Abstract
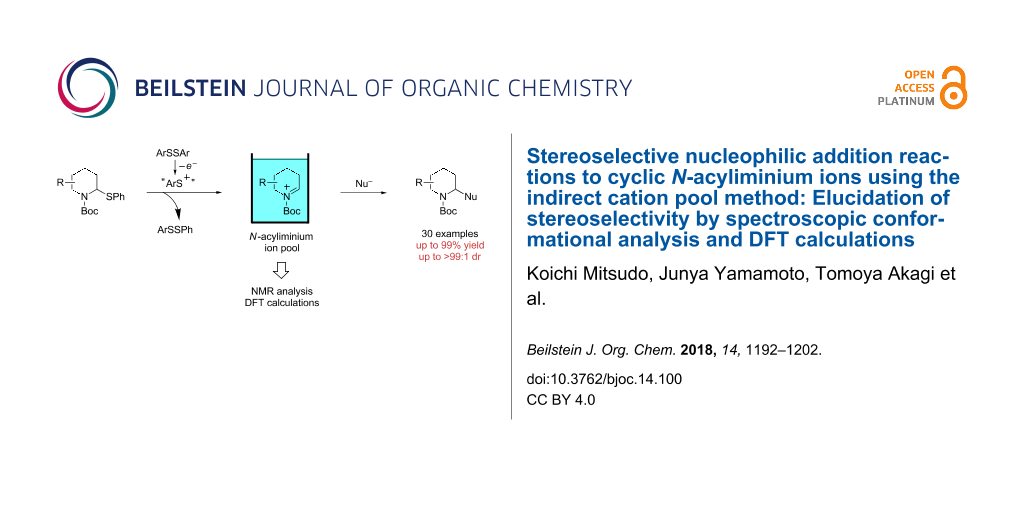
In this study, six-membered N-acyliminium ions were generated by the “indirect cation pool” method and reacted with several nucleophiles. These reactions afforded disubstituted piperidine derivatives with high diastereoselectivities and good to excellent yields. The conformations of the obtained N-acyliminium ions were studied by low temperature NMR analyses and DFT calculations and were found to be consistent with the Steven’s hypothesis.

Graphical Abstract
Introduction
Cyclic amines are significant key motifs in pharmaceutical and natural products because a variety of compounds bearing those moieties exhibit physiological and pharmacological activities [1-3]. As many of these compounds feature asymmetric carbon atoms at the α-position of the cyclic amine, the stereoselective carbon–carbon bond formation at this position is of great importance from the perspective of drug discovery [4,5]. Meanwhile the “cation pool” method can realize the generation and accumulation of highly reactive cationic species such as N-acyliminium ions in relatively high concentrations by low temperature electrolysis [6-10]. The N-acyliminium ions thus generated can directly react with a variety of carbon nucleophiles to very efficiently yield carbon–carbon bond-formation products. In addition, this research lead to the development of an indirect cation pool method that enables the creation of the cation pool by reacting a cation precursor having a C–S bond with an electrochemically generated ArS(ArSSAr)+ as the cation-generating reagent. For example, a pool of alkoxycarbenium ions can be rapidly generated by the reaction of thioacetals and ArS(ArSSAr)+ (Ar = p-FC6H4, Scheme 1) [11].
The cation pool method is advantageous over the conventional Lewis acid promoted generation of carbocation species because the reactive cationic species can be detected by spectroscopy such as NMR and IR measurements. For this research, the reaction of six-membered N-acyliminium ions having a substituent in the 4-, 5-, or 6-position were generated by the indirect cation pool method and examined in the reaction with several nucleophiles to give the disubstituted piperidine derivatives in an excellent diastereoselective manner (Scheme 1). This result inspired us to investigate the stereochemistry of the N-acyliminium ions by means of NMR spectroscopy, which is challenging to achieve without using cation pool methods. The current report presents the diastereoselective synthesis of disubstituted piperidine derivatives by the indirect cation pool method and the elucidation of the stereoselectivity based on both NMR analyses of the cyclic N-acyliminium ions and DFT calculations.
![[1860-5397-14-100-i1]](/bjoc/content/inline/1860-5397-14-100-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Generation and reaction of cationic species generated by “indirect cation pool” methods.
