Abstract
Large amplitude conformational change is one of the features of biomolecular recognition and is also the basis for allosteric effects and signal transduction in functional biological systems. However, synthetic receptors with controllable conformational changes are rare. In this article, we present a thorough study on the host–guest chemistry of a conformationally adaptive macrocycle, namely per-O-ethoxyzorb[4]arene (ZB4). Similar to per-O-ethoxyoxatub[4]arene, ZB4 is capable of accommodating a wide range of organic cations. However, ZB4 does not show large amplitude conformational responses to the electronic substituents on the guests. Instead of a linear free-energy relationship, ZB4 follows a parabolic free-energy relationship. This is explained by invoking the influence of secondary C–H···O hydrogen bonds on the primary cation···π interactions based on the information obtained from four representative crystal structures. In addition, heat capacity changes (ΔCp) and enthalpy–entropy compensation phenomena both indicate that solvent reorganization is also involved during the binding. This research further deepens our understanding on the binding behavior of ZB4 and lays the basis for the construction of stimuli-responsive materials with ZB4 as a major component.
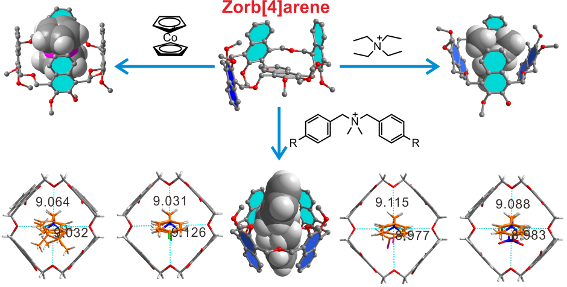
Graphical Abstract
Introduction
Macrocyclic receptors are the principal workhorses used in supramolecular chemistry [1]. A myriad of synthetic macrocycles have sprouted during the past decade, greatly enriching the arsenal of supramolecular chemists [2-11]. The majority of artificial macrocycles are featured with rigid backbones as it is widely accepted that preorganization [12] is crucial for minimizing the entropy cost in molecular recognition. In contrast, bioreceptors often possess flexible backbone structures and even undergo large amplitude conformational changes upon binding substrates [13,14]. This conformational adaptivity is the basis of the allosteric effects [15,16] and signal transduction [17] observed with bioreceptors. However, similar conformationally adaptive synthetic macrocyclic receptors are relatively rare in the literature [18-23].
During the last five years, we have developed two classes of macrocyclic receptors with biomimetic structures [24]: endo-functionalized molecular tubes [25-30] and conformationally adaptive macrocycles [31-37]. Among the conformationally adaptive macrocycles two types were reported: oxatub[n]arenes [31-36] and zorb[n]arenes [37]. These macrocycles possess multiple conformers due to the naphthalene flipping in analogy with the phenyl-ring flipping seen in the more common calixarenes. The conformers so formed undergo quick interconversion and each one has a slightly different cavity. Thus, these conformers consist of a complex conformational network. We have carefully looked into the properties of oxatub[n]arenes and found that the macrocycles have many unique properties. For example, oxatub[4]arene has a wide guest scope and can bind almost all of the common organic cations [32]. It also shows conformational responses to solvent change [33] and remote electronic substituents on the guests [34]. In addition, different alkyl side chains on oxatub[4]arenes lead to different macroscopic self-assembly behaviors [36]. Zorb[4]arene was first synthesized, reported and so named by the Georghiou group in 2005. The derivatives per-O-methoxy- and per-O-ethoxyzorb[4]arene were shown to be effective tetramethylammonium ion receptors [38]. The per-O-n-butoxyzorb[4]arene (ZB4, Scheme 1a) was only recently further studied by us with respect to its rich conformational properties and the consequence on macroscopic self-assembly [37]. In the present research, we report the binding behavior of ZB4 to a much wider guest scope. We found that the guest-binding ability and conformational adaptivity of ZB4 are quite different from that of per-O-n-butoxyoxatub[4]arene (TA4).
![[1860-5397-14-134-i1]](/bjoc/content/inline/1860-5397-14-134-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (a) Chemical structures of ZB4 and the guests involved in this research. The counterions are PF6−. (b) The four representative conformers of ZB4 resulting from naphthalene flipping. Numberings on the structures are used to assign NMR signals.
Scheme 1: (a) Chemical structures of ZB4 and the guests involved in this research. The counterions are PF6−. ...
Results and Discussion
Conformational adaptability enables oxatub[4]arenes to host a wide range of organic cations [32]. ZB4 is also a conformationally adaptive macrocycle. We wondered whether ZB4 has a wide guest binding scope. It was reported that quaternary ammonium-based organic cations (1+–3+) can be hosted by zorb[4]arenes [37,38]. Quaternary ammonium cations 4+ and 5+ and other types of organic cations hosted by TA4 (6+–10+) were tested with ZB4. Most of these guests can indeed be complexed. But there are some exceptions. Changing the core quaternary ammonium structure of 3+ completely shuts down the binding, because no obvious complexation-induced shifts were detected in the 1:1 mixture of ZB4 with 4+ or 5+ (Figures S1 and S2 in Supporting Information File 1). This indicates the importance of the core quaternary ammonium ions in the host−guest complexation. All other guests can be encapsulated in the cavity of ZB4, and significant chemical shifts on both the guests and ZB4 were observed in the NMR spectra (Figures S3–S7 in Supporting Information File 1). The ESI mass spectra of equimolar mixtures of guests 9+ and 10+ and ZB4 were obtained (Figures S9 and S10 in Supporting Information File 1) and the predominant peaks were assigned to 1:1 complexes after losing PF6−.
