 13 to 14, whilst elevated temperatures appear to accelerate the rate. Increasing the residence time allows more time for 13 to be converted into 14 and hence the higher selectivity for it and the appearance of trace amounts of mono- and bis-ethylated 14.
We have studied the incorporation of CO2 further by investigating the reaction of N-(2-aminoethyl)ethanolamine 15. The use of 15 as a starting material might be expected to produce high selectivity for the corresponding imidazolidinone 16 via the incorporation of CO2. The competing oxazolidinone formation should be limited as the nucleophilicity of nitrogen is more than that of the oxygen. Furthermore, the formation of dimers were expected to be supressed as 16 does not contain a “CO2 unit” that can serve as a leaving group. This was indeed the case as, at 250 °C, 85% selectivity, 70% yield for 16 was observed when the reaction was run in scCO2 (Scheme 3a). In the absence of CO2 as a solvent the formation of imidazolidinone 16 was not observed. When the starting solution was pre-saturated with CO2 and run in the absence of CO2 as a solvent, 16 was formed in 62% selectivity, 15% yield from 24% conversion of the starting material. This poor conversion suggests that CO2 is needed in an excess for the reaction to be successful, and the use of CO2 as the solvent as well as a reagent in this case provides the highest possible concentration of CO2. To establish whether any dimers are formed when 16 is exposed to the catalyst bed for an extended time or to higher reaction temperatures, a solution of 16 in iPrOH (0.5 M) was flowed at 250 and 275 °C, but no dimers were detected and unreacted 16 was the main product observed. The reaction of 15 with CO2 could be supressed using higher temperatures, for example at 380 °C in methanol the intramolecular cyclisation is favoured and N,N’-dimethylpiperazine (10) is obtained as the major product in 68% yield (Scheme 3b, 380 °C at 1 mL min−1), and no imidazolidinone 16 was detected.
![[1860-5397-13-36-i3]](/bjoc/content/inline/1860-5397-13-36-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: a) Reactions highlighting the incorporation of CO2 in to 16. b) High temperature reaction of 15 yielding N,N’-dimethylpiperazine (10).
Scheme 3: a) Reactions highlighting the incorporation of CO2 in to 16. b) High temperature reaction of 15 yie...
Conclusion
Using a self-optimising reactor and a simple heterogeneous catalyst, γ-Al2O3, moderate to high yields of several alkylated cyclic amines, formed in a two-step intramolecular cyclisation/N-alkyation reaction, using amino alcohols and simple alcohols has been achieved (Scheme 4).
![[1860-5397-13-36-i4]](/bjoc/content/inline/1860-5397-13-36-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Summary of products obtained from the reactions of amino alcohols over γ-Al2O3 in scCO2.
Scheme 4: Summary of products obtained from the reactions of amino alcohols over γ-Al2O3 in scCO2.
Using scCO2 as the solvent proved to be beneficial to the yield of cyclic N-alkylated amines, in particular for the N-alkylation step which was arrested in the absence of scCO2. The intramolecular cyclisation of the amino alcohols was favoured at higher temperatures in both the presence and absence of scCO2. Increasing the primary alcohol length led to slightly lower yields of the target products whereas secondary alcohols did not react with the amines at all. Varying the chain length of the amino alcohol produced the corresponding N-alkylated five- (4) and seven-membered ring (6), three-membered aziridine rings were not detected. Competing N- and O-alkylation was observed at higher temperatures with ethanolamine (8) and 6-amino-1-hexanol (5), suggesting ring closure is slower in these cases. Ethanolamine (8) produced dimers as the major products, mainly via the amination pathway; however, some esterification/deamination was observed as N-methylmorpholine (9) was also detected. CO2 incorporation in 12 and 15 was perhaps the most surprising result as this occurred at lower temperatures compared to the cyclisation, however at higher temperatures intramolecular reactions were favoured. The formation of oxazolidinones was shown to be reversible releasing CO2 as dimers are formed. Imidazolidinones were shown to be stable to further reaction and no release of CO2 was observed under the conditions studied. Further optimisation and investigations into the incorporation of CO2 are in progress.
Experimental
CAUTION! The described reactions involve high pressures and require equipment (Figure 3) with appropriate pressure ratings.
