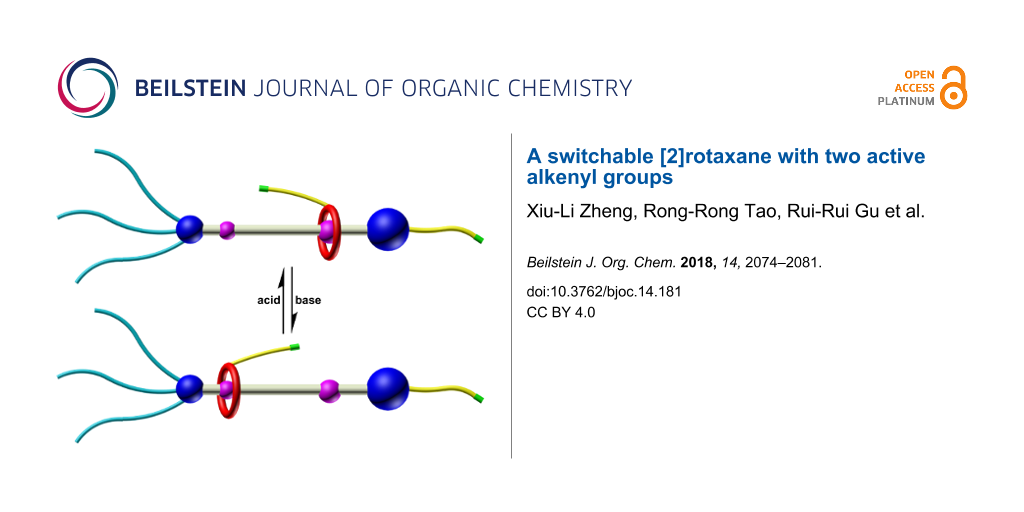
Abstract
A novel functional [2]rotaxane containing two alkenyl bonds was designed, synthesized and characterized by 1H, 13C NMR spectroscopy and HRESI mass spectrometry. The introduction of alkenyl bonds endowed the [2]rotaxane a fascinating ability to react with versatile functional groups such as alkenyl and thiol functional groups. The reversible shuttling movement of the macrocycle between two different recognition sites on the molecular thread can be driven by external acid and base. This kind of rotaxane bearing functional groups provides a powerful platform for preparing stimuli-responsive polymers.
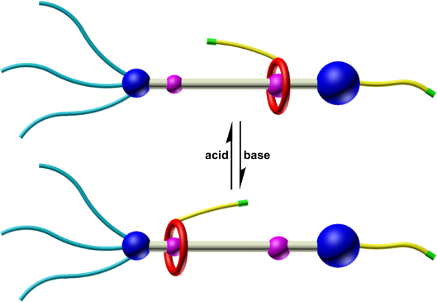
Graphical Abstract
Introduction
Along with the development of supramolecular chemistry, much attention has been paid to the design and synthesis of novel and complicated mechanical interlocked molecules (MIMs) [1-7]. During the past years, a large number of different MIMs has been constructed, such as handcuff catenane [8], molecular elevators [9,10], molecular muscles [11,12], trefoil necklace [13-15], molecular walkers [16-18] and molecular pumps [19,20]. Combining the stimuli-responsive microscopic units with traditional materials to achieve morphological changes or other novel properties in smart materials is continuously receiving wide attention. Hence, in recent years, scientists have put more interests on the macroscopic changes caused by the stimuli-responsiveness of constituent units, which act as important precursors for constructing stimuli-responsive supramolecular materials [21,22].
Rotaxanes [23-31], as one of the most important MIMs, have been deeply investigated because of their excellent properties and convenient synthesis. By introducing various functional groups, such kind of MIMs has been used to construct stimuli-responsive materials, which diversified and improved the functions of traditional polymers. Up to now, the repertoire of available functional rotaxanes as building blocks for the fabrication of stimuli-responsive polymers remains limited because of the fact that the induction of active and functional groups make the preparation of rotaxanes more difficult and complicated. Therefore, it is urgent to enrich both the family of stimuli-responsive units and the methods for constructing this kind of smart materials.
In this paper, we report the design, synthesis, characterization and shuttling motion of a [2]rotaxane R1, which is modified with two naked alkenyl bonds. In this [2]rotaxane, a functional dibenzo-24-crown-8 (DB24C8) macrocycle is interlocked with an flexible chain and could perform reversible shuttling between two different recognition sites under the stimuli of external acid and base. The alkenyl bonds were chosen as the functional group due to their considerable stability during the synthesis and remarkable reaction activity. The latter indicates the ability of reacting with different functional groups such as alkenyl bonds in the presence of Grubbs’ catalyst and thiol groups irradiated by UV light. Besides, the shuttling movement of the DB24C8 macrocyclic ring was confirmed by 1H NMR spectroscopy.