NMR titrations and isothermal titration microcalorimetry (ITC) were then performed to obtain the association constants. For small guests such as 1+, 2+, 6+–9+, NMR titration experiments with ZB4 have been performed due to the fast equilibrium of the free and ZB4-complexed guests on the NMR time scale. All titration curves agreed well with a 1:1 stoichiometry (Figures S11–S14 in Supporting Information File 1). In case of guests 3+ and 10+, the binding heats were high enough to be measured. Thus the binding parameters were determined by ITC titrations (Figure S15 in Supporting Information File 1) and the results are shown in Table 1. Generally, ZB4 shows weaker binding affinities to these guests than TA4 does with the same counterions. For example, ZB4 and cation 3+, with a binding constant of 5.4 × 104 M−1, was the best guest among the studied ones. However, the corresponding association constant with TA4 has been 1.7 × 105 M−1. Similar differences were also observed for cations 9+ and 10+, their binding affinities with ZB4 were lower by 1–2 orders of magnitude than those with TA4. However, the small guests, such as 2+ and 6+–8+ share rather similar binding affinities to both ZB4 and TA4.
Table 1: Association constants (M−1) and other thermodynamic parameters as determined by 1H NMR titrations (400 MHz, CD2Cl2/CD3CN 1:1, 298 K) or by ITC titrations in a 1:1 mixture of 1,2-dichloroethane and MeCN at 298 K.
| guestsa | Ka (M−1) | guestsa | Ka (M−1) | |
|---|---|---|---|---|
| 1+b | 4700 ± 600 | 7+ | 349 ± 29 | |
| 2+b | 590 ± 30 | 8+ | 468 ± 31 | |
| 6+ | 524 ± 48 | 9+ | 1300 ± 100 | |
| guestsc | Ka (M−1) | ΔG (kJ∙mol−1) | ΔH (kJ∙mol−1) | −TΔS (kJ∙mol−1) |
| 3+b | (5.4 ± 1.2) × 104 | −27.0 ± 0.8 | −31.6 | 4.6 |
| 10+ | (4.3 ± 1.0) × 104 | −26.5 ± 0.7 | −18.1 | −8.4 |
aThe association constants were determined by NMR titrations; bthe binding parameters of these guests have been reported (see ref. [37]); cthe association constants were determined by ITC titrations.
The X-ray crystal structure of free ZB4 shows it to exist as a self-inclusion conformation in the solid state (Figure 1a). This conformation is different from the ones containing different lower-rim alkyl groups reported earlier [37,38]. Crystals were obtained by slow evaporation of the compounds’ CH3CN solutions and the different conformations in the solid state may result from the packing of the different lower-rim alkyl groups. For the conformers with cavities (Scheme 1b), three out of the four have been predominantly selected by three different guests. For example, guests 2+ and 3+ induced conformers IV and III, respectively, to achieve optimal binding [37]. This has been unambiguously confirmed by X-ray single crystallography (Figure 1b and 1c). Guest 10+ is a strong binder and its induction on the conformations of ZB4 was further analyzed. The guest exchange in solution of 10+@ZB4 is fast/intermediate on the NMR timescale, as witnessed by broadening of all signals in the spectrum at 25 °C (Figure S7b in Supporting Information File 1). Thus, a 1H NMR experiment at −20 °C was performed to slow down the guest exchange. Indeed, the protons a and b are clearly separated, suggesting that the guest exchange is now slow on the NMR timescale (Figure S7c in Supporting Information File 1). Only two signals for the aromatic protons of the host are observed, suggesting that ZB4 predominantly exists as either conformer I or IV in the complex 10+@ZB4. However, it has been not clear which one ZB4 adopts. Fortunately, a single crystal suitable for X-ray diffraction could be obtained by slow evaporation of the solution of 10+ and ZB4 in a mixture of CH2Cl2 and CH3CN. The crystal structure clearly shows that conformer I (Figure 1d) is the selected conformation by guest 10+.
![[1860-5397-14-134-1]](/bjoc/content/figures/1860-5397-14-134-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray single crystal structure of ZB4 and the host–guest complexes. a) ZB4, b) 2+@ZB4-IV, c) 3+@ZB4-IV, d) 10+@ZB4-I. Hydrogen atoms of the host are removed and butyl groups are shortened to methyl groups for viewing clarity. The X-ray single crystal structures of 2+@ZB4-IV (b) and 3+@ZB4-III (c) have been reported previously (see [37]).
Figure 1: X-ray single crystal structure of ZB4 and the host–guest complexes. a) ZB4, b) 2+@ZB4-IV, c) 3+@ZB4...