All reagents and solvents were purchased from commercial sources and used as received. CO2 was supplied by BOC Gases (99.8%). The γ-alumina (PURALOX NWa155) was supplied by SASOL. It was sieved before use, to obtain the desired particle size (125–170 μm), which was used as the catalyst. Reaction mixtures were analysed using GC, GC–MS, 1H and 13C NMR. Compounds 1a–c, 4, 9, 10, 13, 14, 16 were obtained from Aldrich and used as standards. 1d,e [50], 6 [51], 7 [52], and 11 [53] were identified as previously described in the literature.
GC analysis was carried out using the following instrument and conditions: Online Shimadzu GC-2014 with a high pressure sample loop and an OPTIMA delta-3 column (30 m, 0.25 mm ID, 0.25 µm FT): hold 50 °C 4 min, ramp to 100 °C at 25 °C/min, ramp to 250 °C at 10 °C/min, hold for 2 min, pressure 132.1 kPa, purge 3.0 mL/min split ratio 40.
The high pressure continuous set-up (Figure 3) employed in the described reactions consisted of a HPLC pump through which a solution of the desired amino alcohol in an alcoholic solvent was delivered. A stainless steel reactor (1/4’’ tube, 1.83 mL volume) was packed with γ-alumina (approx. 2 g) and attached below a pre-heater column (1/4’’ tube, 1.83 mL volume) that was packed with sand to increase mixing. A crosspiece was used to mix the CO2 and reagent flows before the reactors and the resulting product mixture was collected downstream of the back pressure regulator. The sampling to the on-line GC was done with a high pressure sample loop (Vici, 0.5 μL), which allowed a sample to be taken from the reaction flow. During optimisations a sample was taken once the conditions had been changed and stable state had been reached (10 min).
![[1860-5397-13-36-3]](/bjoc/content/figures/1860-5397-13-36-3.png?scale=1.7&max-width=1024&background=FFFFFF)
Figure 3: Diagram of the high pressure equipment used in the experiments.
Figure 3: Diagram of the high pressure equipment used in the experiments.
Some experiments were carried out by using a self-optimising reactor which has been described in detail previously [34,35,37]. All SNOBFIT [47] optimisations were performed within the following limits: Temperature 250–380 °C and flow rate 0.1–1.0 mL min−1. The number of points produced by each call to SNOBFIT (nreq) was 6, and 10% of all the points were requested as global points (p = 0.1). The results at each condition were determined by GC analysis (programme time 20–23 min) and the pressure of the system was controlled by a back-pressure regulator at the outlet and was adjusted manually.
Supporting Information
| Supporting Information File 1: Experimental data. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgements
We thank the Erasmus Mundus Joint Doctorate SINCHEM (FPA 2013-0037) for funding E. Streng’s grant. We thank W. Leitner and J. Klankermayer for hosting E. Streng’s visit to the ITMC, RWTH Aachen, which prompted this work. We thank M. Guyler, P. Fields, R. Wilson, K. Hind, D. Litchfield and J. Warren for their technical support at Nottingham.
References
-
Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/B703488C
Return to citation in text: [1] -
Lawrence, S. A. Amines: Synthesis, Properties and Applications; Cambridge University Press: Cambridge, 2004.