Results and Discussion
The structures of the two states of [2]rotaxane R1 are shown in Scheme 1. In the target structure [2]rotaxane R1, the DB24C8 macrocycle is functionalized with an alkenyl unit on one of the benzene groups. Two distinguishable recognition sites, a dibenzylammonium (DBA) and an amide binding site, are introduced to the axle and linked with a long alkyl chain. The amide moiety could act as another combining station when the DBA site is deprotonated by external base. Besides, in the structure, one side of the chain is terminated by a bulky stopper bearing three long alkyl chains to prevent the macrocyclic moiety from slipping out the thread. Meanwhile, the long alkyl chains make it easy to form gels when the alkenyl units are reacted to generate polymers. On the other side, a naked alkenyl bond is introduced in the para position of the aromatic stopper. In the presence of external acid–base stimuli, the macrocyclic ring could be driven back and forth along the linear thread. Deprotonation of the DBA site by base makes the DB24C8 moiety slide to the amide station and the macrocycle could move back to the DBA recognition site when acid was added (Scheme 1).
![[1860-5397-14-181-i1]](/bjoc/content/inline/1860-5397-14-181-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Chemical structures and acid-base stimuli responsiveness of target [2]rotaxane R1 and deprotonated [2]rotaxane R1-1.
Scheme 1: Chemical structures and acid-base stimuli responsiveness of target [2]rotaxane R1 and deprotonated ...
The synthesis routes of the target compound [2]rotaxane R1 are shown in Scheme 2. Compounds 1 [32], 2 [33], 6 [34], 7 [35] and functional crown ether 9 [36] were synthesized according to the previous literature. The key intermediate compound 5, containing a prior DBA recognition station and possessing alkynyl and ethylenic groups at two terminals respectively, was prepared from aldehyde 1 and amine 2. A “Schiff base” reaction was introduced at first and treated with NaBH4, protected by di-tert-butyl pyrocarbonate to get compound 3. The esterification reaction was carried out subsequently between hydroxy compound 3 and allylacetic acid to afford compound 4, which was further reacted with CF3COOH and followed by the anion exchange in the methyl alcohol with saturated NH4PF6 solution to obtain compound 5. For another key intermediate 8, containing an amide site for stabilizing the DB24C8 macrocycle, a 1,2,3-tris(dodecyloxy) benzene group as a stopper and an azide group which is used for the reaction with other moieties, was prepared through the amide reaction between compound 6 and 6-azidohexan-1-amine in the presence of 2-chloro-4,6-dimethoxy-1,3,5-triazine and N-methylmorpholine. Finally, the classical and effective “Click” reaction was used to produce the target compound [2]rotaxane R1. The crown ether 9 and dibenzylammonium ion (R2NH2+) containing intermediate compound 5 were assembled in dichloromethane through host–guest interaction and capped with compound 8 under Cu(I)-catalyzed azide–alkyne cycloaddition to get the final [2]rotaxane with two distinguishable recognition sites.
![[1860-5397-14-181-i2]](/bjoc/content/inline/1860-5397-14-181-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syntheses of key intermediates 5, 8 and target [2]rotaxane R1.
Scheme 2: Syntheses of key intermediates 5, 8 and target [2]rotaxane R1.
The target [2]rotaxane R1 was then characterized by 1H NMR spectroscopy and high-resolution electrospray ionization (HRESI) mass spectrometry. The HRESI mass spectra of [2]rotaxane R1 showed a major peak at m/z 2083.3862, corresponding to the species of R1 losing one PF6− anion, i.e., [M − PF6−]+. The 1H NMR spectrum also fitted the R1 structure well and reveals that the DB24C8 macrocycle 9 prefers encompassing the DBA recognition site. Comparing the 1H NMR spectrum of the rotaxane product R1 with the reactants 8 and 5 (Figure 1), the resonances of protons H5 and H7 were both shifted downfield (ΔδH5 = 1.09 ppm and ΔδH7 = 0.51 ppm) while a new peak appeared at 7.56 ppm. This new peak corresponds to H6 coming from the alkynyl–azide cycloaddition. Meanwhile, the signal of H12 and H13 split into two signals and underwent downfield shifts (ΔδH12 = 0.73 ppm and ΔδH13 = 0.87 ppm) while those of H11 and H10 moved upfield (ΔδH11 = −0.08 ppm and ΔδH10 = −0.20 ppm). Nevertheless, there were no obvious changes in protons of H3, H4 and H2, indicating that the macrocycle stayed at the DBA recognition site. These results were consistent with the previous literature report by our group [37]. All the evidences discussed above demonstrate that the axle compound successfully threaded into the macrocycle and consequently formed the target [2]rotaxane R1.
![[1860-5397-14-181-1]](/bjoc/content/figures/1860-5397-14-181-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Partial 1H NMR spectra (400 MHz, CDCl3, 298 K). (a) Compound 5, (b) target [2]rotaxane R1, (c) azide compound 8.
Figure 1: Partial 1H NMR spectra (400 MHz, CDCl3, 298 K). (a) Compound 5, (b) target [2]rotaxane R1, (c) azid...
