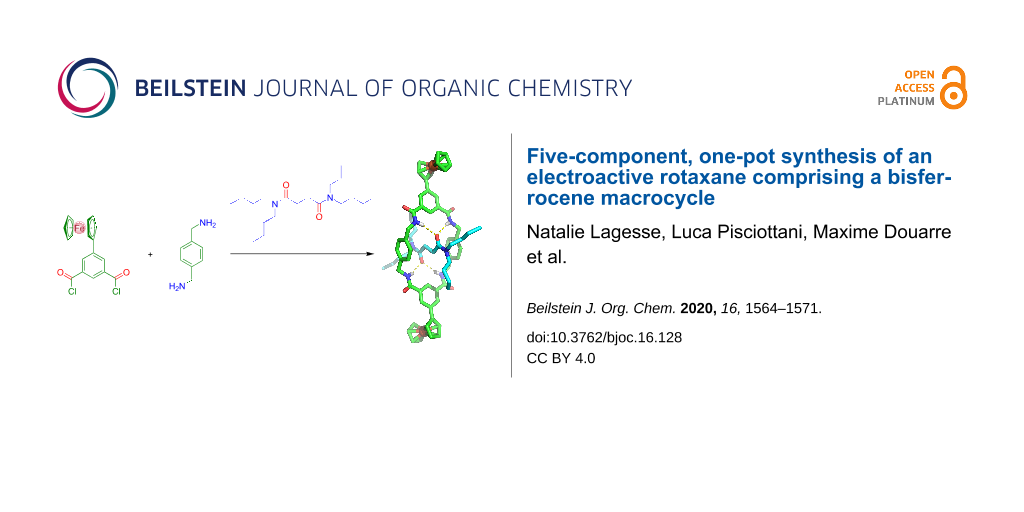
Abstract
The templated clipping of a ferrocene-grafted isophthalic acid derivative to encircle a hydrogen-bonding axle through the reaction with 1,4-bis(aminomethyl)benzene is described. The constituent electroactive macrocycle of the resultant [2]rotaxane is a homologue of the versatile benchmark tetraamide variant developed by Leigh and co-workers. The relative templating effect of different hydrogen-bonding motifs in rotaxane and pseudorotaxane generation is compared, with yields varying from 0 to 41%. The electrochemical properties and single crystal X-ray structure of a doubly ferrocene-decorated [2]rotaxane are further reported.

Graphical Abstract
Introduction
The development of interlocked molecules with tailored properties allowed the preparation of molecular machines able to perform several functions as artificial molecular switches [1]. The template-directed synthesis of such sophisticated catenane and rotaxane molecular architectures allowed the expansion of chemical diversity and properties. Among these architectures, electroactive rotaxanes have been described to act as stimuli-responsive molecular “shuttles” with potential applications to prepare nanoscale devices for computing and biomimetic engineering [2,3]. The highly efficient rotaxane formation developed by Leigh allowed the generation of a tetraamide macrocycle on a fumaramide or succinamide thread in high yields. This methodology consisted of a 4-component macrocyclization reaction, templated by the thread to obtain the corresponding interlocked molecule (Figure 1a) [4,5]. This class of macrocycle has proved extremely versatile, having given rise to a wealth of functional architectures [6-14]. In this context, we report the synthesis of a rotaxane, where a “clipping” reaction generates a tetraamide macrocycle with two peripheral ferrocene moieties on a preformed thread (Figure 1b). The resulting versatile and easily accessible electroactive macrocycle is anticipated to prove a valuable component for the construction of novel redox-active supramolecular systems, such as rotaxanes with strongly binding H-bonding templating sites, or indeed juxtaposed into existing functional architectures and benchmark variants.
![[1860-5397-16-128-1]](/bjoc/content/figures/1860-5397-16-128-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Non-functionalized rotaxanes previously described in the literature. (b) The redox-active rotaxane developed in this work.
Figure 1: (a) Non-functionalized rotaxanes previously described in the literature. (b) The redox-active rotax...
Results and Discussion
Synthesis
Our first approach to the synthesis of macrocycle 2 was carried out using N,N'-dihexyl-1,4-butanediamide as the template, as represented in Figure 2. While the formation of some macrocyclic product was identified by 1H NMR and MS, it could not be completely separated from impurities (<29% yield after chromatographic purification). The isolated material had low solubility, which is likely due to self-aggregation via complementary amide hydrogen bonds. This prompted us to use a thread with bulky stopper groups to prepare the corresponding rotaxane compounds, which would increase the solubility by the formation of intramolecular macrocycle-thread hydrogen bonds, thereby reducing self-aggregation. To this end, a five-component clipping strategy was adopted using different tetrabutylsuccinamide threads (Figure 2) with varying hydrogen-bond basicity (amides > esters) [4,15]. Threads containing an ester group were selected as esters could potentially be hydrolysed post-clipping to allow isolation of the macrocycle. The diamide/bisamide thread had flexible stoppers, which may enable dethreading by slippage in a polar solvent as previously observed [5,16].