TA4 shows a large amplitude of conformational change in response to the remote electronic substituents on the guests [34]. We wondered whether a similar behavior would be observed for ZB4. Consequently, a series of guests with different substituents in the para-positions of guest 3+ were employed to study the electronic substituent effect of the guests on the binding behavior of ZB4. As the guest exchange is slow on the NMR timescale the experiments were performed by 1H NMR spectroscopy. Surprisingly, all the 1H NMR spectra of the complexes (Figure S8 in Supporting Information File 1) shared similar peak patterns as 3+@ZB4, suggesting conformer III (C2h symmetry) [37] to be the most favored conformation for all complexes. Obviously the conformational network of ZB4 shows no response to the electronic substituents on the guests. This is quite different from TA4.
In addition, there has been a linear free energy relationship between electronic properties of substituents present in the guests and their binding affinities with TA4, indicating that the binding affinities are affected by substituents through a field/inductive effect [34]. However, this is again quite different for ZB4. The association constants of ZB4 to these guests were determined by ITC titrations (Figures S15–S26 in Supporting Information File 1), and the data are shown in Table 2. The logarithm of the corresponding association constants of 11+–21+ over 3+ were parabolic as the function of Hammett parameter (σp) [39] (Figure 2). For substituents C(Me)3, OMe, Me, SMe, F, Cl, Br, and I, the binding affinities increase with increasing σp. However, the binding affinities decrease with further increasing σp (CF3, NO2, and CN). The guest with iodo substituent (18+) is the best, with an association constant of 4.9 × 105 M−1 at 25 °C. It is interesting to note that although the substituents of guests 12+ and 19+ are quite different in view of their electronic properties, they share very similar binding affinities.
Table 2: Association constants (M−1) and other thermodynamic parameters of ZB4 with guests 11+–21+ as determined by ITC titrations in a 1:1 mixture of 1,2-dichloroethane and MeCN at 298 K.
| guests | R3 | Ka | ΔG (kJ∙mol−1) | ΔH (kJ∙mol−1) | −TΔS (kJ∙mol−1) |
|---|---|---|---|---|---|
| 11+ | CH3 | (6.4 ± 0.5) × 104 | −27.4 ± 0.5 | −37.0 | 9.6 |
| 12+ | OMe | (8.8 ± 1.1) × 104 | −28.2 ± 0.7 | −38.1 | 9.8 |
| 13+ | SMe | (2.3 ± 0.2) × 105 | −30.6 ± 0.7 | −50.1 | 19.5 |
| 14+ | t-Bu | (2.3 ± 0.6) × 104 | −24.9 ± 0.5 | −40.7 | 15.8 |
| 15+ | F | (1.2 ± 0.1) × 105 | −29.0 ± 0.7 | −35.6 | 6.6 |
| 16+ | Cl | (2.2 ± 0.3) × 105 | −30.6 ± 0.8 | −37.3 | 6.7 |
| 17+ | Br | (3.4 ± 0.2) × 105 | −31.5 ± 0.8 | −39.0 | 7.5 |
| 18+ | I | (4.9 ± 0.7) × 105 | −32.5 ± 0.9 | −40.0 | 7.5 |
| 19+ | CN | (8.6 ± 1.5) × 104 | −28.2 ± 0.7 | −32.4 | 4.2 |
| 20+ | CF3 | (1.9 ± 0.2) × 105 | −30.1 ± 0.9 | −37.4 | 7.3 |
| 21+ | NO2 | (1.2 ± 0.1) × 105 | −30.4 ± 0.5 | −32.6 | 3.2 |
![[1860-5397-14-134-2]](/bjoc/content/figures/1860-5397-14-134-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Parabolic free-energy relationship between log(KR/KH) and Hammett parameter σp. KR: guests 11+–21+; KH: guest 3+.
Figure 2: Parabolic free-energy relationship between log(KR/KH) and Hammett parameter σp. KR: guests 11+–21+; ...
What is the underlying reason for the response of binding affinities to the electronic substituent effect on the guests? Luckily, single crystals of complexes 14+@ZB4-III, 16+@ZB4-III, 18+@ZB4-III, and 21+@ZB4-III suitable for X-ray single crystallography, were obtained and their crystal structures are shown in Figure 3. The substituents of these four guests located at three representative positions in Figure 2. Therefore, a closer look at their crystal structures may provide an explanation for their surprising binding behaviors. Multiple non-covalent interactions, including C–H···O hydrogen bonds, cation···π, C–H···π and π···π interactions, are involved in all the cases. Undoubtedly, cation···π interactions between the core quaternary ammonium ions of the guests and the four naphthalene rings of the host should still be the major driving force as mentioned above. However, it was noticed that the distances between diagonal linker oxygen atoms in the backbone of the host are slightly different for the four complexes. These interactions may be tuned by the size of the host cavity. As shown in Figure 3 (bottom), the vertical and horizontal distances between the diagonal oxygen atoms are different for all the four complexes. This distance is presumably tuned through the C–H···O hydrogen bonds between the CH2–O–CH2 oxygen atoms and the aromatic protons of the guests. The acidities of aromatic protons on the guests are, however, influenced by the substituents. Indeed, the electron-withdrawing nitro group and the electron-donating tert-butyl group both result in shorter C–H···O hydrogen bonds than the chloro and iodo groups do. The cavity sizes of ZB4 in complexes 16+@ZB4-III and 18+@ZB4-III may be better suited than those of 14+@ZB4-III and 21+@ZB4-III to host the quaternary ammonium and maximize all the non-covalent interactions. Any deviation from these cavity sizes weakens the binding. That is, the secondary C–H···O hydrogen bonds can be tuned through the substituents to leverage the primary cation···π interactions and thus the final binding affinities. This may explain the parabolic distribution of binding affinities over the Hammett parameters (σp) of the substituents as shown in Figure 2. The conformational adaptivity or flexibility allows ZB4 to adapt according to the need of the guests. Simultaneously, the guest may also conformationally adapt to better interact with ZB4. As shown in Figure 4, the crystal structure of 18+ in 18+@ZB4-III is slightly different in shape from free cation 18+.