The reversible shuttling motion of the functional DB24C8 macrocycle between the two different recognition sites on the axle was also confirmed by the 1H NMR spectroscopy. After addition of 2 equiv1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to [2]rotaxane R1 in CDCl3 to deprotonate the ammonium moiety, the DB24C8 macrocycle moved to the amide station. As shown in Figure 2, the protons H12, H13 shifted upfield (ΔδH12 = −0.99 ppm and ΔδH13 = −0.75 ppm) and incorporated into one signal peak from two while the H11 and H10 shifted slightly downfield (ΔδH11 = 0.05 ppm and Δδ10 = 0.19 ppm) due to the deprotonation of the R2NH2+ and the migration of macrocycle. At the same time, the peaks for the protons around the amide site changed, for H4, H5, H6 with ΔδH4 = −0.26 ppm, ΔδH5 = −0.14 ppm and ΔδH6 = −0.09 ppm, respectively and H3 with a ΔδH3 = 0.51 ppm due to the association with the DB24C8 macrocycle through hydrogen bonding. All the evidences reveal that the functionalized macrocycle migrated from the DBA recognition site to the amide station when an external base was added to the solution of rotaxane R1. Then, 3 equiv trifluoroacetic acid were added to reprotonate the -NH- moiety, the 1H NMR spectrum restored to the initial state, showing that the macrocyclic compound 9 moved back to the DBA recognition site. Therefore, the reversible shuttling movement of DB24C8 moiety along the thread between two recognition sites driven by acid-base stimuli has been confirmed.
![[1860-5397-14-181-2]](/bjoc/content/figures/1860-5397-14-181-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Partial 1H NMR spectra (400 MHz, CDCl3, 298 K). (a) [2]Rotaxane R1, (b) deprotonation by the addition of 2.0 equiv of DBU to (a), (c) reprotonation with addition of 3.0 equiv of TFA to (b).
Figure 2: Partial 1H NMR spectra (400 MHz, CDCl3, 298 K). (a) [2]Rotaxane R1, (b) deprotonation by the additi...
To further demonstrate the binding mode of the functional macrocycle with the axle, 2D ROESY NMR spectra of rotaxane R1 before and after the deprotonation in CDCl3 were obtained. As shown in Figure 3a, the cross-peaks of phenyl protons H10 (peak A), H14 (peak B), H11 (peak C) (around the DBA recognition site) and methylene protons on the functional crown ether indicate that the DB24C8 macrocycle was located on the DBA site, corresponding to the structure of [2]rotaxane R1. After addition of 2 equiv DBU to the solution of R1, the NOE correlations between the phenyl proton H2 (peak D) near the amide station, H5 (peak E) on the thread and methylene proton on DB24C8 could be observed (see Figure 3b), illustrating the position of the DB24C8 ring on the amide recognition site.
![[1860-5397-14-181-3]](/bjoc/content/figures/1860-5397-14-181-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Partial 2D ROESY NMR spectra (500 MHz, CDCl3, 298 K). (a) [2]Rotaxane R1, (b) deprotonation with addition of 2.0 equiv of DBU to (a).
Figure 3: Partial 2D ROESY NMR spectra (500 MHz, CDCl3, 298 K). (a) [2]Rotaxane R1, (b) deprotonation with ad...
Conclusion
In summary, a novel functional [2]rotaxane R1 with two alkenyl bonds on the DB24C8 macrocycle and axle, respectively, was prepared and well characterized. The shuttling movement of the functionalized DB24C8 macrocycle between the DBA recognition site and amide moiety could be realized by stimuli of external acid-base and was investigated by 1H NMR and 2D ROESY NMR spectroscopy. The naked alkenyl groups could react with other functional groups such as alkenyl and thiol groups to prepare stimuli-responsive polymers. Besides, the introduction of long alkyl chains makes the polymers easier to form gels. This kind of functional rotaxane enriches the species of building blocks to construct stimuli-responsive polymers and smart materials.
Experimental
General and materials
1H NMR and 13C NMR spectra were measured on a Bruker AV-400 spectrometer. The electronic spray ionization (ESI) mass spectra were tested on a LCT Premier XE mass spectrometer.
Chemicals were used as received from Acros, Aldrich, Fluka, or Merck. All solvents were reagent grade, which were dried and distilled prior to use according to standard procedures. The molecular structures were confirmed via 1H NMR, 13C NMR and high-resolution ESI mass spectroscopy. The synthesis of compound 1, 2, 6, 7, 9 have already been reported.
Synthesis
Compound 8: The mixture of compound 6 (5.0 g, 6.83 mmol), compound 7 (2.9 g, 20.49 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT, 3.6 g, 20.49 mmol) was placed in a 250 mL round-bottom flask and dissolved by 100 mL methylene chloride. Then, N-methylmorpholine (NMM, 3.5 g, 34.15 mmol) was added into the solution slowly under ice bath cooling. Afterwards, the mixture was stirred at 40 °C for 12 h under an argon atmosphere. The reaction mixture was cooled to room temperature, the solution was evaporated under reduced pressure and the residue was purified via column chromatography (SiO2, PE/EA = 3:1) to give compound 8 (4.9 g, 83%) as a white solid. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.30–7.23 (m, 1H), 7.02 (s, 2H), 6.67–6.65 (m, 1H), 4.28 (d, J = 5.2 Hz, 2H), 4.02–3.93 (m, 6H), 3.31–3.19 (m, 4H), 1.80–1.72 (m, 6H), 1.61–1.49 (m, 4H), 1.48–1.40 (m, 6H), 1.36–1.23 (m, 52H), 0.87 (t, J = 6.8 Hz, 9H); 13C NMR (CDCl3, 100 MHz, 298 K) δ 169.2, 167.8, 153.1, 141.3, 128.1, 105.6, 73.5, 69.2, 51.2, 44.1, 39.5, 31.9, 30.2, 29.70, 29.68, 29.66, 29.62, 29.56, 29.39, 29.36, 29.34, 29.31, 28.7, 26.4, 26.3, 26.07, 26.05, 22.7, 14.1; HRMS–ESI–TOF (m/z): [M + K]+ calcd for C51H93N5O5K+, 894.6808; found, 894.6813.