![[1860-5397-16-128-2]](/bjoc/content/figures/1860-5397-16-128-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesis of the redox-active rotaxanes 1 and macrocycle 2.
Figure 2: Synthesis of the redox-active rotaxanes 1 and macrocycle 2.
When the reaction was performed with the double ester template 4c, it was not possible to identify any proton signals corresponding to the rotaxane in the crude 1H NMR (see Supporting Information File 1). The ester-amide template 4b also proved inefficient in rotaxane formation, indeed only weak signals of the rotaxane were observed in the crude 1H NMR of the reaction mixture (<5% yield) and the product could not be isolated. Then, a more efficient template was employed with a double amide bonding moiety and the reaction of tetrabutylsuccinamide thread 4a and p-xylylenediamine (6) and ferrocene isophthaloyl chloride 5 yielded the corresponding rotaxane 1a in 41% yield. Unlike the free ferrocene macrocycle, the ferrocene rotaxane 1a was soluble in CDCl3 suggesting a complementary macrocycle-thread hydrogen-bonding interaction. While the rotaxane 1a was insoluble at room temperature in DMSO-d6, heating at 160 °C dissolved the rotaxane. Macrocycle dethreading in DMSO-d6 at 160 °C was monitored by 1H NMR (Figures S12 and S13 in Supporting Information File 1). After 1 h of heating at 160 °C, the integration of the NMR signals showed circa 58% of rotaxane 1a, along with free macrocycle 2 and thread 4a. After 23 h of reaction, no starting material remained and only 34% of the macrocycle was present, as judged by proton resonances in the 1H NMR spectrum, which along with the reaction mixture turning dark brown and the presence of a black precipitate suggested degradation.
In order to evaluate the templating effect of the N,N’-dihexyl-1,4-butanediamide (3), molecular modelling studies of the 4-component macrocycle precursor were carried out. To reduce computational cost a model compound was considered, replacing the ferrocene by a hydrogen atom and replacing the hexyl chains of the template by methyl groups. Monte Carlo conformational searches were performed on the model structures (7 and 8) to obtain the most stable conformers (Figure 3) [17,18]. A comparison of the geometries of the most stable conformers indicated that the presence of the template produces a favourable preorganisation of the precursor, not only placing the amino and acid chloride groups in proximity, but also arranging the molecule in an appropriate reactive position favouring the intramolecular macrocyclization reaction (Figure 3b) [19]. Despite the theoretically favourable preorganization, the observed experimental templating effect was modest as the macrocyclization yield was only ≈29% of impure material. In contrast, in the absence of the template both reactive groups were not positioned in a geometry favouring macrocycle formation, which may prove conducive to oligomerization or catenane formation (Figure 3a).
![[1860-5397-16-128-3]](/bjoc/content/figures/1860-5397-16-128-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Most stable conformers obtained by Monte Carlo conformational search using model compounds. (a) Model macrocyclization reaction intermediate in the absence of a template; (b) model macrocyclization reaction intermediate in the presence of a template.
Figure 3: Most stable conformers obtained by Monte Carlo conformational search using model compounds. (a) Mod...
The 1H NMR spectrum of the rotaxane 1a in CDCl3 (Figure 4) showed a characteristic upfield shift (1.57 ppm) of the succinic protons i by the macrocycle phenyl rings providing evidence of the interlocked nature of the structure. An analysis of the signal shapes showed a broadening (peak width of 55 Hz in the 300 MHz 1H NMR) of the macrocycle CH2 protons g, suggesting a dynamic process of macrocycle motion around the thread close to the coalescence temperature, in contrast with the other rotaxane proton signals, which had a peak width in the range of 2–5 Hz in the 300 MHz 1H NMR spectrum. A similar macrocycle pirouetting behaviour was observed in related 1-station [2]rotaxane systems [20]. An analysis of the thread signals in the 1H NMR of the rotaxane showed a non-equivalency of both butyl chains indicating a slow rotation of the amide bond in the NMR timescale (Figure 4). One of these butyl chains is in close proximity to the macrocycle aromatic rings producing a shielding of the signals (j, k, l, and m).
![[1860-5397-16-128-4]](/bjoc/content/figures/1860-5397-16-128-4.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 4: 1H NMR spectrum (300 MHz) of rotaxane 1a (top) and thread 4a (bottom) in CDCl3 (a designation of the signals is described in Figure 2).