Scheme 1: Generation and reaction of cationic species generated by “indirect cation pool” methods.
Results and Discussion
N-Acyliminium ions obtained from N-Boc-piperidines are attractive intermediates because they easily can be transformed to a variety of alkaloids having a piperidine skeleton. In our preliminary study, we found the indirect cation pool method suitable for the generation and accumulation of N-acyliminium ions. Therefore, the reaction of N-Boc-4-phenyl-2,3,4,5-tetrahydropyridin-1-ium (C1) derived from precursor 1a and ArS(ArSSAr)+ (Ar = p-FC6H4) generated by the low temperature electrolysis was first performed with several nucleophiles (Table 1). The starting 1a and other N-acyliminium ion precursors were synthesized by the modified Beak’s protocol [12] (see Supporting Information File 1). As observed in our previous studies, the Boc protecting group was found not suitable for the direct cation pool method due to its cleavage by protic acid generated during the electrolysis of the cation precursor. However, it can be utilized in the indirect cation pool method as diaryldisulfides are the exclusive side products formed during carbocation generation. Thus, the reaction of acyliminium ion C1 with Me3Al afforded trans-1-(tert-butoxycarbonyl)-2-methyl-4-phenylpiperidine (2aa) in a good yield and virtually complete diastereoselectivity (cis/trans = <1:99). Similarly, the reaction of C1 with diethylzinc or allyltributylstannane gave piperidine derivatives 2ab and 2ac with high yields and trans-selectivity (Table 1, entries 2 and 3). Also, the introduction of phenyl or cyano groups in the piperidine core proceeded in a highly regioselective manner and good to high yields (Table 1, entries 4 and 5).
Table 1: Reaction of N-acyliminium ion C1 with several nucleophiles.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-100-i2.svg?max-width=637&scale=1.0)
|
||||
| entry | Nu− | 2 | yield (%)a | cis/transb |
|---|---|---|---|---|
| 1 |
Me3Al
(5.0 equiv) |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-100-i3.svg?max-width=637&scale=1.0)
2aa |
85 | <1:99 |
| 2 |
Et2Zn
(2.5 equiv) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-100-i4.svg?max-width=637&scale=1.0)
2ab |
89 | 3:97 |
| 3 |
(allyl)SnBu3
(3.0 equiv) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-100-i5.svg?max-width=637&scale=1.0)
2ac |
>99 | <1:99 |
| 4 |
PhMgBr
(2.0 equiv) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-100-i6.svg?max-width=637&scale=1.0)
2ad |
87 | 15:85 |
| 5 |
TMSCN
(5.0 equiv) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-100-i7.svg?max-width=637&scale=1.0)
2ae |
99 | <1:99 |
aIsolated yield. bDetermined by GC analysis.
Next, we examined the reaction of N-Boc-4-methyl-2,3,4,5-tetrahydropyridin-1-ium (C2) with nucleophiles (Table 2). Both, the reactivity and diastereoselectivity of the reactions were similar to those of C1 and the disubstituted piperidine derivatives 2ba–be were obtained in good to high yields with excellent diastereoselectivities.
Table 2: Reaction of N-acyliminium ion C2 with several nucleophiles.
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-100-i8.svg?max-width=637&scale=1.0)
|
||||
| entry | Nu− | 2 | yield (%)a | cis:transb |
|---|---|---|---|---|
| 1 |
Me3Al
(2.5 equiv) |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-100-i9.svg?max-width=637&scale=1.0)
2ba |
86 | <1:99 |
| 2 |
Et2Zn
(5.0 equiv) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-100-i10.svg?max-width=637&scale=1.0)
2bb |
95 | <1:99 |
| 3 |
(allyl)SnBu3
(2.5 equiv) |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-100-i11.svg?max-width=637&scale=1.0)
2bc |
95 | <1:99 |
| 4 |
PhMgBr
(2.5 equiv) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-100-i12.svg?max-width=637&scale=1.0)
2bd |
93 | 3:97 |
| 5 |
TMSCN
(5.0 equiv) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-100-i13.svg?max-width=637&scale=1.0)
2be |
89 | <1:99 |
aIsolated yield. bDetermined by GC analysis.