Compound 3: A mixture of 1 (3.0 g, 10.63 mmol) and 2 (4.0 g, 12.75 mmol) in methyl alcohol (100 mL) was refluxed overnight under a nitrogen atmosphere. After the reaction mixture was cooled to room temperature, NaBH4 (1.6 g, 30.5 mmol) was then added to the solution in portions while colling in an ice bath. After the mixture was stirred overnight, the solvent was removed under vacuum, and the residue was extracted by dichloromethane. The organic layer was washed by brine, dried with Na2SO4, and then concentrated to give the free amine compound. To the solution of the amine in dry DCM (50 mL) was added di-tert-butyldicarbonate (3.5 g, 15.95 mmol) and the mixture was stirred for 5 h at room temperature. Then, the reaction mixture was washed with water, dried over Na2SO4 and evaporated in vacuo to give a crude product, which was purified by column chromatography (SiO2, PE/EA = 2:1) to afford product 3 (4.4 g, 60%) as a yellow oil. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.20–7.01 (m, 2H), 6.83 (d, J = 8.4 Hz, 2H), 6.37 (d, J = 26.0 Hz, 2H), 4.29 (dd, J = 32.8 Hz, J = 18.4 Hz, 4H), 4.12 (s, 2H), 3.97–3.87 (m, 4H), 3.78 (s, 6H), 3.62 (t, J = 6.4 Hz, 2H), 3.49 (t, J = 6.4 Hz, 2H), 2.41 (s, 1H), 1.79–1.70 (m, 4H), 1.61–1.54 (m, 4H), 1.52–1.35 (m, 15H), 1.37–1.27 (m, 10H); 13C NMR (CDCl3, 100 MHz, 298 K) δ 171.2, 158.3, 153.2, 129.6, 129.3, 128.7, 114.3, 104.9, 104.3, 79.9, 74.0, 73.2, 70.2, 67.9, 62.7, 60.3, 57.9, 56.0, 32.6, 29.9, 29.6, 29.30, 29.27, 29.2, 28.4, 26.0, 25.9, 25.5, 25.4; HRMS–ESI–TOF (m/z): [M + Na]+ calcd for C40H61NO8Na+, 706.4289; found, 706.4295.
Compound 4: To the mixture of compound 3 (2.0 g, 2.92 mmol) and allylacetic acid (0.88 g, 8.77 mmol) in DCM (50 mL) was added EDCI (2.24 g, 11.68 mmol) and DMAP (0.36 g, 2.92 mmol) while cooling with an ice bath. After that the mixture was stirred overnight at room temperature and then washed the mixture with brine (3 × 50 mL). The organic layer was dried over anhydrous sodium sulfate. The combined organic layer was evaporated, and the residue was purified via column chromatography (SiO2, PE/EA = 15:1) to give compound 4 (1.97 g, 88%) as a yellow oil. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.21–7.03 (m, 2H), 6.84 (d, J = 8.4 Hz, 2H), 6.38 (d, J = 22.8 Hz, 2H), 5.87–5.76 (m, 1H), 5.09–4.95 (m, 2H), 4.29 (dd, J = 32.0 Hz, J = 16.4 Hz, 4H), 4.13 (d, J = 2.4 Hz, 2H), 4.08 (t, J = 6.8 Hz, 2H), 3.94 (t, J = 6.4 Hz, 4H), 3.78 (s, 6H), 3.50 (t, J = 6.8 Hz, 2H), 5.18–2.33 (m, 5H), 1.81–1.70 (m, 4H), 1.68–1.39 (m, 21H), 1.34–1.28 (m, 8H); 13C NMR (CDCl3, 100 MHz, 298 K) δ 173.1, 158.4, 155.9, 153.4, 136.7, 129.7, 129.3, 128.7, 115.4, 114.4, 104.9, 104.3, 80.0, 74.0, 73.1, 70.4, 70.2, 67.9, 64.4, 57.9, 56.0, 33.5, 29.9, 29.4, 29.33, 29.30, 29.2, 28.8, 28.6, 28.4; HRMS–ESI–TOF (m/z): [M + Na]+ calcd for C45H67NO9Na+, 788.4708; found, 788.4732.