Figure 4: 1H NMR spectrum (300 MHz) of rotaxane 1a (top) and thread 4a (bottom) in CDCl3 (a designation of th...
Further evidence of the interlocked nature of rotaxane 1a could be obtained from the 1H,1H-ROESY NMR spectrum. Multiple through-space cross-coupling correlation between the thread and the macrocycle protons were observed between the mechanically-bonded macrocycle and the thread (Figure 5). Additional proof of the mechanical bond was provided by 1H DOSY NMR showing the same diffusion for thread and macrocycle signals with a diffusion coefficient of −9.33 m2/s and a hydrodynamic radius of 8.7 Å (see Figure S4 in Supporting Information File 1).
![[1860-5397-16-128-5]](/bjoc/content/figures/1860-5397-16-128-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: 1H,1H-ROESY NMR spectrum (600 MHz) of the rotaxane 1a in CDCl3.
Figure 5: 1H,1H-ROESY NMR spectrum (600 MHz) of the rotaxane 1a in CDCl3.
Electrochemistry
The ferrocene-containing rotaxane 1a was studied by cyclic voltammetry in CH2Cl2/CH3CN 1:5 (v/v) using a three-electrode cell, with a glassy carbon working electrode, a silver wire counter electrode and an Ag/AgCl (3 M KCl) reference electrode. Tetrabutylammonium hexafluorophosphate was used as the supporting electrolyte. Rotaxane 1a presents a reversible redox transition at E1/2 = 0.51 V (that corresponds to E1/2 = +0.47 V vs SCE), where E1/2 = (Epa + Epc)/2 (Figure 6). As only one oxidation wave was observed, this showed that the two metal centres were electronically decoupled, while the oxidation potential of ferrocene was E1/2 = +0.380 V vs SCE showing that rotaxane 1a retained the redox properties of the parent ferrocene [21,22]. Importantly, the full reversibility of the one electron oxidation–reduction process attested to the stability of the electroactive system.
![[1860-5397-16-128-6]](/bjoc/content/figures/1860-5397-16-128-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Cyclic voltammogram of ferrocene rotaxane 1a (0.67 mM) in CH2Cl2/CH3CN 1:5 (TBAPF6 0.10 M, scan rate = 10 mV/s).
Figure 6: Cyclic voltammogram of ferrocene rotaxane 1a (0.67 mM) in CH2Cl2/CH3CN 1:5 (TBAPF6 0.10 M, scan rat...
Solid-state X-ray structure
The solid-state structure of rotaxane 1a was determined by single crystal X-ray diffraction of crystals obtained by slow evaporation of a dichloromethane solution. The analysis of the X-ray solid-state structure of the rotaxane 1a showed a significant difference to Leigh’s rotaxane I [4]. Rotaxane I only presented two macrocycle–thread hydrogen bonds, with the other two macrocycle amides forming hydrogen bonds with the competitive crystallization solvent DMF. In contrast, rotaxane 1a presented four thread–macrocycle hydrogen bonds (Figure 7 and Table 1). The strength of the hydrogen bonds could be estimated by the donor–acceptor distances: 2.2–2.5 Å “strong, mostly covalent”, 2.5–3.2 Å “moderate, mostly electrostatic”, and 3.2–4.0 Å “weak, electrostatic” [23]. The corresponding stabilization energies were estimated to be in the ranges 40–14 kcal/mol, 15–4 kcal/mol, and < 4 kcal/mol, respectively. The macrocycle conformation in the rotaxane placed the two ferrocene groups with a metal centre-to-metal centre distance of 20.7 Å.
![[1860-5397-16-128-7]](/bjoc/content/figures/1860-5397-16-128-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Single crystal X-ray structures of (a) rotaxane 1a and (b) Leigh’s rotaxane I [4].
Figure 7: Single crystal X-ray structures of (a) rotaxane 1a and (b) Leigh’s rotaxane I [4].
Conclusion
A five-component, one-pot reaction gave rise to an electroactive [2]rotaxane, through four amidination reactions between two ferrocene-grafted isophthalic acid derivatives and two 1,4-diaminomethylbenzene molecules, in the presence of a suitable hydrogen-bonding template. Among the different templates employed, 4a proved to be the most successful giving rise to rotaxane 1a in 41% yield. Meanwhile, 4b and 4c gave little or no rotaxane formation. Cyclic voltammetry showed that the electroactive ferrocene units were electronically decoupled and retained the reversible oxidation properties of the parent compound, while the single crystal X-ray structure of a doubly ferrocene-decorated [2]rotaxane indicated four relatively long and seemingly weak NH···CO hydrogen bonds. As the [2]rotaxane could be efficiently constructed in an analogous clipping fashion to the versatile benchmark tetraamide variant developed by Leigh and co-workers, one might anticipate that this doubly ferrocene-decorated motif could be directly transposed into a range of functional electroactive interlocked architectures.