Then, the reaction of 5-phenyl-substituted N-acyliminium cation C3 was performed (Table 3). Interestingly, all reactions led to the 2,5-disubstituted piperidine derivatives 2ca–ce in very good yields and high cis-selective manner. A similar trend is observed for the reactions of the 5-methyl-substituted cation C4 and all products 2da–dc were obtained in high to excellent cis-diastereoselectivities (Table 4).
Table 3: Reaction of N-acyliminium ion C3 with several nucleophiles.
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-100-i14.svg?max-width=637&scale=1.0)
|
||||
| entry | Nu− | 2 | yield (%)a | cis:transb |
|---|---|---|---|---|
| 1c |
Me3Al
(2.5 equiv) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-100-i15.svg?max-width=637&scale=1.0)
2ca |
89 | 94:6 |
| 2 |
Et2Zn
(2.5 equiv) |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-100-i16.svg?max-width=637&scale=1.0)
2cb |
99 | 96:4 |
| 3c |
(allyl)SnBu3
(2.5 equiv) |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-100-i17.svg?max-width=637&scale=1.0)
2cc |
97 | 98:2 |
| 4 |
PhMgBr
(2.5 equiv) |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-100-i18.svg?max-width=637&scale=1.0)
2cd |
97 | 96:4c |
| 5 |
TMSCN
(5.0 equiv) |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-100-i19.svg?max-width=637&scale=1.0)
2ce |
96 | 98:2 |
aIsolated yield. bDetermined by GC analysis. cDetermined by 1H NMR analysis.
Table 4: Reaction of N-acyliminium ion C4 with several nucleophiles.
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-100-i20.svg?max-width=637&scale=1.0)
|
||||
| entry | Nu− | 2 | yield (%)a | cis:transb |
|---|---|---|---|---|
| 1 | Me3Al |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-100-i21.svg?max-width=637&scale=1.0)
2da |
73 | 98:2 |
| 2 | Et2Zn |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-100-i22.svg?max-width=637&scale=1.0)
2db |
82 | 95:5 |
| 3 | (allyl)SnBu3 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-100-i23.svg?max-width=637&scale=1.0)
2dc |
90 | 98:2 |
| 4 | PhMgBr |
![[Graphic 23]](/bjoc/content/inline/1860-5397-14-100-i24.svg?max-width=637&scale=1.0)
2dd |
83 | 91:9 |
| 5 | TMSCN |
![[Graphic 24]](/bjoc/content/inline/1860-5397-14-100-i25.svg?max-width=637&scale=1.0)
2de |
98 | 96:4 |
aIsolated yield. bDetermined by GC analysis.
The tendency of the reactions of the 6-phenyl-substituted N-acyliminium cation C5 with nucleophiles was slightly different compared to those of C1–C4 (Table 5). The reactions proceeded in a cis-selective manner, however, the selectivity was not high except in case of product 2ed (Table 5, entry 4). The low yield of 2ed was due to the generation of an enamine as a byproduct. While the reason of the low selectivity has not been clear, it is probably due to steric repulsion between the phenyl and Boc groups. A distortion of the Boc group in C5 was also suggested by DFT calculation (see Supporting Information File 1). The reaction of 6-methyl-substituted N-acyliminium cation C6 bearing a methyl group, which is smaller than a phenyl group, gave the corresponding cis-products with high diastereoselectivities (Table 6).
Table 5: Reaction of N-acyliminium ion C5 with several nucleophiles.
![[Graphic 25]](/bjoc/content/inline/1860-5397-14-100-i26.svg?max-width=637&scale=1.0)
|
||||
| entry | Nu− | 2 | yield (%)a | cis:transb |
|---|---|---|---|---|
| 1 | Me3Al |
![[Graphic 26]](/bjoc/content/inline/1860-5397-14-100-i27.svg?max-width=637&scale=1.0)
2ea |
40 | 63:37 |
| 2 | Et2Zn |
![[Graphic 27]](/bjoc/content/inline/1860-5397-14-100-i28.svg?max-width=637&scale=1.0)
2eb |
90 | 76:24 |
| 3 | (allyl)SnBu3 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-14-100-i29.svg?max-width=637&scale=1.0)
2ec |
94 | 72:28 |
| 4 | PhMgBr |
![[Graphic 29]](/bjoc/content/inline/1860-5397-14-100-i30.svg?max-width=637&scale=1.0)
2ed |
56 | 92:8 |
| 5 | TMSCN |
![[Graphic 30]](/bjoc/content/inline/1860-5397-14-100-i31.svg?max-width=637&scale=1.0)
2ee |
85 | 64:36 |
aIsolated yield. bDetermined by GC analysis.