![[1860-5397-14-134-3]](/bjoc/content/figures/1860-5397-14-134-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray single crystal structures of 14+@ZB4-III, 16+@ZB4-III, 18+@ZB4-III and 21+@ZB4-III. Butyl groups of the host are removed for viewing clarity.
Figure 3: X-ray single crystal structures of 14+@ZB4-III, 16+@ZB4-III, 18+@ZB4-III and 21+@ZB4-III. Butyl gro...
![[1860-5397-14-134-4]](/bjoc/content/figures/1860-5397-14-134-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray single crystal structures of 18+@ZB4-III and 18+ in 18+@ZB4-III.
Figure 4: X-ray single crystal structures of 18+@ZB4-III and 18+ in 18+@ZB4-III.
In addition, thermodynamic parameters at different temperatures for the complex between ZB4 and 18+ were determined by ITC experiments (Figure S27 in Supporting Information File 1). The data are compiled in Table 3. The heat capacity change (ΔCp) for the formation of 18+@ZB4 is −0.13 kJ mol−1 K−1 as determined from the slope of the linear fitting in plots of ΔH versus temperature from 283 to 313 K (Figure 5). The release of solvent molecules upon complex formation may account for the negative heat capacity change and similar heat capacity changes were also reported for the fullerenes recognition [40]. Meanwhile, the changes of ΔG for the formation of 18+@ZB4 complex over the temperature range 283–313 K is very small (0.31 kJ mol−1), while the changes in ΔH and −TΔS are much larger (ca. 4–4.3 kJ mol−1). The changes in ΔH and −TΔS are opposite in signs and perfectly compensate each other. The enthalpy–entropy compensation phenomenon may be explained by invoking a solvent reorganization during the formation of 18+@ZB4 complex, which is common for reactions taking place in aqueous solution [41].
Table 3: Thermodynamic parameters for the complex between 18+ and ZB4 as determined by ITC in a 1:1 mixture of 1,2-dichloroethane and MeCN at different temperatures.
| T (K) | Ka (M−1) | ΔG (kJ∙mol−1) | ΔH (kJ∙mol−1) | −TΔS (kJ∙mol−1) |
|---|---|---|---|---|
| 283 | 8.4 × 105 | −32.09 | −38.59 | 6.50 |
| 293 | 5.2 × 105 | −32.04 | −39.96 | 7.92 |
| 298 | 4.2 × 105 | −32.10 | −40.72 | 8.62 |
| 303 | 3.3 × 105 | −32.01 | −41.19 | 9.18 |
| 308 | 2.6 × 105 | −31.88 | −42.08 | 10.2 |
| 313 | 2.0 × 105 | −31.78 | −42.58 | 10.8 |
![[1860-5397-14-134-5]](/bjoc/content/figures/1860-5397-14-134-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Linear relationships of ΔH with temperature (left, slope = −0.13, R2 = 0.9956) and TΔS (right, slope = 0.93, R2 = 0.9976).
Figure 5: Linear relationships of ΔH with temperature (left, slope = −0.13, R2 = 0.9956) and TΔS (right, slop...
Conclusion
In summary, we systematically studied the guest binding scope, electronic substituent effects and thermodynamic origin on the molecular recognition of ZB4 using NMR, ITC titration and X-ray crystallography. Similar to TA4, ZB4 is able to host a wide range of organic cations. However, in contrast to TA4, ZB4 shows no large amplitude of conformational response to the electronic nature of substituents on the guests, and its binding affinities follow a parabolic rather than a linear free energy relationship. A closer look at four representative crystal structures suggested that the parabolic free energy relationship may be caused through influencing the major interactions in the host–guest complexes by tuning the weak C–H∙∙∙O hydrogen bonds. Heat capacity changes and enthalpy–entropy compensation indicate that solvent reorganization is also involved during the host–guest binding. Generally, ZB4 is quite different from TA4, and further enriches the arsenal of conformationally adaptive macrocycles. With these model systems, we may further understand the importance of conformational adaptivity in biomolecular recognition and even design stimuli-responsive materials by harnessing this large amplitude of conformational changes.