Return to citation in text: [1] -
Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337–2347. doi:10.1039/b602413k
Return to citation in text: [1] -
Salvatore, R. N.; Yoon, C. H.; Jung, K. W. Tetrahedron 2001, 57, 7785–7811. doi:10.1016/S0040-4020(01)00722-0
Return to citation in text: [1] -
Marichev, K. O.; Takacs, J. M. ACS Catal. 2016, 6, 2205–2210. doi:10.1021/acscatal.6b00175
Return to citation in text: [1] -
Bhawal, B. N.; Morandi, B. ACS Catal. 2016, 6, 7528–7535. doi:10.1021/acscatal.6b02333
Return to citation in text: [1] -
Shimizu, K. Catal. Sci. Technol. 2015, 5, 1412–1427. doi:10.1039/C4CY01170H
Return to citation in text: [1] -
Li, Q.-Q.; Xiao, Z.-F.; Yao, C.-Z.; Zheng, H.-X.; Kang, Y.-B. Org. Lett. 2015, 17, 5328–5331. doi:10.1021/acs.orglett.5b02685
Return to citation in text: [1] -
Leonard, J.; Blacker, A. J.; Marsden, S. P.; Jones, M. F.; Mulholland, K. R.; Newton, R. Org. Process Res. Dev. 2015, 19, 1400–1410. doi:10.1021/acs.oprd.5b00199
Return to citation in text: [1] -
Yan, T.; Feringa, B. L.; Barta, K. Nat. Commun. 2014, 5, No. 5602. doi:10.1038/ncomms6602
Return to citation in text: [1] -
Obora, Y. ACS Catal. 2014, 4, 3972–3981. doi:10.1021/cs501269d
Return to citation in text: [1] -
Watson, A. J. A.; Williams, J. M. J. Science 2010, 329, 635–636. doi:10.1126/science.1191843
Return to citation in text: [1] -
Lamb, G. W.; Al Badran, F. A.; Williams, J. M. J.; Kolaczkowski, S. T. Chem. Eng. Res. Des. 2010, 88, 1533–1540. doi:10.1016/j.cherd.2010.04.005
Return to citation in text: [1] -
Hamid, M. H. S. A.; Allen, C. L.; Lamb, G. W.; Maxwell, A. C.; Maytum, H. C.; Watson, A. J. A.; Williams, J. M. J. J. Am. Chem. Soc. 2009, 131, 1766–1774. doi:10.1021/ja807323a
Return to citation in text: [1] -
Del Zotto, A.; Baratta, W.; Sandri, M.; Verardo, G.; Rigo, P. Eur. J. Inorg. Chem. 2004, 524–529. doi:10.1002/ejic.200300518
Return to citation in text: [1] -
Fujita, K.; Li, Z.; Ozeki, N.; Yamaguchi, R. Tetrahedron Lett. 2003, 44, 2687–2690. doi:10.1016/S0040-4039(03)00371-X
Return to citation in text: [1] -
Watanabe, Y.; Tsuji, Y.; Ohsugi, Y. Tetrahedron Lett. 1981, 22, 2667–2670. doi:10.1016/S0040-4039(01)92965-X
Return to citation in text: [1] -
Grigg, R.; Mitchell, T. R. B.; Sutthivaiyakit, S.; Tongpenyai, N. J. Chem. Soc., Chem. Commun. 1981, 611–612. doi:10.1039/c39810000611
Return to citation in text: [1] -
Bui-The-Khai; Concilio, C.; Porzi, G. J. Org. Chem. 1981, 46, 1759–1760. doi:10.1021/jo00321a056
Return to citation in text: [1] -
Su, J.; Li, X.; Chen, Y.; Cui, Y.; Xu, J.; Qian, C.; Chen, X. RSC Adv. 2016, 6, 55643–55649. doi:10.1039/C6RA07998A
Return to citation in text: [1] -
Li, Y.-Q.; Chen, Y.-B.; Huang, Z.-Z. Chin. Chem. Lett. 2014, 25, 1540–1544. doi:10.1016/j.cclet.2014.07.006
Return to citation in text: [1] -
Yang, J.-M.; Jiang, R.; Wu, L.; Xu, X.-P.; Wang, S.-Y.; Ji, S.-J. Tetrahedron 2013, 69, 7988–7994. doi:10.1016/j.tet.2013.07.010