Experimental
Materials and methods
All chemicals and solvents were obtained from commercial sources and used without further purification. Dry solvents were obtained using a standard solvent purification system, except for dry CHCl3, which was purchased from Sigma-Aldrich. Mass spectra and NMR spectra were performed in the CESAMO analytical facilities (Bordeaux, France).
NMR: 1H and 13C NMR spectra were recorded using a Bruker Avance 300 or a Bruker Avance III 600 spectrometer. Chemical shifts were reported in ppm and referenced to the residual solvent peaks. NMR chemical shifts (δ) were reported in parts per million (ppm) and coupling constants J are given in hertz (Hz). Multiplicity for each signal is indicated as follows: s = singlet, br s = broad singlet, d = doublet, t = triplet, and m = multiplet.
Mass spectrometry: Field desorption spectra (FD) were recorded on an AccuTOF (JEOL) mass spectrometer using an FD emitter with an emitter voltage of 10 kV. The sample (1–2 µL) was deposited on a 13 μm emitter wire. Electrospray spectra (ESI) were recorded on a Qexactive (Thermo) mass spectrometer. The instrument was equipped with an ESI source and spectra were recorded in the positive mode. The spray voltage was maintained at 3200 V and the capillary temperature set to 320 °C. Samples were introduced by injection through a 20 µL loop into a 300 µL/min flow of methanol from the LC pump.
Electrochemistry: Cyclic voltammetry was carried out in a Metrohm Autolab PGSTAT302N potentiostat using a three-electrode cell, with a glassy carbon working electrode, a silver wire counter electrode, and an Ag/AgCl (3 M KCl) reference electrode. The rotaxane 1a (4.0 mg) was dissolved in dry dichloromethane (0.8 mL), sonicated for 5 min then diluted in acetonitrile (4 mL) to give a final concentration of 0.67 mM. Tetrabutylammonium hexafluorophosphate (0.10 M) was used as supporting electrolyte. Samples were degassed with argon for 5 min prior to measurement. In analogous conditions, ferrocene was used as internal reference.
Synthetic procedures
The tetrabutylsuccinamide thread was prepared using a literature procedure [5]. 5-Ferrocenylisophthalic chloride was prepared using literature procedures [24,25]. N,N’-Dihexyl-1,4-butanediamide was prepared using a literature procedure [26].
Synthesis of rotaxane 1a
A solution of p-xylylenediamine (6, 272 mg, 2 mmol) in chloroform (15 mL) and a solution of 5-ferrocenylisophthaloyl chloride (5, 774 mg, 2 mmol) in chloroform (15 mL) were simultaneously added with a syringe pump during 5 hours to a solution of tetrabutylsuccinamide thread 4a (85 mg, 0.25 mmol) and dry triethylamine (0.84 mL, 6 mmol) in chloroform (50 mL). The solution was stirred at room temperature overnight. The reaction mixture was filtered through Celite and the filtrate was washed with hydrochloric acid solution (1 M, 10 mL), saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL). The organic layer was dried over MgSO4, filtered and the solvent removed in vacuo. Purification by silica gel column chromatography (DCM/ethyl acetate 9:1 (v/v)) afforded 1a as beige solid (129 mg, 41% yield). 1H NMR (300 MHz, CDCl3) δ 8.66 (s, 2H, He), 8.46 (d, J = 1.3 Hz, 4H, Hd), 7.76 (t, J = 5.8 Hz, 4H, Hf), 7.15 (s, 8H, Hh + Hi), 5.45 (br s, 4H, Hg), 4.87 (t, J = 1.8 Hz, 4H, Ha or b), 4.44 (t, J = 1.7 Hz, 4H, Hb or a), 4.09 (s, 10H, Hc), 3.80 (br s, 4H, Hg), 3.31–3.20 (m, 4H, Hj’), 2.81–2.70 (m, 4H, Hj), 1.58–1.50 (m, 4H, Hk’), 1.40–1.30 (m, 4H, Hl’), 1.28–1.20 (m, 4H, Hk), 1.12 (s, 4H, Hi), 0.97 (t, J = 7.3 Hz, 6H, Hm’), 0.68–0.60 (m, 10H, Hl + Hm); 13C NMR (151 MHz, CDCl3) δ 172.72, 165.46, 141.76, 138.71, 133.46, 129.25, 128.96, 119.70, 83.34, 69.75, 69.67, 66.89, 48.24, 46.63, 43.25, 30.79, 30.27, 28.15, 20.25, 19.83, 13.89, 13.77; HRMS–FD (m/z): [M]+ calcd for C72H84N6O6Fe2, 1240.5151; found, 1240.5152.