Supporting Information
| Supporting Information File 1: Experimental procedures, NMR spectra, mass spectra, determination of association constants and X-ray single crystal data. | ||
| Format: PDF | Size: 2.3 MB | Download |
Acknowledgements
This research was financially supported by the National Natural Science foundation of China (WJ: Nos. 21572097 and 21772083), Thousand Young Talents Program (WJ), and SZSTI (WJ: Nos. JCYJ20160226192118056, JCYJ20170307105848463, and KQJSCX20170728162528382). Academy of Finland (AV: project no. 314343) is also acknowledged for funding. We thank SUSTech-MCPC for the support with instruments.
References
-
Schrader, T.; Hamilton, A. D. Functional Synthetic Receptors; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/352760572X
Return to citation in text: [1] -
Rambo, B. M.; Gong, H.-Y.; Oh, M.; Sessler, J. L. Acc. Chem. Res. 2012, 45, 1390–1401. doi:10.1021/ar300076b
Return to citation in text: [1] -
Lee, S.; Chen, C.-H.; Flood, A. H. Nat. Chem. 2013, 5, 704–710. doi:10.1038/nchem.1668
Return to citation in text: [1] -
Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765
Return to citation in text: [1] -
Chen, H.; Fan, J.; Hu, X.; Ma, J.; Wang, S.; Li, J.; Yu, Y.; Jia, X.; Li, C. Chem. Sci. 2015, 6, 197–202. doi:10.1039/C4SC02422B
Return to citation in text: [1] -
Liu, H.-B.; Zhang, Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2018, 57, 6536–6540. doi:10.1002/anie.201802650
Return to citation in text: [1] -
Zhang, G.-W.; Li, P.-F.; Meng, Z.; Wang, H.-X.; Han, Y.; Chen, C.-F. Angew. Chem., Int. Ed. 2016, 55, 5304–5308. doi:10.1002/anie.201600911
Return to citation in text: [1] -
Svec, J.; Necas, M.; Sindelar, V. Angew. Chem., Int. Ed. 2010, 49, 2378–2381. doi:10.1002/anie.201000420
Return to citation in text: [1] -
Gao, B.; Tan, L.-L.; Song, N.; Li, K.; Yang, Y.-W. Chem. Commun. 2016, 52, 5804–5807. doi:10.1039/C6CC01892K
Return to citation in text: [1] -
Zhu, H.; Shi, B.; Chen, K.; Wei, P.; Xia, D.; Mondal, J. H.; Huang, F. Org. Lett. 2016, 18, 5054–5057. doi:10.1021/acs.orglett.6b02500
Return to citation in text: [1] -
Jiang, B.; Wang, W.; Zhang, Y.; Lu, Y.; Zhang, C.-W.; Yin, G.-Q.; Zhao, X.-L.; Xu, L.; Tan, H.; Li, X.; Jin, G.-X.; Yang, H.-B. Angew. Chem., Int. Ed. 2017, 56, 14438–14442. doi:10.1002/anie.201707209
Return to citation in text: [1] -
Cram, D. J. Angew. Chem., Int. Ed. Engl. 1986, 25, 1039–1057. doi:10.1002/anie.198610393
Return to citation in text: [1] -
Koshland, D. E., Jr. Nat. Med. 1998, 4, 1112–1114. doi:10.1038/2605
Return to citation in text: [1] -
Tama, F.; Sanejouand, Y.-H. Protein Eng. 2001, 14, 1–6. doi:10.1093/protein/14.1.1
Return to citation in text: [1] -
Kremer, C.; Lützen, A. Chem. – Eur. J. 2013, 19, 6162–6196. doi:10.1002/chem.201203814
Return to citation in text: [1] -
Yuan, Y.; Tam, M. F.; Simplaceanu, V.; Ho, C. Chem. Rev. 2015, 115, 1702–1724. doi:10.1021/cr500495x
Return to citation in text: [1] -
Changeux, J.-P.; Edelstein, S. J. Science 2005, 308, 1424–1428. doi:10.1126/science.1108595
Return to citation in text: [1] -
Ikeda, A.; Shinkai, S. Chem. Rev. 1997, 97, 1713–1734. doi:10.1021/cr960385x
Return to citation in text: [1] -
Wang, M.-X. Acc. Chem. Res. 2012, 45, 182–195. doi:10.1021/ar200108c
Return to citation in text: [1] -
Kim, D. S.; Sessler, J. L. Chem. Soc. Rev. 2015, 44, 532–546. doi:10.1039/C4CS00157E
Return to citation in text: [1] -
Talotta, C.; Gaeta, C.; Qi, Z.; Schalley, C. A.; Neri, P. Angew. Chem., Int. Ed. 2013, 52, 7437–7441. doi:10.1002/anie.201301570