Return to citation in text: [1] -
Pathare, S. P.; Akamanchi, K. G. Appl. Catal., A 2013, 452, 29–33. doi:10.1016/j.apcata.2012.11.017
Return to citation in text: [1] -
Zhao, Y.; Foo, S. W.; Saito, S. Angew. Chem., Int. Ed. 2011, 50, 3006–3009. doi:10.1002/anie.201006660
Return to citation in text: [1] -
Motokura, K.; Nakagiri, N.; Mizugaki, T.; Ebitani, K.; Kaneda, K. J. Org. Chem. 2007, 72, 6006–6015. doi:10.1021/jo070416w
Return to citation in text: [1] -
Ko, A.-N.; Yang, C.-L.; Zhu, W.-d.; Lin, H.-e. Appl. Catal., A 1996, 134, 53–66. doi:10.1016/0926-860X(95)00209-X
Return to citation in text: [1] -
Brown, A. B.; Reid, E. E. J. Am. Chem. Soc. 1924, 46, 1836–1839. doi:10.1021/ja01673a011
Return to citation in text: [1] -
Frankland, P. F.; Challenger, F.; Nicholls, N. A. J. Chem. Soc., Trans. 1919, 115, 198–205. doi:10.1039/CT9191500198
Return to citation in text: [1] -
Oku, T.; Ikariya, T. Angew. Chem., Int. Ed. 2002, 41, 3476–3479. doi:10.1002/1521-3773(20020916)41:18<3476::AID-ANIE3476>3.0.CO;2-5
Return to citation in text: [1] -
Oku, T.; Arita, Y.; Tsuneki, H.; Ikariya, T. J. Am. Chem. Soc. 2004, 126, 7368–7377. doi:10.1021/ja048557s
Return to citation in text: [1] -
Gray, W. K.; Smail, F. R.; Hitzler, M. G.; Ross, S. K.; Poliakoff, M. J. Am. Chem. Soc. 1999, 121, 10711–10718. doi:10.1021/ja991562p
Return to citation in text: [1] [2] -
Walsh, B.; Hyde, J. R.; Licence, P.; Poliakoff, M. Green Chem. 2005, 7, 456–463. doi:10.1039/b413890b
Return to citation in text: [1] [2] -
Gooden, P. N.; Bourne, R. A.; Parrott, A. J.; Bevinakatti, H. S.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2010, 14, 411–416. doi:10.1021/op900307w
Return to citation in text: [1] [2] -
Parrott, A. J.; Bourne, R. A.; Akien, G. R.; Irvine, D. J.; Poliakoff, M. Angew. Chem., Int. Ed. 2011, 50, 3788–3792. doi:10.1002/anie.201100412
Return to citation in text: [1] [2] [3] [4] -
Bourne, R. A.; Skilton, R. A.; Parrott, A. J.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2011, 15, 932–938. doi:10.1021/op200109t
Return to citation in text: [1] [2] [3] [4] -
Jumbam, D. N.; Skilton, R. A.; Parrott, A. J.; Bourne, R. A.; Poliakoff, M. J. Flow Chem. 2012, 2, 24–27. doi:10.1556/jfchem.2012.00019
Return to citation in text: [1] [2] -
Skilton, R. A.; Parrott, A. J.; George, M. W.; Poliakoff, M.; Bourne, R. A. Appl. Spectrosc. 2013, 67, 1127–1131. doi:10.1366/13-06999
Return to citation in text: [1] [2] [3] [4] -
Skilton, R. A.; Bourne, R. A.; Amara, Z.; Horvath, R.; Jin, J.; Scully, M. J.; Streng, E.; Tang, S. L. Y.; Summers, P. A.; Wang, J.; Pérez, E.; Asfaw, N.; Aydos, G. L. P.; Dupont, J.; Comak, G.; George, M. W.; Poliakoff, M. Nat. Chem. 2015, 7, 1–5. doi:10.1038/nchem.2143
Return to citation in text: [1] [2] -
Hammerschmidt, W.; Baiker, A.; Wokaun, A.; Fluhr, W. Appl. Catal. 1986, 20, 305–312. doi:10.1016/0166-9834(86)80022-7