Compound 5: TFA (1.9 mL, 25.72 mmol) was added to a solution of the product 4 (1.97 g, 25.72 mmol) in dichloromethane (50 mL) and the mixture was stirred for 10 h at room temperature. The organic solvent was evaporated under reduced pressure to get a yellow solid, which was dissolved in MeOH (50 mL) and 20 mL saturated aqueous solution of NH4PF6 was added. After stirring for 5 h at room temperature, the mixture was diluted with CH2Cl2 (100 mL), the organic layer was separated and evaporated under reduced pressure to get the crude product, which was purified by column chromatography (SiO2, CH2Cl2/MeOH = 50:1) to afford product 5 (1.67 g, 80%) as a yellow solid. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.26 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 6.57 (s, 2H), 5.90–5.74 (m, 1H), 5.13–4.89 (m, 2H), 4.13 (d, J = 2.4 Hz, 2H), 4.08 (t, J = 6.8 Hz, 2H), 3.97–3.88 (m, 4H), 3.84 (s, 6H), 3.74 (d, J = 6.0 Hz, 4H), 3.51 (t, J = 6.4 Hz, 2H), 2.48–2.32 (m, 5H), 1.81–1.70 (m, 4H), 1.69–1.55 (m, 4H), 1.53–1.38 (m, 6H), 1.36–1.28 (m, 10H); 13C NMR (CDCl3, 100 MHz, 298 K) δ 173.3, 160.3, 153.4, 136.8, 136.5, 131.4, 125.2, 121.4, 115.4, 115.0, 106.6, 79.9, 74.1, 73.1, 70.2, 68.0, 64.3, 57.9, 56.0, 51.6, 51.2, 33.4, 29.43, 29.38, 29.30, 29.28, 29.1, 28.8, 28.4, 26.0, 25.9, 25.5, 25.1; HRMS–ESI–TOF (m/z): [M − PF6−]+ calcd for C40H60NO7+, 666.4364; found, 666.4361.
Compound R1: A mixture of 5 (300 mg, 0.370 mmol) and crown ether 9 (414 mg, 0.739 mmol) was dissolved in dry CH2Cl2 (10 mL) and stirred for 0.5 h at room temperature. Then compound 8 (475 mg, 0.555 mmol) and [Cu(CH3CN)4]PF6 (138 mg, 0.370 mmol) were added to the solution. The mixture was stirred for two days under an argon atmosphere at room temperature. After removal of the solvent, the residue was purified via column chromatography (SiO2, CH2Cl2/MeOH = 80:1) to give compound R1 (495 mg, 60%) as a yellow solid. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.61–7.42 (m, 3H), 7.33 (t J = 4.8 Hz, 1H), 7.16 (d J = 8.8 Hz, 2H), 7.06 (s, 2H), 6.91–6.83 (m, 3H), 6.82–6.72 (m, 4H), 6.63 (d J = 8.8 Hz, 2H), 6.58–6.45 (m, 3H), 5.87–5.71 (m, 2H), 5.09–4.91 (m, 6H), 4.68–4.53 (m, 4H), 4.51–4.40 (m, 2H), 4.31 (t, J = 7.2 Hz, 2H), 4.13–4.04 (m, 10H), 4.00–3.93 (m, 6H), 3.85–3.70 (m, 12H), 3.56 (s, 6H), 3.53–3.46 (m, 8H), 3.23 (dd, J = 12.8 Hz, J = 6.8 Hz, 2H), 2.46–2.32 (m, 8H), 2.07–1.99 (m, 2H), 1.92–1.83 (m, 2H), 1.78–1.58 (m, 12H), 1.51–1.36 (m, 14H), 1.31–1.22 (m, 62H), 0.86 (t, J = 6.4 Hz, 9H); 13C NMR (CDCl3, 100 MHz, 298 K) δ 173.1, 172.8, 169.2, 167.5, 159.8, 153.4, 153.0, 147.3, 147.1, 145.2, 141.0, 137.4, 136.6, 136.5, 130.6, 129.4, 128.3, 127.4, 122.8, 122.5, 121.8, 121.7, 115.5, 115.4, 114.4, 112.8, 112.33, 112.26, 105.7, 105.5, 73.4, 73.2, 70.8, 70.4, 70.1, 70.0, 69.1, 68.1, 68.0, 67.9, 65.8, 64.4, 64.1, 55.9, 52.3, 52.1, 50.0, 43.8, 39.1, 33.5, 33.4, 31.9, 30.3, 29.9, 29.7, 29.64, 29.62, 29.58, 29.53, 29.36, 29.32, 29.30, 29.27, 29.1, 28.9, 28.8, 28.7, 28.5, 26.03, 26.01, 25.9, 25.8, 25.7, 25.6, 25.4, 22.6, 14.1; HRMS–ESI–TOF (m/z): [M −PF6−]+ calcd for C121H193N6O22+, 2083.4196; found, 2083.3862.