Synthesis of thread 4c
Succinyl chloride (395 mg, 2.55 mmol) was added slowly to a cooled (0 °C) solution of 2,2-diphenylethanol (100.0 g, 5.1 mmol) in dry CH2Cl2 (50 mL). The brown solution was stirred at room temperature for 36 h under a N2 atmosphere. The solution was diluted with CH2Cl2 (50 mL) and an aqueous work up was performed with sat. NaHCO3 (2 × 100 mL), H2O (1 × 200 mL), and brine (1 × 100 mL). The organic extract was dried using Na2SO4, filtered, and the solvent was removed in vacuo. Finally, the product was purified by column chromatography using a solvent gradient (hexane/ethyl acetate 9:1 to 1:1, v/v) to obtain the pure product as white crystalline powder (622 mg, 51%). 1H NMR (300 MHz, CDCl3) δ 7.41–7.32 (m, 8H, Ph), 7.32–7.23 (m, 12H, Ph), 4.67 (d, J = 7.6 Hz, 4H, CH), 4.40 (t, J = 7.6 Hz, 2H, OCH2CH), 2.50 (s, 4H, CH2); 13C NMR (75 MHz, CDCl3) δ 172.1, 141.1, 128.6, 128.3, 126.9, 66.9, 49.8, 29.1; HRMS–ESI (m/z): [M + Na]+ calcd for C32H30O4Na, 501.20363; found, 501.20295.
Crystal structure determination
Single crystals of rotaxane 1a (C36H42FeN3O3) were obtained by slow evaporation of a dichloromethane solution. A suitable crystal was mounted on a cryoloop with paratone®-N oil on an AFC11 partial Chi goniometer. The data were collected at 120 K on Rigaku FRX® high flux rotating anode with a Pilatus 200K hybrid pixel detector. Using Olex2 [27], the structure was solved with the ShelXT [28] structure solution program using Intrinsic Phasing and refined with the ShelXL [29] refinement package using the full least-squares correlation matrix. The non-hydrogen atoms were located in successive difference Fourier maps and refined with anisotropic thermal parameters on F2. All hydrogen atoms were generated theoretically at the specific atoms positions and refined isotropically with fixed thermal factors.
Crystal data for rotaxane 1a C36H42FeN3O3 (M = 620.57 g/mol): triclinic, space group P-1 (no. 2), a = 9.5366(5) Å, b = 11.6279(7) Å, c = 14.9396(9) Å, α = 99.002(5), β = 108.001(5), γ = 90.007(4), V = 1554.09(16) Å3, Z = 2, T = 120 K, μ(Cu Kα) = 4.208 mm−1, Dcalc = 1.326 g/cm3, 14633 reflections measured (9.124° ≤ 2Θ ≤ 111.358°), 3928 unique (Rint = 0.0450, Rsigma = 0.0402) which were used in all calculations. The final R1 was 0.0454 (I > 2σ(I)) and wR2 was 0.1180 (all data). CCDC Deposition Number 1968472.
Molecular modelling
Monte Carlo conformational searches with the MMFF force field were carried out with the Spartan ‘18 software [30]. For each structure 2025 conformers were examined (see text) and the obtained conformers were ordered by relative energy to obtain the most stable one.
Acknowledgements
We thank the analytical facilities CESAMO (NMR and HRMS) of the Institut des Sciences Moléculaires, University of Bordeaux.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 796612. Equally, financial support from the Agence Nationale de la Recherche (project ANR-16-CE29-0011, PETIMIT), University of Bordeaux; CNRS and Ministère de la Recherche et de l’Enseignement Supérieur (L.P. and N.L.) is gratefully acknowledged.