Return to citation in text: [1] -
Bai, G. Y.; Li, Y.; Yan, X.; He, F.; Chen, L. React. Kinet. Catal. Lett. 2004, 82, 33–39. doi:10.1023/B:REAC.0000028802.66602.0f
Return to citation in text: [1] -
Nagaiah, K.; Rao, A. S.; Kulkarni, S. J.; Subrahmanyam, M.; Rao, A. V. R. J. Catal. 1994, 147, 349–351. doi:10.1006/jcat.1994.1147
Return to citation in text: [1] -
Wu, Z.; Wang, H.; Sun, M.; Du, X.; Chen, L.; Li, Y. Res. Chem. Intermed. 2012, 38, 1149–1157. doi:10.1007/s11164-011-0450-4
Return to citation in text: [1] -
Wu, Z.; Yang, F.; Wang, H.; Ma, J.; Chen, L.; Li, Y. React. Kinet., Mech. Catal. 2012, 106, 485–493. doi:10.1007/s11144-012-0447-z
Return to citation in text: [1] -
Amara, Z.; Streng, E. S.; Skilton, R. A.; Jin, J.; George, M. W.; Poliakoff, M. Eur. J. Org. Chem. 2015, 6141–6145. doi:10.1002/ejoc.201500980
Return to citation in text: [1] [2] -
Reizman, B. J.; Jensen, K. F. Acc. Chem. Res. 2016, 49, 1786–1796. doi:10.1021/acs.accounts.6b00261
Return to citation in text: [1] -
Fabry, D. C.; Sugiono, E.; Rueping, M. Isr. J. Chem. 2014, 54, 341–350. doi:10.1002/ijch.201300080
Return to citation in text: [1] -
Huyer, W.; Neumaier, A. ACM Trans. Math. Software 2008, 35, No. 9. doi:10.1145/1377612.1377613
Return to citation in text: [1] [2] -
Freundlich, H.; Salomon, G. Ber. Dtsch. Chem. Ges. A 1933, 66, 355–357. doi:10.1002/cber.19330660308
Return to citation in text: [1] -
Arrowood, T.; MacDonald, J. Preparation of Dihydroxyethyl Piperazine. U.S. Pat. Appl. US20150274682 A1, Oct 1, 2015.
Return to citation in text: [1] -
Katritzky, A. R.; Fan, W.-Q. J. Org. Chem. 1990, 55, 3205–3209. doi:10.1021/jo00297a041
Return to citation in text: [1] -
Reeves, J. T.; Tan, Z.; Marsini, M. A.; Han, Z. S.; Xu, Y.; Reeves, D. C.; Lee, H.; Lu, B. Z.; Senanayake, C. H. Adv. Synth. Catal. 2013, 355, 47–52. doi:10.1002/adsc.201200835
Return to citation in text: [1] -
Barbry, D.; Hasiak, B. Collect. Czech. Chem. Commun. 1983, 48, 1734–1744. doi:10.1135/cccc19831734
Return to citation in text: [1] -
Remenar, J. F.; Lucht, B. L.; Collum, D. B. J. Am. Chem. Soc. 1997, 119, 5567–5572. doi:10.1021/ja970029b
Return to citation in text: [1]
| 34. | Parrott, A. J.; Bourne, R. A.; Akien, G. R.; Irvine, D. J.; Poliakoff, M. Angew. Chem., Int. Ed. 2011, 50, 3788–3792. doi:10.1002/anie.201100412 |
| 35. | Bourne, R. A.; Skilton, R. A.; Parrott, A. J.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2011, 15, 932–938. doi:10.1021/op200109t |
| 37. | Skilton, R. A.; Parrott, A. J.; George, M. W.; Poliakoff, M.; Bourne, R. A. Appl. Spectrosc. 2013, 67, 1127–1131. doi:10.1366/13-06999 |
| 52. | Barbry, D.; Hasiak, B. Collect. Czech. Chem. Commun. 1983, 48, 1734–1744. doi:10.1135/cccc19831734 |
| 53. | Remenar, J. F.; Lucht, B. L.; Collum, D. B. J. Am. Chem. Soc. 1997, 119, 5567–5572. doi:10.1021/ja970029b |