Supporting Information
| Supporting Information File 1: 1H, 13C NMR spectra and HRESI mass spectra of compounds 3, 4, 5, 8 and [2]rotaxane R1. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Saha, S.; Stoddart, J. F. Chem. Soc. Rev. 2007, 36, 77–92. doi:10.1039/B607187B
Return to citation in text: [1] -
Dasgupta, S.; Wu, J.-S. Chem. Sci. 2012, 3, 425–432. doi:10.1039/C1SC00613D
Return to citation in text: [1] -
Qu, D.-H.; Tian, H. Chem. Sci. 2011, 2, 1011–1015. doi:10.1039/c0sc00653j
Return to citation in text: [1] -
Zhu, L.-L.; Yan, H.; Nguyen, K. T.; Tian, H.; Zhao, Y.-L. Chem. Commun. 2012, 48, 4290–4292. doi:10.1039/c2cc17114g
Return to citation in text: [1] -
Dzyuba, E. V.; Baytekin, B.; Sattler, D.; Schalley, C. A. Eur. J. Org. Chem. 2012, 1171–1178. doi:10.1002/ejoc.201101231
Return to citation in text: [1] -
Rao, S.-J.; Zhang, Q.; Mei, J.; Ye, X.-H.; Gao, C.; Wang, Q.-C.; Qu, D.-H.; Tian, H. Chem. Sci. 2017, 8, 6777–6783. doi:10.1039/C7SC03232C
Return to citation in text: [1] -
Gao, C.; Luan, Z.-L.; Zhang, Q.; Rao, S.-J.; Qu, D.-H.; Tian, H. Org. Lett. 2017, 19, 3931–3934. doi:10.1021/acs.orglett.7b01853
Return to citation in text: [1] -
Hartlieb, K. J.; Blackburn, A. K.; Schneebeli, S. T.; Forgan, R. S.; Sarjeant, A. A.; Stern, C. L.; Cao, D.; Stoddart, J. F. Chem. Sci. 2014, 5, 90–100. doi:10.1039/C3SC52106K
Return to citation in text: [1] -
Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845–1849. doi:10.1126/science.1094791
Return to citation in text: [1] -
Badjić, J. D.; Ronconi, C. M.; Stoddart, J. F.; Balzani, V.; Silvi, S.; Credi, A. J. Am. Chem. Soc. 2006, 128, 1489–1499. doi:10.1021/ja0543954
Return to citation in text: [1] -
Goujon, A.; Du, G.; Moulin, E.; Fuks, G.; Maaloum, M.; Buhler, E.; Giuseppone, N. Angew. Chem., Int. Ed. 2016, 55, 703–707. doi:10.1002/anie.201509813
Return to citation in text: [1] -
Goujon, A.; Lang, T.; Mariani, G.; Moulin, E.; Fuks, G.; Raya, J.; Buhler, E.; Giuseppone, N. J. Am. Chem. Soc. 2017, 139, 14825–14828. doi:10.1021/jacs.7b06710
Return to citation in text: [1] -
Roh, S.-G.; Park, K.-M.; Park, G.-J.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Angew. Chem., Int. Ed. 1999, 38, 637–641. doi:10.1002/(SICI)1521-3773(19990301)38:5<637::AID-ANIE637>3.0.CO;2-4
Return to citation in text: [1] -
Kim, K. Chem. Soc. Rev. 2002, 31, 96–107. doi:10.1039/a900939f
Return to citation in text: [1] -
Chang, C.-F.; Chuang, C.-J.; Lai, C.-C.; Liu, Y.-H.; Peng, S.-M.; Chiu, S.-H. Angew. Chem., Int. Ed. 2012, 51, 10094–10098. doi:10.1002/anie.201205498
Return to citation in text: [1] -
von Delius, M.; Geertsema, E. M.; Leigh, D. A. Nat. Chem. 2009, 2, 96–101. doi:10.1038/nchem.481
Return to citation in text: [1] -
Lewandowski, B.; De Bo, G.; Ward, J. W.; Papmeyer, M.; Kuschel, S.; Aldegunde, M. J.; Gramlich, P. M. E.; Heckmann, D.; Goldup, S. M.; D’Souza, D. M.; Fernandes, A. E.; Leigh, D. A. Science 2013, 339, 189–193. doi:10.1126/science.1229753
Return to citation in text: [1] -
Qu, D.-H.; Tian, H. Chem. Sci. 2013, 4, 3031–3035. doi:10.1039/c3sc51160j
Return to citation in text: [1] -
Cheng, C.; McGonigal, P. R.; Schneebeli, S. T.; Li, H.; Vermeulen, N. A.; Ke, C.; Stoddart, J. F. Nat. Nanotechnol. 2015, 10, 547–553. doi:10.1038/nnano.2015.96
Return to citation in text: [1] -
Meng, Z.; Xiang, J.-F.; Chen, C.-F. J. Am. Chem. Soc. 2016, 138, 5652–5658. doi:10.1021/jacs.6b01852
Return to citation in text: [1] -
Brunsveld, L.; Folmer, B. J. B.; Meijer, E. W.; Sijbesma, R. P. Chem. Rev. 2001, 101, 4071–4098. doi:10.1021/cr990125q
Return to citation in text: [1] -
Cao, Z.-Q.; Miao, Q.; Zhang, Q.; Li, H.; Qu, D.-H.; Tian, H. Chem. Commun. 2015, 51, 4973–4976. doi:10.1039/C4CC09976A
Return to citation in text: [1] -
Raymo, F. M.; Stoddart, J. F. Chem. Rev. 1999, 99, 1643–1664. doi:10.1021/cr970081q
Return to citation in text: [1] -
Balzani, V.; Credi, A.; Silvi, S.; Venturi, M. Chem. Soc. Rev. 2006, 35, 1135–1149. doi:10.1039/b517102b
Return to citation in text: [1] -
Tian, H.; Wang, Q.-C. Chem. Soc. Rev. 2006, 35, 361–374. doi:10.1039/b512178g
Return to citation in text: [1] -
Champin, B.; Mobian, P.; Sauvage, J.-P. Chem. Soc. Rev. 2007, 36, 358–366. doi:10.1039/B604484K
Return to citation in text: [1] -
Crowley, J. D.; Goldup, S. M.; Lee, A.-L.; Leigh, D. A.; McBurney, R. T. Chem. Soc. Rev. 2009, 38, 1530–1541. doi:10.1039/b804243h
Return to citation in text: [1] -
Hänni, K. D.; Leigh, D. A. Chem. Soc. Rev. 2010, 39, 1240–1251. doi:10.1039/B901974J
Return to citation in text: [1] -
Zhu, L.-L.; Yan, H.; Wang, X.-J.; Zhao, Y.-L. J. Org. Chem. 2012, 77, 10168–10175. doi:10.1021/jo301807y
Return to citation in text: [1] -
Cui, J.-S.; Ba, Q.-K.; Ke, H.; Valkonen, A.; Rissanen, K.; Jiang, W. Angew. Chem., Int. Ed. 2018, 57, 7809–7814. doi:10.1002/anie.201803349
Return to citation in text: [1] -
Gao, C.; Luan, Z.-L.; Zhang, Q.; Yang, S.; Rao, S.-J.; Qu, D.-H.; Tian, H. Org. Lett. 2017, 19, 1618–1621. doi:10.1021/acs.orglett.7b00393
Return to citation in text: [1] -
Guidry, E. N.; Li, J.; Stoddart, J. F.; Grubbs, R. H. J. Am. Chem. Soc. 2007, 129, 8944–8945. doi:10.1021/ja0725100
Return to citation in text: [1] -
Jiang, Y.; Guo, J.-B.; Chen, C.-F. Org. Lett. 2010, 12, 4248–4251. doi:10.1021/ol101920b
Return to citation in text: [1] -
Kawakami, H.; Toma, K. Diamide type gelling agent. Jpn. Kokai Tokkyo Koho JP2004262809A, Sept 24, 2004.