References
-
Bruns, C. J.; Stoddart, J. F. The Nature of the Mechanical Bond: From Molecules to Machines; John Wiley and Sons: Hoboken, NJ, USA, 2017. doi:10.1002/9781119044123
Return to citation in text: [1] -
Bissell, R. A.; Córdova, E.; Kaifer, A. E.; Stoddart, J. F. Nature 1994, 369, 133–137. doi:10.1038/369133a0
Return to citation in text: [1] -
Credi, A.; Venturi, M. Electroactive rotaxanes and catenanes. In Electrochemistry of Functional Supramolecular Systems; Ceroni, P.; Credi, A.; Venturi, M., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2010; pp 377–424. doi:10.1002/9780470583463.ch13
Return to citation in text: [1] -
Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D. A.; Wong, J. K. Y.; Zerbetto, F. Angew. Chem., Int. Ed. 2003, 42, 2296–2300. doi:10.1002/anie.200250745
Return to citation in text: [1] [2] [3] [4] -
Martinez-Cuezva, A.; Rodrigues, L. V.; Navarro, C.; Carro-Guillen, F.; Buriol, L.; Frizzo, C. P.; Martins, M. A. P.; Alajarin, M.; Berna, J. J. Org. Chem. 2015, 80, 10049–10059. doi:10.1021/acs.joc.5b01651
Return to citation in text: [1] [2] [3] -
Leigh, D. A.; Marcos, V.; Nalbantoglu, T.; Vitorica-Yrezabal, I. J.; Yasar, F. T.; Zhu, X. J. Am. Chem. Soc. 2017, 139, 7104–7109. doi:10.1021/jacs.7b03307
Return to citation in text: [1] -
Panman, M. R.; Bakker, B. H.; den Uyl, D.; Kay, E. R.; Leigh, D. A.; Buma, W. J.; Brouwer, A. M.; Geenevasen, J. A. J.; Woutersen, S. Nat. Chem. 2013, 5, 929–934. doi:10.1038/nchem.1744
Return to citation in text: [1] -
Fernandes, A.; Viterisi, A.; Aucagne, V.; Leigh, D. A.; Papot, S. Chem. Commun. 2012, 48, 2083–2085. doi:10.1039/c2cc17458h
Return to citation in text: [1] -
Tron, A.; Pianet, I.; Martinez-Cuezva, A.; Tucker, J. H. R.; Pisciottani, L.; Alajarin, M.; Berna, J.; McClenaghan, N. D. Org. Lett. 2017, 19, 154–157. doi:10.1021/acs.orglett.6b03457
Return to citation in text: [1] -
Martinez-Cuezva, A.; Marin-Luna, M.; Alonso, D. A.; Ros-Ñiguez, D.; Alajarin, M.; Berna, J. Org. Lett. 2019, 21, 5192–5196. doi:10.1021/acs.orglett.9b01791
Return to citation in text: [1] -
Hernandez, J. V.; Kay, E. R.; Leigh, D. A. Science 2004, 306, 1532–1537. doi:10.1126/science.1103949
Return to citation in text: [1] -
Leigh, D. A.; Wong, J. K. Y.; Dehez, F.; Zerbetto, F. Nature 2003, 424, 174–179. doi:10.1038/nature01758
Return to citation in text: [1] -
Berná, J.; Leigh, D. A.; Lubomska, M.; Mendoza, S. M.; Pérez, E. M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Nat. Mater. 2005, 4, 704–710. doi:10.1038/nmat1455
Return to citation in text: [1] -
Brouwer, A. M.; Frochot, C.; Gatti, F. G.; Leigh, D. A.; Mottier, L.; Paolucci, F.; Roffia, S.; Wurpel, G. W. H. Science 2001, 291, 2124–2128. doi:10.1126/science.1057886
Return to citation in text: [1] -
Gatti, F. G.; Leigh, D. A.; Nepogodiev, S. A.; Slawin, A. M. Z.; Teat, S. J.; Wong, J. K. Y. J. Am. Chem. Soc. 2001, 123, 5983–5989. doi:10.1021/ja001697r
Return to citation in text: [1] -
Martinez-Cuezva, A.; Morales, F.; Marley, G. R.; Lopez-Lopez, A.; Martinez-Costa, J. C.; Bautista, D.; Alajarin, M.; Berna, J. Eur. J. Org. Chem. 2019, 3480–3488. doi:10.1002/ejoc.201900073
Return to citation in text: [1] -
Martí-Centelles, V.; Burguete, M. I.; Luis, S. V. J. Org. Chem. 2016, 81, 2143–2147. doi:10.1021/acs.joc.5b02676
Return to citation in text: [1] -
Martí-Centelles, V.; Burguete, M. I.; Cativiela, C.; Luis, S. V. J. Org. Chem. 2014, 79, 559–570. doi:10.1021/jo4022309
Return to citation in text: [1] -
Martí-Centelles, V.; Pandey, M. D.; Burguete, M. I.; Luis, S. V. Chem. Rev. 2015, 115, 8736–8834. doi:10.1021/acs.chemrev.5b00056
Return to citation in text: [1] -
Berná, J.; Alajarín, M.; Martínez-Espín, J. S.; Buriol, L.; Martins, M. A. P.; Orenes, R.-Á. Chem. Commun. 2012, 48, 5677–5679. doi:10.1039/c2cc32092d
Return to citation in text: [1] -
Pavlishchuk, V. V.; Addison, A. W. Inorg. Chim. Acta 2000, 298, 97–102. doi:10.1016/s0020-1693(99)00407-7
Return to citation in text: [1] -
Pisciottani, L.; Douarre, M.; Bibal, B.; Davies, C.; Roberts, H.; Kauffmann, B.; Horswell, S. L.; Tucker, J. H. R.; McClenaghan, N. D. Supramol. Chem. 2018, 30, 869–875. doi:10.1080/10610278.2018.1426856
Return to citation in text: [1] -
Jeffrey, G. A. An introduction to hydrogen bonding; Oxford University Press: New York, Oxford, 1997.