| 1. | Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/B703488C |
| 2. | Lawrence, S. A. Amines: Synthesis, Properties and Applications; Cambridge University Press: Cambridge, 2004. |
| 3. | Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337–2347. doi:10.1039/b602413k |
| 29. | Oku, T.; Ikariya, T. Angew. Chem., Int. Ed. 2002, 41, 3476–3479. doi:10.1002/1521-3773(20020916)41:18<3476::AID-ANIE3476>3.0.CO;2-5 |
| 30. | Oku, T.; Arita, Y.; Tsuneki, H.; Ikariya, T. J. Am. Chem. Soc. 2004, 126, 7368–7377. doi:10.1021/ja048557s |
| 50. | Katritzky, A. R.; Fan, W.-Q. J. Org. Chem. 1990, 55, 3205–3209. doi:10.1021/jo00297a041 |
| 20. | Su, J.; Li, X.; Chen, Y.; Cui, Y.; Xu, J.; Qian, C.; Chen, X. RSC Adv. 2016, 6, 55643–55649. doi:10.1039/C6RA07998A |
| 21. | Li, Y.-Q.; Chen, Y.-B.; Huang, Z.-Z. Chin. Chem. Lett. 2014, 25, 1540–1544. doi:10.1016/j.cclet.2014.07.006 |
| 22. | Yang, J.-M.; Jiang, R.; Wu, L.; Xu, X.-P.; Wang, S.-Y.; Ji, S.-J. Tetrahedron 2013, 69, 7988–7994. doi:10.1016/j.tet.2013.07.010 |
| 23. | Pathare, S. P.; Akamanchi, K. G. Appl. Catal., A 2013, 452, 29–33. doi:10.1016/j.apcata.2012.11.017 |
| 24. | Zhao, Y.; Foo, S. W.; Saito, S. Angew. Chem., Int. Ed. 2011, 50, 3006–3009. doi:10.1002/anie.201006660 |
| 25. | Motokura, K.; Nakagiri, N.; Mizugaki, T.; Ebitani, K.; Kaneda, K. J. Org. Chem. 2007, 72, 6006–6015. doi:10.1021/jo070416w |
| 26. | Ko, A.-N.; Yang, C.-L.; Zhu, W.-d.; Lin, H.-e. Appl. Catal., A 1996, 134, 53–66. doi:10.1016/0926-860X(95)00209-X |
| 27. | Brown, A. B.; Reid, E. E. J. Am. Chem. Soc. 1924, 46, 1836–1839. doi:10.1021/ja01673a011 |
| 28. | Frankland, P. F.; Challenger, F.; Nicholls, N. A. J. Chem. Soc., Trans. 1919, 115, 198–205. doi:10.1039/CT9191500198 |
| 51. | Reeves, J. T.; Tan, Z.; Marsini, M. A.; Han, Z. S.; Xu, Y.; Reeves, D. C.; Lee, H.; Lu, B. Z.; Senanayake, C. H. Adv. Synth. Catal. 2013, 355, 47–52. doi:10.1002/adsc.201200835 |
| 5. | Marichev, K. O.; Takacs, J. M. ACS Catal. 2016, 6, 2205–2210. doi:10.1021/acscatal.6b00175 |
| 6. | Bhawal, B. N.; Morandi, B. ACS Catal. 2016, 6, 7528–7535. doi:10.1021/acscatal.6b02333 |
| 7. | Shimizu, K. Catal. Sci. Technol. 2015, 5, 1412–1427. doi:10.1039/C4CY01170H |
| 8. | Li, Q.-Q.; Xiao, Z.-F.; Yao, C.-Z.; Zheng, H.-X.; Kang, Y.-B. Org. Lett. 2015, 17, 5328–5331. doi:10.1021/acs.orglett.5b02685 |
| 9. | Leonard, J.; Blacker, A. J.; Marsden, S. P.; Jones, M. F.; Mulholland, K. R.; Newton, R. Org. Process Res. Dev. 2015, 19, 1400–1410. doi:10.1021/acs.oprd.5b00199 |
| 10. | Yan, T.; Feringa, B. L.; Barta, K. Nat. Commun. 2014, 5, No. 5602. doi:10.1038/ncomms6602 |
| 11. | Obora, Y. ACS Catal. 2014, 4, 3972–3981. doi:10.1021/cs501269d |
| 12. | Watson, A. J. A.; Williams, J. M. J. Science 2010, 329, 635–636. doi:10.1126/science.1191843 |
| 13. | Lamb, G. W.; Al Badran, F. A.; Williams, J. M. J.; Kolaczkowski, S. T. Chem. Eng. Res. Des. 2010, 88, 1533–1540. doi:10.1016/j.cherd.2010.04.005 |
