Abstract
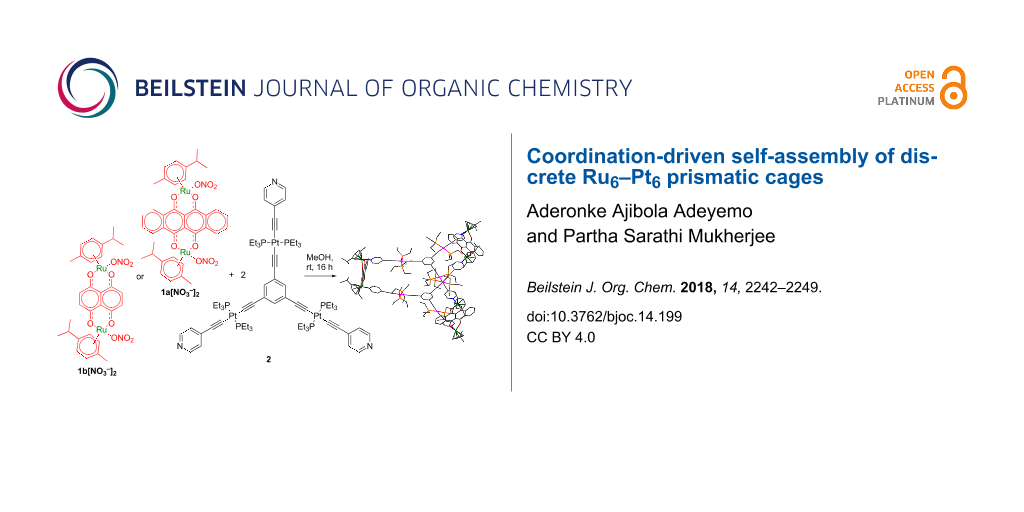
The coordination-driven self-assembly of two new Ru6–Pt6 hexanuclear trigonal prismatic cages comprising arene–ruthenium(II) clips (1a(NO3)2 and 1b(NO3)2) and a tritopic platinum(II) metalloligand 2 has been performed in methanol at room temperature. The [3 + 2] hexanuclear cages 3a and 3b were isolated in good yields and characterized by well-known spectroscopic techniques including multinuclear NMR, mass spectrometry, UV–vis and infrared studies. Geometry optimization revealed the shapes and sizes of these hexanuclear prismatic cages. The combination of ruthenium and platinum metal center in a one-pot self-assembly reaction showcases the construction of aesthetically elegant heterometallic structures in supramolecular chemistry leading to the formation of a single major product.

Graphical Abstract
Introduction
Coordination-driven self-assembly of discrete architectures has evolved as a unique protocol to construct elegant supramolecular architectures of different shapes, sizes and functionalities over the last two decades [1-23]. These 2D and 3D-supramolecular architectures mostly comprise pure organic ligands as electron-rich donors and transition metals as electron-deficient acceptors. Diverse functionalities embedded in these homometallic architectures have found useful applications in chemical sensing [24-35], catalysis [11,36-46], drug delivery [47-51] and host–guest chemistry [52-56] among others. The cardinal prerequisites to obtain these self-assembled supramolecular architectures include stoichiometry and conformational complementarity on the binding sites of the building blocks [57-61]. However, the use of a single metal and a single organic ligand design may limit the structural diversity as well as the functionality of homometallic supramolecular architectures. In the last few years, enormous efforts have been channeled towards multicomponent self-assembly involving the construction of sophisticated heterobimetallic supramolecular architectures in a one-pot reaction and their functional properties are currently being explored [55,62-70]. The incorporation of two different metal centers in a supramolecular architecture can impart different functional properties arising from each of the metals and/or the organic components which is quite interesting.
The metalloligand synthetic approach has been efficiently used to achieve the construction of heterobimetallic supramolecular architectures in order to minimize the formation of two independent homometallic architectures or a mixture of products [71-78]. The metalloligand is a kinetically stable coordination complex with a metal center and one or more appended donor site(s) which can further coordinate to another metal center [79-83]. Such metalloligands are predesigned and encrypted with the desired functional properties before they are used in self-assembly reactions so as to induce the desired functional properties into the final supramolecular architecture [84-92]. Hence, the metalloligand becomes the electron-rich building block which offers structural rigidity while the second metal is the electron-acceptor building block. The facile self-assembly of 2D-heterobimetallic supramolecular architectures are well reported in the literature [24,93,94], however, 3D-heterobimetallic systems are still less explored [95-97].