Return to citation in text: [1] -
Evans, N. H.; Rahman, H.; Leontiev, A. V.; Greenham, N. D.; Orlowski, G. A.; Zeng, Q.; Jacobs, R. M. J.; Serpell, C. J.; Kilah, N. L.; Davis, J. J.; Beer, P. D. Chem. Sci. 2012, 3, 1080–1089. doi:10.1039/c2sc00909a
Return to citation in text: [1] -
Liu, W.; Zheng, H.; Du, L.; Chen, B.; Song, M. Z. Anorg. Allg. Chem. 2010, 636, 236–241. doi:10.1002/zaac.200900270
Return to citation in text: [1] -
Dellinger, J. A.; George, G. D.; Roberts, C. W. J. Polym. Sci., Polym. Lett. Ed. 1978, 16, 357–365. doi:10.1002/pol.1978.130160708
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Spartan 18, build 1.3.0; Wavefunction Inc., 2019.
Return to citation in text: [1]
| 29. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 27. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 28. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 1. | Bruns, C. J.; Stoddart, J. F. The Nature of the Mechanical Bond: From Molecules to Machines; John Wiley and Sons: Hoboken, NJ, USA, 2017. doi:10.1002/9781119044123 |
| 4. | Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D. A.; Wong, J. K. Y.; Zerbetto, F. Angew. Chem., Int. Ed. 2003, 42, 2296–2300. doi:10.1002/anie.200250745 |
| 15. | Gatti, F. G.; Leigh, D. A.; Nepogodiev, S. A.; Slawin, A. M. Z.; Teat, S. J.; Wong, J. K. Y. J. Am. Chem. Soc. 2001, 123, 5983–5989. doi:10.1021/ja001697r |
| 24. | Evans, N. H.; Rahman, H.; Leontiev, A. V.; Greenham, N. D.; Orlowski, G. A.; Zeng, Q.; Jacobs, R. M. J.; Serpell, C. J.; Kilah, N. L.; Davis, J. J.; Beer, P. D. Chem. Sci. 2012, 3, 1080–1089. doi:10.1039/c2sc00909a |
| 25. | Liu, W.; Zheng, H.; Du, L.; Chen, B.; Song, M. Z. Anorg. Allg. Chem. 2010, 636, 236–241. doi:10.1002/zaac.200900270 |
| 6. | Leigh, D. A.; Marcos, V.; Nalbantoglu, T.; Vitorica-Yrezabal, I. J.; Yasar, F. T.; Zhu, X. J. Am. Chem. Soc. 2017, 139, 7104–7109. doi:10.1021/jacs.7b03307 |
| 7. | Panman, M. R.; Bakker, B. H.; den Uyl, D.; Kay, E. R.; Leigh, D. A.; Buma, W. J.; Brouwer, A. M.; Geenevasen, J. A. J.; Woutersen, S. Nat. Chem. 2013, 5, 929–934. doi:10.1038/nchem.1744 |
| 8. | Fernandes, A.; Viterisi, A.; Aucagne, V.; Leigh, D. A.; Papot, S. Chem. Commun. 2012, 48, 2083–2085. doi:10.1039/c2cc17458h |
| 9. | Tron, A.; Pianet, I.; Martinez-Cuezva, A.; Tucker, J. H. R.; Pisciottani, L.; Alajarin, M.; Berna, J.; McClenaghan, N. D. Org. Lett. 2017, 19, 154–157. doi:10.1021/acs.orglett.6b03457 |
| 10. | Martinez-Cuezva, A.; Marin-Luna, M.; Alonso, D. A.; Ros-Ñiguez, D.; Alajarin, M.; Berna, J. Org. Lett. 2019, 21, 5192–5196. doi:10.1021/acs.orglett.9b01791 |
| 11. | Hernandez, J. V.; Kay, E. R.; Leigh, D. A. Science 2004, 306, 1532–1537. doi:10.1126/science.1103949 |
| 12. | Leigh, D. A.; Wong, J. K. Y.; Dehez, F.; Zerbetto, F. Nature 2003, 424, 174–179. doi:10.1038/nature01758 |
| 13. | Berná, J.; Leigh, D. A.; Lubomska, M.; Mendoza, S. M.; Pérez, E. M.; Rudolf, P.; Teobaldi, G.; Zerbetto, F. Nat. Mater. 2005, 4, 704–710. doi:10.1038/nmat1455 |