| 14. | Hamid, M. H. S. A.; Allen, C. L.; Lamb, G. W.; Maxwell, A. C.; Maytum, H. C.; Watson, A. J. A.; Williams, J. M. J. J. Am. Chem. Soc. 2009, 131, 1766–1774. doi:10.1021/ja807323a |
| 15. | Del Zotto, A.; Baratta, W.; Sandri, M.; Verardo, G.; Rigo, P. Eur. J. Inorg. Chem. 2004, 524–529. doi:10.1002/ejic.200300518 |
| 16. | Fujita, K.; Li, Z.; Ozeki, N.; Yamaguchi, R. Tetrahedron Lett. 2003, 44, 2687–2690. doi:10.1016/S0040-4039(03)00371-X |
| 17. | Watanabe, Y.; Tsuji, Y.; Ohsugi, Y. Tetrahedron Lett. 1981, 22, 2667–2670. doi:10.1016/S0040-4039(01)92965-X |
| 18. | Grigg, R.; Mitchell, T. R. B.; Sutthivaiyakit, S.; Tongpenyai, N. J. Chem. Soc., Chem. Commun. 1981, 611–612. doi:10.1039/c39810000611 |
| 19. | Bui-The-Khai; Concilio, C.; Porzi, G. J. Org. Chem. 1981, 46, 1759–1760. doi:10.1021/jo00321a056 |
| 48. | Freundlich, H.; Salomon, G. Ber. Dtsch. Chem. Ges. A 1933, 66, 355–357. doi:10.1002/cber.19330660308 |
| 4. | Salvatore, R. N.; Yoon, C. H.; Jung, K. W. Tetrahedron 2001, 57, 7785–7811. doi:10.1016/S0040-4020(01)00722-0 |
| 49. | Arrowood, T.; MacDonald, J. Preparation of Dihydroxyethyl Piperazine. U.S. Pat. Appl. US20150274682 A1, Oct 1, 2015. |
| 45. | Reizman, B. J.; Jensen, K. F. Acc. Chem. Res. 2016, 49, 1786–1796. doi:10.1021/acs.accounts.6b00261 |
| 46. | Fabry, D. C.; Sugiono, E.; Rueping, M. Isr. J. Chem. 2014, 54, 341–350. doi:10.1002/ijch.201300080 |
| 34. | Parrott, A. J.; Bourne, R. A.; Akien, G. R.; Irvine, D. J.; Poliakoff, M. Angew. Chem., Int. Ed. 2011, 50, 3788–3792. doi:10.1002/anie.201100412 |
| 35. | Bourne, R. A.; Skilton, R. A.; Parrott, A. J.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2011, 15, 932–938. doi:10.1021/op200109t |
| 37. | Skilton, R. A.; Parrott, A. J.; George, M. W.; Poliakoff, M.; Bourne, R. A. Appl. Spectrosc. 2013, 67, 1127–1131. doi:10.1366/13-06999 |
| 44. | Amara, Z.; Streng, E. S.; Skilton, R. A.; Jin, J.; George, M. W.; Poliakoff, M. Eur. J. Org. Chem. 2015, 6141–6145. doi:10.1002/ejoc.201500980 |
| 44. | Amara, Z.; Streng, E. S.; Skilton, R. A.; Jin, J.; George, M. W.; Poliakoff, M. Eur. J. Org. Chem. 2015, 6141–6145. doi:10.1002/ejoc.201500980 |
| 31. | Gray, W. K.; Smail, F. R.; Hitzler, M. G.; Ross, S. K.; Poliakoff, M. J. Am. Chem. Soc. 1999, 121, 10711–10718. doi:10.1021/ja991562p |
| 32. | Walsh, B.; Hyde, J. R.; Licence, P.; Poliakoff, M. Green Chem. 2005, 7, 456–463. doi:10.1039/b413890b |
| 33. | Gooden, P. N.; Bourne, R. A.; Parrott, A. J.; Bevinakatti, H. S.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2010, 14, 411–416. doi:10.1021/op900307w |
| 34. | Parrott, A. J.; Bourne, R. A.; Akien, G. R.; Irvine, D. J.; Poliakoff, M. Angew. Chem., Int. Ed. 2011, 50, 3788–3792. doi:10.1002/anie.201100412 |
| 35. | Bourne, R. A.; Skilton, R. A.; Parrott, A. J.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2011, 15, 932–938. doi:10.1021/op200109t |