Return to citation in text: [1] -
Talotta, C.; Gaeta, C.; De Rosa, M.; Ascenso, J. R.; Marcos, P. M.; Neri, P. Eur. J. Org. Chem. 2016, 158–167. doi:10.1002/ejoc.201501319
Return to citation in text: [1] -
Galan, A.; Escudero-Adán, E. C.; Frontera, A., II; Ballester, P. J. Org. Chem. 2014, 79, 5545–5557. doi:10.1021/jo5007224
Return to citation in text: [1] -
Yang, L.-P.; Liu, W.-E.; Jiang, W. Tetrahedron Lett. 2016, 57, 3978–3985. doi:10.1016/j.tetlet.2016.07.077
Return to citation in text: [1] -
Huang, G.; He, Z.; Cai, C.-X.; Pan, F.; Yang, D.; Rissanen, K.; Jiang, W. Chem. Commun. 2015, 51, 15490–15493. doi:10.1039/C5CC06768E
Return to citation in text: [1] -
Huang, G.; Valkonen, A.; Rissanen, K.; Jiang, W. Chem. Commun. 2016, 52, 9078–9081. doi:10.1039/C6CC00349D
Return to citation in text: [1] -
Huang, G.-B.; Wang, S.-H.; Ke, H.; Yang, L.-P.; Jiang, W. J. Am. Chem. Soc. 2016, 138, 14550–14553. doi:10.1021/jacs.6b09472
Return to citation in text: [1] -
Wang, L.-L.; Chen, Z.; Liu, W.-E.; Ke, H.; Wang, S.-H.; Jiang, W. J. Am. Chem. Soc. 2017, 139, 8436–8439. doi:10.1021/jacs.7b05021
Return to citation in text: [1] -
Huang, G.-B.; Liu, W.-E.; Valkonen, A.; Yao, H.; Rissanen, K.; Jiang, W. Chin. Chem. Lett. 2018, 29, 91–94. doi:10.1016/j.cclet.2017.07.005
Return to citation in text: [1] -
Ma, Y.-L.; Ke, H.; Valkonen, A.; Rissanen, K.; Jiang, W. Angew. Chem., Int. Ed. 2018, 57, 709–713. doi:10.1002/anie.201711077
Return to citation in text: [1] -
Jia, F.; He, Z.; Yang, L.-P.; Pan, Z.-S.; Yi, M.; Jiang, R.-W.; Jiang, W. Chem. Sci. 2015, 6, 6731–6738. doi:10.1039/C5SC03251B
Return to citation in text: [1] [2] -
Jia, F.; Wang, H.-Y.; Li, D.-H.; Yang, L.-P.; Jiang, W. Chem. Commun. 2016, 52, 5666–5669. doi:10.1039/C6CC01052K
Return to citation in text: [1] [2] [3] [4] -
Yang, L.-P.; Liu, H.; Lu, S.-B.; Jia, F.; Jiang, W. Org. Lett. 2017, 19, 1212–1215. doi:10.1021/acs.orglett.7b00181
Return to citation in text: [1] [2] [3] -
Jia, F.; Yang, L.-P.; Li, D.-H.; Jiang, W. J. Org. Chem. 2017, 82, 10444–10449. doi:10.1021/acs.joc.7b01914
Return to citation in text: [1] [2] [3] [4] [5] -
Jia, F.; Li, D.-H.; Yang, T.-L.; Yang, L.-P.; Dang, L.; Jiang, W. Chem. Commun. 2017, 53, 336–339. doi:10.1039/C6CC09038A
Return to citation in text: [1] [2] -
Yang, L.-P.; Jia, F.; Pan, F.; Pan, Z.-S.; Rissanen, K.; Jiang, W. Chem. Commun. 2017, 53, 12572–12575. doi:10.1039/C7CC07630D
Return to citation in text: [1] [2] [3] -
Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Tran, A. H.; Miller, D. O.; Georghiou, P. E. J. Org. Chem. 2005, 70, 1115–1121. doi:10.1021/jo0484427
Return to citation in text: [1] [2] [3] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Le, V. H.; Yanney, M.; McGuire, M.; Sygula, A.; Lewis, E. A. J. Phys. Chem. B 2014, 118, 11956–11964. doi:10.1021/jp5087152
Return to citation in text: [1] -
Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Escudero-Adán, E. C.; Ballester, P. J. Am. Chem. Soc. 2014, 136, 3208–3218. doi:10.1021/ja412098v
Return to citation in text: [1]
| 39. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 40. | Le, V. H.; Yanney, M.; McGuire, M.; Sygula, A.; Lewis, E. A. J. Phys. Chem. B 2014, 118, 11956–11964. doi:10.1021/jp5087152 |
| 41. | Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Escudero-Adán, E. C.; Ballester, P. J. Am. Chem. Soc. 2014, 136, 3208–3218. doi:10.1021/ja412098v |