Table 6: Reaction of N-acyliminium ion C6 with several nucleophiles.
![[Graphic 31]](/bjoc/content/inline/1860-5397-14-100-i32.svg?max-width=637&scale=1.0)
|
||||
| entry | Nu− | 2 | yield (%)a | cis:transb |
|---|---|---|---|---|
| 1 | Me3Al |
![[Graphic 32]](/bjoc/content/inline/1860-5397-14-100-i33.svg?max-width=637&scale=1.0)
2fa |
49 | 82:18 |
| 2 | Et2Zn |
![[Graphic 33]](/bjoc/content/inline/1860-5397-14-100-i34.svg?max-width=637&scale=1.0)
2fb |
46 | 96:4 |
| 3 | (allyl)SnBu3 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-14-100-i35.svg?max-width=637&scale=1.0)
2fc |
87 | 96:4 |
| 4 | PhMgBr |
![[Graphic 35]](/bjoc/content/inline/1860-5397-14-100-i36.svg?max-width=637&scale=1.0)
2fd |
87 | 96:4 |
| 5 | TMSCN |
![[Graphic 36]](/bjoc/content/inline/1860-5397-14-100-i37.svg?max-width=637&scale=1.0)
2fe |
90 | >99:1 |
aIsolated yield. bDetermined by GC analysis.
As mentioned, the reaction between the cyclic N-acyliminium ions C1–C6 and several nucleophiles proceeded diastereoselectively. To clarify the reason of the observed selectivity, 1H NMR analyses of C1–C6 were performed at low temperature (Figures 1–3, see also Supporting Information File 1). All protons on the piperidine ring in C1 (Ha–Hh) have been assigned by H–H COSY (Figure 1). In addition, the signal of Hd (3.10 ppm) exhibited an axial–axial coupling (J > 10 Hz) and 7.1% ROE was observed between Hd and Hg. These results suggest that Hd adopts an axial and the phenyl group a pseudo-equatorial position at low temperature.
![[1860-5397-14-100-1]](/bjoc/content/figures/1860-5397-14-100-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) 1H NMR of N-acyliminium ion C1 in CD2Cl2 at −80 °C (600 MHz). (b) Preferred conformation of C1.
Figure 1: (a) 1H NMR of N-acyliminium ion C1 in CD2Cl2 at −80 °C (600 MHz). (b) Preferred conformation of C1.
The low temperature NMR analysis of C3, which has a phenyl group at 5-position, was next performed and the 1H NMR spectrum is depicted in Figure 2. An axial–axial coupling between He and Hf was observed, suggesting that these two protons were located at axial positions. This result indicates that the phenyl group of C3 was located in the pseudo-equatorial position.
![[1860-5397-14-100-2]](/bjoc/content/figures/1860-5397-14-100-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) 1H NMR spectrum of C3 in CD2Cl2 at −60 °C (400 MHz). (b) Preferred conformation of C3.
Figure 2: (a) 1H NMR spectrum of C3 in CD2Cl2 at −60 °C (400 MHz). (b) Preferred conformation of C3.
The conformation of C5, an N-acyliminium ion bearing a phenyl group in the 6-position, was also examined by NMR analysis (Figure 3). The signal of Hf was observed as a triplet due to the axial–axial coupling with He and the geminal coupling with Hg, and these results indicate that the phenyl group of C5 is placed in the pseudo-axial position. The low temperature NMR measurements of N-acyliminium ions C2, C4, and C6 were also performed. The conformations of C2 and C6 were similar to those of C1 and C5, respectively (C2: pseudo-equatorial, C6: pseudo-axial). Although the conformation of C4 could not be determined by the NMR analysis, the conformation is assumed to be similar to that of C3 (pseudo-equatorial), because both ions led to similar stereoselectivities in the reactions with nucleophiles. As mentioned, the conformation of N-acyliminium cation C1–C4 was pseudo-equatorial and that of C5 and C6 was pseudo-equatorial (Figure 4).