Return to citation in text: [1] -
Li, F.-F.; Li, Y.-J.; Zhou, Z.-C.; Lv, S.-X.; Deng, Q.-R.; Xu, X.; Yin, L.-C. ACS Appl. Mater. Interfaces 2017, 9, 23586–23601. doi:10.1021/acsami.7b08534
Return to citation in text: [1] -
Candrill, S. J.; Grubbs, R. H.; Lanari, D.; Leung, K. C.-F.; Nelson, A.; Poulin-Kerstien, K. G.; Smidt, S. P.; Stoddart, J. F.; Tirrell, D. A. Org. Lett. 2005, 7, 4213–4216. doi:10.1021/ol051599g
Return to citation in text: [1] -
Zhang, H.; Liu, Q.; Li, J.; Qu, D.-H. Org. Lett. 2013, 15, 338–341. doi:10.1021/ol3032686
Return to citation in text: [1]
| 1. | Saha, S.; Stoddart, J. F. Chem. Soc. Rev. 2007, 36, 77–92. doi:10.1039/B607187B |
| 2. | Dasgupta, S.; Wu, J.-S. Chem. Sci. 2012, 3, 425–432. doi:10.1039/C1SC00613D |
| 3. | Qu, D.-H.; Tian, H. Chem. Sci. 2011, 2, 1011–1015. doi:10.1039/c0sc00653j |
| 4. | Zhu, L.-L.; Yan, H.; Nguyen, K. T.; Tian, H.; Zhao, Y.-L. Chem. Commun. 2012, 48, 4290–4292. doi:10.1039/c2cc17114g |
| 5. | Dzyuba, E. V.; Baytekin, B.; Sattler, D.; Schalley, C. A. Eur. J. Org. Chem. 2012, 1171–1178. doi:10.1002/ejoc.201101231 |
| 6. | Rao, S.-J.; Zhang, Q.; Mei, J.; Ye, X.-H.; Gao, C.; Wang, Q.-C.; Qu, D.-H.; Tian, H. Chem. Sci. 2017, 8, 6777–6783. doi:10.1039/C7SC03232C |
| 7. | Gao, C.; Luan, Z.-L.; Zhang, Q.; Rao, S.-J.; Qu, D.-H.; Tian, H. Org. Lett. 2017, 19, 3931–3934. doi:10.1021/acs.orglett.7b01853 |
| 13. | Roh, S.-G.; Park, K.-M.; Park, G.-J.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Angew. Chem., Int. Ed. 1999, 38, 637–641. doi:10.1002/(SICI)1521-3773(19990301)38:5<637::AID-ANIE637>3.0.CO;2-4 |
| 14. | Kim, K. Chem. Soc. Rev. 2002, 31, 96–107. doi:10.1039/a900939f |
| 15. | Chang, C.-F.; Chuang, C.-J.; Lai, C.-C.; Liu, Y.-H.; Peng, S.-M.; Chiu, S.-H. Angew. Chem., Int. Ed. 2012, 51, 10094–10098. doi:10.1002/anie.201205498 |
| 37. | Zhang, H.; Liu, Q.; Li, J.; Qu, D.-H. Org. Lett. 2013, 15, 338–341. doi:10.1021/ol3032686 |
| 11. | Goujon, A.; Du, G.; Moulin, E.; Fuks, G.; Maaloum, M.; Buhler, E.; Giuseppone, N. Angew. Chem., Int. Ed. 2016, 55, 703–707. doi:10.1002/anie.201509813 |
| 12. | Goujon, A.; Lang, T.; Mariani, G.; Moulin, E.; Fuks, G.; Raya, J.; Buhler, E.; Giuseppone, N. J. Am. Chem. Soc. 2017, 139, 14825–14828. doi:10.1021/jacs.7b06710 |
| 9. | Badjić, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F. Science 2004, 303, 1845–1849. doi:10.1126/science.1094791 |
| 10. | Badjić, J. D.; Ronconi, C. M.; Stoddart, J. F.; Balzani, V.; Silvi, S.; Credi, A. J. Am. Chem. Soc. 2006, 128, 1489–1499. doi:10.1021/ja0543954 |
| 35. | Li, F.-F.; Li, Y.-J.; Zhou, Z.-C.; Lv, S.-X.; Deng, Q.-R.; Xu, X.; Yin, L.-C. ACS Appl. Mater. Interfaces 2017, 9, 23586–23601. doi:10.1021/acsami.7b08534 |