| 14. | Brouwer, A. M.; Frochot, C.; Gatti, F. G.; Leigh, D. A.; Mottier, L.; Paolucci, F.; Roffia, S.; Wurpel, G. W. H. Science 2001, 291, 2124–2128. doi:10.1126/science.1057886 |
| 26. | Dellinger, J. A.; George, G. D.; Roberts, C. W. J. Polym. Sci., Polym. Lett. Ed. 1978, 16, 357–365. doi:10.1002/pol.1978.130160708 |
| 4. | Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D. A.; Wong, J. K. Y.; Zerbetto, F. Angew. Chem., Int. Ed. 2003, 42, 2296–2300. doi:10.1002/anie.200250745 |
| 5. | Martinez-Cuezva, A.; Rodrigues, L. V.; Navarro, C.; Carro-Guillen, F.; Buriol, L.; Frizzo, C. P.; Martins, M. A. P.; Alajarin, M.; Berna, J. J. Org. Chem. 2015, 80, 10049–10059. doi:10.1021/acs.joc.5b01651 |
| 4. | Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D. A.; Wong, J. K. Y.; Zerbetto, F. Angew. Chem., Int. Ed. 2003, 42, 2296–2300. doi:10.1002/anie.200250745 |
| 2. | Bissell, R. A.; Córdova, E.; Kaifer, A. E.; Stoddart, J. F. Nature 1994, 369, 133–137. doi:10.1038/369133a0 |
| 3. | Credi, A.; Venturi, M. Electroactive rotaxanes and catenanes. In Electrochemistry of Functional Supramolecular Systems; Ceroni, P.; Credi, A.; Venturi, M., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2010; pp 377–424. doi:10.1002/9780470583463.ch13 |
| 5. | Martinez-Cuezva, A.; Rodrigues, L. V.; Navarro, C.; Carro-Guillen, F.; Buriol, L.; Frizzo, C. P.; Martins, M. A. P.; Alajarin, M.; Berna, J. J. Org. Chem. 2015, 80, 10049–10059. doi:10.1021/acs.joc.5b01651 |
| 20. | Berná, J.; Alajarín, M.; Martínez-Espín, J. S.; Buriol, L.; Martins, M. A. P.; Orenes, R.-Á. Chem. Commun. 2012, 48, 5677–5679. doi:10.1039/c2cc32092d |
| 4. | Altieri, A.; Bottari, G.; Dehez, F.; Leigh, D. A.; Wong, J. K. Y.; Zerbetto, F. Angew. Chem., Int. Ed. 2003, 42, 2296–2300. doi:10.1002/anie.200250745 |
| 19. | Martí-Centelles, V.; Pandey, M. D.; Burguete, M. I.; Luis, S. V. Chem. Rev. 2015, 115, 8736–8834. doi:10.1021/acs.chemrev.5b00056 |
| 23. | Jeffrey, G. A. An introduction to hydrogen bonding; Oxford University Press: New York, Oxford, 1997. |
| 17. | Martí-Centelles, V.; Burguete, M. I.; Luis, S. V. J. Org. Chem. 2016, 81, 2143–2147. doi:10.1021/acs.joc.5b02676 |
| 18. | Martí-Centelles, V.; Burguete, M. I.; Cativiela, C.; Luis, S. V. J. Org. Chem. 2014, 79, 559–570. doi:10.1021/jo4022309 |
| 5. | Martinez-Cuezva, A.; Rodrigues, L. V.; Navarro, C.; Carro-Guillen, F.; Buriol, L.; Frizzo, C. P.; Martins, M. A. P.; Alajarin, M.; Berna, J. J. Org. Chem. 2015, 80, 10049–10059. doi:10.1021/acs.joc.5b01651 |
| 16. | Martinez-Cuezva, A.; Morales, F.; Marley, G. R.; Lopez-Lopez, A.; Martinez-Costa, J. C.; Bautista, D.; Alajarin, M.; Berna, J. Eur. J. Org. Chem. 2019, 3480–3488. doi:10.1002/ejoc.201900073 |
| 21. | Pavlishchuk, V. V.; Addison, A. W. Inorg. Chim. Acta 2000, 298, 97–102. doi:10.1016/s0020-1693(99)00407-7 |
| 22. | Pisciottani, L.; Douarre, M.; Bibal, B.; Davies, C.; Roberts, H.; Kauffmann, B.; Horswell, S. L.; Tucker, J. H. R.; McClenaghan, N. D. Supramol. Chem. 2018, 30, 869–875. doi:10.1080/10610278.2018.1426856 |
© 2020 Lagesse et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)