![[1860-5397-14-100-3]](/bjoc/content/figures/1860-5397-14-100-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) 1H NMR spectrum of C5 in CD2Cl2 at −60 °C (400 MHz). (b) Preferred conformation of C5.
Figure 3: (a) 1H NMR spectrum of C5 in CD2Cl2 at −60 °C (400 MHz). (b) Preferred conformation of C5.
![[1860-5397-14-100-4]](/bjoc/content/figures/1860-5397-14-100-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Summary of the conformations of N-acyliminium ions C1–C6.
Figure 4: Summary of the conformations of N-acyliminium ions C1–C6.
It is commonly assumed that the diastereoselectivity of the disubstituted piperidine derivatives 2a–f was influenced by the conformation of the N-acyliminium ions C1–C6. In this context Stevens proposed that the conformation of the six-membered N-acyliminium ion would be a half-chair form and that the attack of the nucleophile proceeds from that side that generates a stable chair form product, because an attack from another side would lead to a more unstable product with a twist form [13]. As a close study, Woerpel reported a diastereoselective substitution reaction for synthesizing disubstituted tetrahydropyrans via six-membered oxocarbenium ions generated in situ from tetrahydropyran acetals [14]. If the Stevens’ hypothesis is true, a nucleophilic reaction with C1, having a pseudo-equatorial phenyl group, should give a trans-2,4-disubstituted product (Figure 5). Similarly, acyliminium ions C3 and C5 should give the cis-2,5- or cis-2,6-disubstituted piperidine derivatives, respectively. These explanations are consistent with above mentioned conformational analyses results, and the hypothesis was in agreement with the experimental results (Figure 6).
![[1860-5397-14-100-5]](/bjoc/content/figures/1860-5397-14-100-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Stevens’ hypothesis on the tendency of the addition of nucleophiles to N-acyliminium ions. The substituent at the nitrogen atom is omitted for clarity.
Figure 5: Stevens’ hypothesis on the tendency of the addition of nucleophiles to N-acyliminium ions. The subs...
![[1860-5397-14-100-6]](/bjoc/content/figures/1860-5397-14-100-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: A plausible mechanism of the observed diastereoselective reaction of the N-acyliminium ions.
Figure 6: A plausible mechanism of the observed diastereoselective reaction of the N-acyliminium ions.
To obtain further insight in the conformation of the cyclic N-acyliminium ions, DFT calculations of C1–C6 were performed (Figure 7). First, ΔG of pseudo-equatorial and pseudo-axial conformations of C1 was calculated at the B3LYP/6-31G(d) level of theory. The pseudo-equatorial conformation of C1 was more stable than the pseudo-axial conformation by 1.27 kcal/mol. Similarly, DFT calculations of C3 and C5 were performed, and the results show that the pseudo-equatorial conformation of C3 and the pseudo-axial conformation of C5 were more stable than the other conformations, respectively. The more stable conformations that the calculations implied are consistent with the conformations determined by the low temperature NMR analyses, and the DFT calculations suggest that the preferred conformations of C2, C4, and C6 were similar to those of C1, C3, and C5.
![[1860-5397-14-100-7]](/bjoc/content/figures/1860-5397-14-100-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Comparison of ΔG for the pseudo-equatorial and pseudo-axial conformations of C1–C6 at the B3LYP/6-31G(d) level.
Figure 7: Comparison of ΔG for the pseudo-equatorial and pseudo-axial conformations of C1–C6 at the B3LYP/6-3...
Conclusion
In this study, we presented an efficient method for the highly diastereoselective synthesis of disubstituted piperidine derivatives through the reaction of N-acyliminium ions with nucleophiles. The starting N-acyliminium ions were generated by the indirect cation pool method and their conformations were confirmed by low temperature NMR analyses for the first time. The experimental results were fully consistent with DFT calculations. The correlation between the stereochemistry of the N-acyliminium ions and the reaction products is in agreement with common interpretation and further synthetic applications and more detailed investigations of the reaction mechanism are in progress in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental details, ORTEP drawings of 1a, 1b, 1d–1f, theoretical calculations of C1–C6, and NMR spectra of all new compounds and C1–C6. | ||
| Format: PDF | Size: 11.2 MB | Download |
| Supporting Information File 2:
X-ray structure analysis data for 1a (CCDC-1813600), 1b (CCDC-1813582), 1d (CCDC-1813594), 1e (CCDC-1813595), 1f (CCDC-1813596). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
X-ray analysis of 1a–f. |
||
| Format: CIF | Size: 4.5 MB | Download |
References
-
Alvarez-Buill, J.; Vaquero, J. J.; Barluenga, J., Eds. Modern Heterocyclic Chemistry; Wiley-VCH: Weinheim, 2011.