| 1. | Schrader, T.; Hamilton, A. D. Functional Synthetic Receptors; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/352760572X |
| 15. | Kremer, C.; Lützen, A. Chem. – Eur. J. 2013, 19, 6162–6196. doi:10.1002/chem.201203814 |
| 16. | Yuan, Y.; Tam, M. F.; Simplaceanu, V.; Ho, C. Chem. Rev. 2015, 115, 1702–1724. doi:10.1021/cr500495x |
| 34. | Jia, F.; Yang, L.-P.; Li, D.-H.; Jiang, W. J. Org. Chem. 2017, 82, 10444–10449. doi:10.1021/acs.joc.7b01914 |
| 13. | Koshland, D. E., Jr. Nat. Med. 1998, 4, 1112–1114. doi:10.1038/2605 |
| 14. | Tama, F.; Sanejouand, Y.-H. Protein Eng. 2001, 14, 1–6. doi:10.1093/protein/14.1.1 |
| 36. | Yang, L.-P.; Jia, F.; Pan, F.; Pan, Z.-S.; Rissanen, K.; Jiang, W. Chem. Commun. 2017, 53, 12572–12575. doi:10.1039/C7CC07630D |
| 12. | Cram, D. J. Angew. Chem., Int. Ed. Engl. 1986, 25, 1039–1057. doi:10.1002/anie.198610393 |
| 32. | Jia, F.; Wang, H.-Y.; Li, D.-H.; Yang, L.-P.; Jiang, W. Chem. Commun. 2016, 52, 5666–5669. doi:10.1039/C6CC01052K |
| 2. | Rambo, B. M.; Gong, H.-Y.; Oh, M.; Sessler, J. L. Acc. Chem. Res. 2012, 45, 1390–1401. doi:10.1021/ar300076b |
| 3. | Lee, S.; Chen, C.-H.; Flood, A. H. Nat. Chem. 2013, 5, 704–710. doi:10.1038/nchem.1668 |
| 4. | Ogoshi, T.; Yamagishi, T.-a.; Nakamoto, Y. Chem. Rev. 2016, 116, 7937–8002. doi:10.1021/acs.chemrev.5b00765 |
| 5. | Chen, H.; Fan, J.; Hu, X.; Ma, J.; Wang, S.; Li, J.; Yu, Y.; Jia, X.; Li, C. Chem. Sci. 2015, 6, 197–202. doi:10.1039/C4SC02422B |
| 6. | Liu, H.-B.; Zhang, Q.; Wang, M.-X. Angew. Chem., Int. Ed. 2018, 57, 6536–6540. doi:10.1002/anie.201802650 |
| 7. | Zhang, G.-W.; Li, P.-F.; Meng, Z.; Wang, H.-X.; Han, Y.; Chen, C.-F. Angew. Chem., Int. Ed. 2016, 55, 5304–5308. doi:10.1002/anie.201600911 |
| 8. | Svec, J.; Necas, M.; Sindelar, V. Angew. Chem., Int. Ed. 2010, 49, 2378–2381. doi:10.1002/anie.201000420 |
| 9. | Gao, B.; Tan, L.-L.; Song, N.; Li, K.; Yang, Y.-W. Chem. Commun. 2016, 52, 5804–5807. doi:10.1039/C6CC01892K |
| 10. | Zhu, H.; Shi, B.; Chen, K.; Wei, P.; Xia, D.; Mondal, J. H.; Huang, F. Org. Lett. 2016, 18, 5054–5057. doi:10.1021/acs.orglett.6b02500 |
| 11. | Jiang, B.; Wang, W.; Zhang, Y.; Lu, Y.; Zhang, C.-W.; Yin, G.-Q.; Zhao, X.-L.; Xu, L.; Tan, H.; Li, X.; Jin, G.-X.; Yang, H.-B. Angew. Chem., Int. Ed. 2017, 56, 14438–14442. doi:10.1002/anie.201707209 |
| 33. | Yang, L.-P.; Liu, H.; Lu, S.-B.; Jia, F.; Jiang, W. Org. Lett. 2017, 19, 1212–1215. doi:10.1021/acs.orglett.7b00181 |
| 25. | Huang, G.; He, Z.; Cai, C.-X.; Pan, F.; Yang, D.; Rissanen, K.; Jiang, W. Chem. Commun. 2015, 51, 15490–15493. doi:10.1039/C5CC06768E |
| 26. | Huang, G.; Valkonen, A.; Rissanen, K.; Jiang, W. Chem. Commun. 2016, 52, 9078–9081. doi:10.1039/C6CC00349D |
| 27. | Huang, G.-B.; Wang, S.-H.; Ke, H.; Yang, L.-P.; Jiang, W. J. Am. Chem. Soc. 2016, 138, 14550–14553. doi:10.1021/jacs.6b09472 |
| 28. | Wang, L.-L.; Chen, Z.; Liu, W.-E.; Ke, H.; Wang, S.-H.; Jiang, W. J. Am. Chem. Soc. 2017, 139, 8436–8439. doi:10.1021/jacs.7b05021 |
| 29. | Huang, G.-B.; Liu, W.-E.; Valkonen, A.; Yao, H.; Rissanen, K.; Jiang, W. Chin. Chem. Lett. 2018, 29, 91–94. doi:10.1016/j.cclet.2017.07.005 |
| 30. | Ma, Y.-L.; Ke, H.; Valkonen, A.; Rissanen, K.; Jiang, W. Angew. Chem., Int. Ed. 2018, 57, 709–713. doi:10.1002/anie.201711077 |
| 31. | Jia, F.; He, Z.; Yang, L.-P.; Pan, Z.-S.; Yi, M.; Jiang, R.-W.; Jiang, W. Chem. Sci. 2015, 6, 6731–6738. doi:10.1039/C5SC03251B |
| 32. | Jia, F.; Wang, H.-Y.; Li, D.-H.; Yang, L.-P.; Jiang, W. Chem. Commun. 2016, 52, 5666–5669. doi:10.1039/C6CC01052K |