| 8. | Hartlieb, K. J.; Blackburn, A. K.; Schneebeli, S. T.; Forgan, R. S.; Sarjeant, A. A.; Stern, C. L.; Cao, D.; Stoddart, J. F. Chem. Sci. 2014, 5, 90–100. doi:10.1039/C3SC52106K |
| 36. | Candrill, S. J.; Grubbs, R. H.; Lanari, D.; Leung, K. C.-F.; Nelson, A.; Poulin-Kerstien, K. G.; Smidt, S. P.; Stoddart, J. F.; Tirrell, D. A. Org. Lett. 2005, 7, 4213–4216. doi:10.1021/ol051599g |
| 23. | Raymo, F. M.; Stoddart, J. F. Chem. Rev. 1999, 99, 1643–1664. doi:10.1021/cr970081q |
| 24. | Balzani, V.; Credi, A.; Silvi, S.; Venturi, M. Chem. Soc. Rev. 2006, 35, 1135–1149. doi:10.1039/b517102b |
| 25. | Tian, H.; Wang, Q.-C. Chem. Soc. Rev. 2006, 35, 361–374. doi:10.1039/b512178g |
| 26. | Champin, B.; Mobian, P.; Sauvage, J.-P. Chem. Soc. Rev. 2007, 36, 358–366. doi:10.1039/B604484K |
| 27. | Crowley, J. D.; Goldup, S. M.; Lee, A.-L.; Leigh, D. A.; McBurney, R. T. Chem. Soc. Rev. 2009, 38, 1530–1541. doi:10.1039/b804243h |
| 28. | Hänni, K. D.; Leigh, D. A. Chem. Soc. Rev. 2010, 39, 1240–1251. doi:10.1039/B901974J |
| 29. | Zhu, L.-L.; Yan, H.; Wang, X.-J.; Zhao, Y.-L. J. Org. Chem. 2012, 77, 10168–10175. doi:10.1021/jo301807y |
| 30. | Cui, J.-S.; Ba, Q.-K.; Ke, H.; Valkonen, A.; Rissanen, K.; Jiang, W. Angew. Chem., Int. Ed. 2018, 57, 7809–7814. doi:10.1002/anie.201803349 |
| 31. | Gao, C.; Luan, Z.-L.; Zhang, Q.; Yang, S.; Rao, S.-J.; Qu, D.-H.; Tian, H. Org. Lett. 2017, 19, 1618–1621. doi:10.1021/acs.orglett.7b00393 |
| 33. | Jiang, Y.; Guo, J.-B.; Chen, C.-F. Org. Lett. 2010, 12, 4248–4251. doi:10.1021/ol101920b |
| 21. | Brunsveld, L.; Folmer, B. J. B.; Meijer, E. W.; Sijbesma, R. P. Chem. Rev. 2001, 101, 4071–4098. doi:10.1021/cr990125q |
| 22. | Cao, Z.-Q.; Miao, Q.; Zhang, Q.; Li, H.; Qu, D.-H.; Tian, H. Chem. Commun. 2015, 51, 4973–4976. doi:10.1039/C4CC09976A |
| 34. | Kawakami, H.; Toma, K. Diamide type gelling agent. Jpn. Kokai Tokkyo Koho JP2004262809A, Sept 24, 2004. |
| 19. | Cheng, C.; McGonigal, P. R.; Schneebeli, S. T.; Li, H.; Vermeulen, N. A.; Ke, C.; Stoddart, J. F. Nat. Nanotechnol. 2015, 10, 547–553. doi:10.1038/nnano.2015.96 |
| 20. | Meng, Z.; Xiang, J.-F.; Chen, C.-F. J. Am. Chem. Soc. 2016, 138, 5652–5658. doi:10.1021/jacs.6b01852 |
| 16. | von Delius, M.; Geertsema, E. M.; Leigh, D. A. Nat. Chem. 2009, 2, 96–101. doi:10.1038/nchem.481 |
| 17. | Lewandowski, B.; De Bo, G.; Ward, J. W.; Papmeyer, M.; Kuschel, S.; Aldegunde, M. J.; Gramlich, P. M. E.; Heckmann, D.; Goldup, S. M.; D’Souza, D. M.; Fernandes, A. E.; Leigh, D. A. Science 2013, 339, 189–193. doi:10.1126/science.1229753 |
| 18. | Qu, D.-H.; Tian, H. Chem. Sci. 2013, 4, 3031–3035. doi:10.1039/c3sc51160j |
| 32. | Guidry, E. N.; Li, J.; Stoddart, J. F.; Grubbs, R. H. J. Am. Chem. Soc. 2007, 129, 8944–8945. doi:10.1021/ja0725100 |
© 2018 Zheng et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)