| 36. | Jumbam, D. N.; Skilton, R. A.; Parrott, A. J.; Bourne, R. A.; Poliakoff, M. J. Flow Chem. 2012, 2, 24–27. doi:10.1556/jfchem.2012.00019 |
| 37. | Skilton, R. A.; Parrott, A. J.; George, M. W.; Poliakoff, M.; Bourne, R. A. Appl. Spectrosc. 2013, 67, 1127–1131. doi:10.1366/13-06999 |
| 38. | Skilton, R. A.; Bourne, R. A.; Amara, Z.; Horvath, R.; Jin, J.; Scully, M. J.; Streng, E.; Tang, S. L. Y.; Summers, P. A.; Wang, J.; Pérez, E.; Asfaw, N.; Aydos, G. L. P.; Dupont, J.; Comak, G.; George, M. W.; Poliakoff, M. Nat. Chem. 2015, 7, 1–5. doi:10.1038/nchem.2143 |
| 39. | Hammerschmidt, W.; Baiker, A.; Wokaun, A.; Fluhr, W. Appl. Catal. 1986, 20, 305–312. doi:10.1016/0166-9834(86)80022-7 |
| 40. | Bai, G. Y.; Li, Y.; Yan, X.; He, F.; Chen, L. React. Kinet. Catal. Lett. 2004, 82, 33–39. doi:10.1023/B:REAC.0000028802.66602.0f |
| 41. | Nagaiah, K.; Rao, A. S.; Kulkarni, S. J.; Subrahmanyam, M.; Rao, A. V. R. J. Catal. 1994, 147, 349–351. doi:10.1006/jcat.1994.1147 |
| 42. | Wu, Z.; Wang, H.; Sun, M.; Du, X.; Chen, L.; Li, Y. Res. Chem. Intermed. 2012, 38, 1149–1157. doi:10.1007/s11164-011-0450-4 |
| 43. | Wu, Z.; Yang, F.; Wang, H.; Ma, J.; Chen, L.; Li, Y. React. Kinet., Mech. Catal. 2012, 106, 485–493. doi:10.1007/s11144-012-0447-z |
| 47. | Huyer, W.; Neumaier, A. ACM Trans. Math. Software 2008, 35, No. 9. doi:10.1145/1377612.1377613 |
| 31. | Gray, W. K.; Smail, F. R.; Hitzler, M. G.; Ross, S. K.; Poliakoff, M. J. Am. Chem. Soc. 1999, 121, 10711–10718. doi:10.1021/ja991562p |
| 32. | Walsh, B.; Hyde, J. R.; Licence, P.; Poliakoff, M. Green Chem. 2005, 7, 456–463. doi:10.1039/b413890b |
| 33. | Gooden, P. N.; Bourne, R. A.; Parrott, A. J.; Bevinakatti, H. S.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2010, 14, 411–416. doi:10.1021/op900307w |
| 34. | Parrott, A. J.; Bourne, R. A.; Akien, G. R.; Irvine, D. J.; Poliakoff, M. Angew. Chem., Int. Ed. 2011, 50, 3788–3792. doi:10.1002/anie.201100412 |
| 35. | Bourne, R. A.; Skilton, R. A.; Parrott, A. J.; Irvine, D. J.; Poliakoff, M. Org. Process Res. Dev. 2011, 15, 932–938. doi:10.1021/op200109t |
| 36. | Jumbam, D. N.; Skilton, R. A.; Parrott, A. J.; Bourne, R. A.; Poliakoff, M. J. Flow Chem. 2012, 2, 24–27. doi:10.1556/jfchem.2012.00019 |
| 37. | Skilton, R. A.; Parrott, A. J.; George, M. W.; Poliakoff, M.; Bourne, R. A. Appl. Spectrosc. 2013, 67, 1127–1131. doi:10.1366/13-06999 |
| 38. | Skilton, R. A.; Bourne, R. A.; Amara, Z.; Horvath, R.; Jin, J.; Scully, M. J.; Streng, E.; Tang, S. L. Y.; Summers, P. A.; Wang, J.; Pérez, E.; Asfaw, N.; Aydos, G. L. P.; Dupont, J.; Comak, G.; George, M. W.; Poliakoff, M. Nat. Chem. 2015, 7, 1–5. doi:10.1038/nchem.2143 |
| 47. | Huyer, W.; Neumaier, A. ACM Trans. Math. Software 2008, 35, No. 9. doi:10.1145/1377612.1377613 |
© 2017 Streng et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)