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. doi:10.1002/anie.200701342
Return to citation in text: [1] -
Carroll, F. I.; Dolle, R. E. ChemMedChem 2014, 9, 1638. doi:10.1002/cmdc.201402142
Return to citation in text: [1] -
McLaughlin, N. P.; Evans, P.; Pines, M. Bioorg. Med. Chem. 2014, 22, 1993. doi:10.1016/j.bmc.2014.02.040
Return to citation in text: [1] -
Källström, S.; Leino, R. Bioorg. Med. Chem. 2008, 16, 601. doi:10.1016/j.bmc.2007.10.018
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Waldvogel, S. R.; Janza, B. Angew. Chem., Int. Ed. 2014, 53, 7122. doi:10.1002/anie.201405082
Return to citation in text: [1] -
Matsumoto, K.; Suga, S.; Yoshida, J.-i. Org. Biomol. Chem. 2011, 9, 2586. doi:10.1039/c0ob01070g
Return to citation in text: [1] -
Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265. doi:10.1021/cr0680843
Return to citation in text: [1] -
Yoshida, J.-i.; Suga, S. Chem. – Eur. J. 2002, 8, 2650. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S
Return to citation in text: [1] -
Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J.-i. J. Am. Chem. Soc. 2006, 128, 7710. doi:10.1021/ja0625778
Return to citation in text: [1] -
Beak, P.; Lee, W.-K. Tetrahedron Lett. 1989, 30, 1197. doi:10.1016/S0040-4039(00)72714-6
Return to citation in text: [1] -
Stevens, R. V. Acc. Chem. Res. 1984, 17, 289. doi:10.1021/ar00104a005
Return to citation in text: [1] -
Ayala, L.; Lucero, C. G.; Romero, J. A. C.; Tabacco, S. A.; Woerpel, K. A. J. Am. Chem. Soc. 2003, 125, 15521. doi:10.1021/ja037935a
Return to citation in text: [1]
| 1. | Alvarez-Buill, J.; Vaquero, J. J.; Barluenga, J., Eds. Modern Heterocyclic Chemistry; Wiley-VCH: Weinheim, 2011. |
| 2. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. doi:10.1002/anie.200701342 |
| 3. | Carroll, F. I.; Dolle, R. E. ChemMedChem 2014, 9, 1638. doi:10.1002/cmdc.201402142 |
| 12. | Beak, P.; Lee, W.-K. Tetrahedron Lett. 1989, 30, 1197. doi:10.1016/S0040-4039(00)72714-6 |
| 11. | Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J.-i. J. Am. Chem. Soc. 2006, 128, 7710. doi:10.1021/ja0625778 |
| 6. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230. doi:10.1021/acs.chemrev.7b00397 |
| 7. | Waldvogel, S. R.; Janza, B. Angew. Chem., Int. Ed. 2014, 53, 7122. doi:10.1002/anie.201405082 |
| 8. | Matsumoto, K.; Suga, S.; Yoshida, J.-i. Org. Biomol. Chem. 2011, 9, 2586. doi:10.1039/c0ob01070g |
| 9. | Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265. doi:10.1021/cr0680843 |
| 10. | Yoshida, J.-i.; Suga, S. Chem. – Eur. J. 2002, 8, 2650. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S |
| 4. | McLaughlin, N. P.; Evans, P.; Pines, M. Bioorg. Med. Chem. 2014, 22, 1993. doi:10.1016/j.bmc.2014.02.040 |
| 5. | Källström, S.; Leino, R. Bioorg. Med. Chem. 2008, 16, 601. doi:10.1016/j.bmc.2007.10.018 |
| 14. | Ayala, L.; Lucero, C. G.; Romero, J. A. C.; Tabacco, S. A.; Woerpel, K. A. J. Am. Chem. Soc. 2003, 125, 15521. doi:10.1021/ja037935a |
© 2018 Mitsudo et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)