| 33. | Yang, L.-P.; Liu, H.; Lu, S.-B.; Jia, F.; Jiang, W. Org. Lett. 2017, 19, 1212–1215. doi:10.1021/acs.orglett.7b00181 |
| 34. | Jia, F.; Yang, L.-P.; Li, D.-H.; Jiang, W. J. Org. Chem. 2017, 82, 10444–10449. doi:10.1021/acs.joc.7b01914 |
| 35. | Jia, F.; Li, D.-H.; Yang, T.-L.; Yang, L.-P.; Dang, L.; Jiang, W. Chem. Commun. 2017, 53, 336–339. doi:10.1039/C6CC09038A |
| 36. | Yang, L.-P.; Jia, F.; Pan, F.; Pan, Z.-S.; Rissanen, K.; Jiang, W. Chem. Commun. 2017, 53, 12572–12575. doi:10.1039/C7CC07630D |
| 24. | Yang, L.-P.; Liu, W.-E.; Jiang, W. Tetrahedron Lett. 2016, 57, 3978–3985. doi:10.1016/j.tetlet.2016.07.077 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 18. | Ikeda, A.; Shinkai, S. Chem. Rev. 1997, 97, 1713–1734. doi:10.1021/cr960385x |
| 19. | Wang, M.-X. Acc. Chem. Res. 2012, 45, 182–195. doi:10.1021/ar200108c |
| 20. | Kim, D. S.; Sessler, J. L. Chem. Soc. Rev. 2015, 44, 532–546. doi:10.1039/C4CS00157E |
| 21. | Talotta, C.; Gaeta, C.; Qi, Z.; Schalley, C. A.; Neri, P. Angew. Chem., Int. Ed. 2013, 52, 7437–7441. doi:10.1002/anie.201301570 |
| 22. | Talotta, C.; Gaeta, C.; De Rosa, M.; Ascenso, J. R.; Marcos, P. M.; Neri, P. Eur. J. Org. Chem. 2016, 158–167. doi:10.1002/ejoc.201501319 |
| 23. | Galan, A.; Escudero-Adán, E. C.; Frontera, A., II; Ballester, P. J. Org. Chem. 2014, 79, 5545–5557. doi:10.1021/jo5007224 |
| 17. | Changeux, J.-P.; Edelstein, S. J. Science 2005, 308, 1424–1428. doi:10.1126/science.1108595 |
| 31. | Jia, F.; He, Z.; Yang, L.-P.; Pan, Z.-S.; Yi, M.; Jiang, R.-W.; Jiang, W. Chem. Sci. 2015, 6, 6731–6738. doi:10.1039/C5SC03251B |
| 32. | Jia, F.; Wang, H.-Y.; Li, D.-H.; Yang, L.-P.; Jiang, W. Chem. Commun. 2016, 52, 5666–5669. doi:10.1039/C6CC01052K |
| 33. | Yang, L.-P.; Liu, H.; Lu, S.-B.; Jia, F.; Jiang, W. Org. Lett. 2017, 19, 1212–1215. doi:10.1021/acs.orglett.7b00181 |
| 34. | Jia, F.; Yang, L.-P.; Li, D.-H.; Jiang, W. J. Org. Chem. 2017, 82, 10444–10449. doi:10.1021/acs.joc.7b01914 |
| 35. | Jia, F.; Li, D.-H.; Yang, T.-L.; Yang, L.-P.; Dang, L.; Jiang, W. Chem. Commun. 2017, 53, 336–339. doi:10.1039/C6CC09038A |
| 36. | Yang, L.-P.; Jia, F.; Pan, F.; Pan, Z.-S.; Rissanen, K.; Jiang, W. Chem. Commun. 2017, 53, 12572–12575. doi:10.1039/C7CC07630D |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 32. | Jia, F.; Wang, H.-Y.; Li, D.-H.; Yang, L.-P.; Jiang, W. Chem. Commun. 2016, 52, 5666–5669. doi:10.1039/C6CC01052K |
| 38. | Tran, A. H.; Miller, D. O.; Georghiou, P. E. J. Org. Chem. 2005, 70, 1115–1121. doi:10.1021/jo0484427 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 34. | Jia, F.; Yang, L.-P.; Li, D.-H.; Jiang, W. J. Org. Chem. 2017, 82, 10444–10449. doi:10.1021/acs.joc.7b01914 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 34. | Jia, F.; Yang, L.-P.; Li, D.-H.; Jiang, W. J. Org. Chem. 2017, 82, 10444–10449. doi:10.1021/acs.joc.7b01914 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 38. | Tran, A. H.; Miller, D. O.; Georghiou, P. E. J. Org. Chem. 2005, 70, 1115–1121. doi:10.1021/jo0484427 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
| 38. | Tran, A. H.; Miller, D. O.; Georghiou, P. E. J. Org. Chem. 2005, 70, 1115–1121. doi:10.1021/jo0484427 |
| 37. | Yang, L.-P.; Jia, F.; Zhou, Q.-H.; Pan, F.; Sun, J.-N.; Rissanen, K.; Chung, L. W.; Jiang, W. Chem. – Eur. J. 2017, 23, 1516–1520. doi:10.1002/chem.201605701 |
© 2018 Yang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)