Dinuclear arene–ruthenium(II) acceptor clips/building blocks have been extensively utilized in supramolecular chemistry because of their rigid directionality toward electron-rich donors due to their restricted coordination sites as a result of the fixed position of the p-cymene moiety [24,93,98-103]. Our group and others have contributed substantially to the chemistry of self-assembled homometallic ruthenium architectures and their applications [100,102-108]. In broadening this research scope, herein, we describe the coordination-driven self-assembly of two new Ru–Pt heterometallic prismatic cages 3a and 3b obtained from the reaction of two arene–ruthenium(II) clips 1a and 1b and tritopic platinum(II) metalloligand 2 in methanol/chloroform mixture in 3:2 ratio (Scheme 1). Both cages were fully characterized by 1H, 31P, 195Pt, 1H,1H COSY, DOSY NMR, electrospray ionization mass spectrometry, and UV–vis analysis. Further structural insights were revealed by computational studies.
![[1860-5397-14-199-i1]](/bjoc/content/inline/1860-5397-14-199-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Self-assembly of the heterometallic prismatic cages.
Scheme 1: Self-assembly of the heterometallic prismatic cages.
Results and Discussion
The triplatinum metalloligand 2 was synthesized through a four-step reaction involving a Sonogashira coupling (Scheme 2) and the crude product was purified by column chromatography to obtain 2 as a yellow powder. The three intermediates A, B and C were also characterized by 1H, 31P, 195Pt and 13C NMR analyses (see Supporting Information File 1, Figures S1–S8). The metalloligand 2 is highly soluble in dichloromethane and chloroform but only partially soluble in methanol and acetonitrile. The 1H, 31P, 195Pt and 13C NMR experiments (Supporting Information File 1, Figures S9–S12) and electrospray ionization mass spectrometry (ESIMS, Supporting Information File 1, Figure S13) of metalloligand 2 evidenced the formation of a pure compound. The 1H NMR spectrum of 2 revealed two doublets and a singlet in the downfield region (8.38–6.99 ppm) corresponding to the pyridyl protons and the central phenyl protons, respectively, while the methylene and methyl protons in the upfield region of the spectrum are observed between 2.16–1.18 ppm (Supporting Information File 1, Figure S9). The 31P NMR spectrum of 2 gave a singlet peak at 11.18 ppm shifting downfield after coordination with the ethynylpyridine moiety, while the singlet 195Pt peak remained almost the same with the precursor compound C (Supporting Information File 1, Figure S12). The mass spectrum of 2 shows a [2 + H]+ peak at m/z 1748.59 (Supporting Information File 1, Figure S13) which is in good agreement with the calculated value of 1748.71 based on the C69H105N3P6Pt3 molecular formula.
![[1860-5397-14-199-i2]](/bjoc/content/inline/1860-5397-14-199-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the platinum metalloligand 2.
Scheme 2: Synthesis of the platinum metalloligand 2.
Self-assembly and characterization of the heterometallic cages
The treatment of the dichloride analogues of 1a and 1b with 2.1 equivalents of silver nitrate in methanol at room temperature for 3 hours gave the dinuclear arene–ruthenium(II) clips 1a,1b(NO3)2. As represented in Scheme 1, the self-assembly reactions of methanolic solutions of 1a,1b(NO3)2 and methanolic solution of the triplatinum metalloligand 2 at room temperature yielded the trigonal prismatic cages 3a and 3b. The heterometallic prismatic cages were isolated as nitrate complexes in good yields. The isolated cages are soluble in methanol, acetonitrile, acetone, nitromethane, dimethyl sulfoxide and partially soluble in chloroform and dichloromethane. The formation of these cages was ascertained by multinuclear NMR experiments and ESIMS analyses.
An upfield shift was observed in the pyridyl protons of cage 3b as compared to the free triplatinum metalloligand 2 in their 1H NMR spectra while cage 3a exhibited a downfield shift in the pyridyl protons (Figure 1 and Supporting Information File 1, Figure S14). This chemical shift is due to the coordination of the pyridyl nitrogen atom to the ruthenium metal center while the upfield shift is due to the shielding effect of the methyl/methylene protons of the p-cymene moiety. In both cages studied, the aromatic protons of the p-cymene moiety in 3a and 3b were slightly shifted downfield while the isopropyl and methyl protons of the p-cymene moiety in all the cages remained almost unchanged as compared to the arene–ruthenium(II) clips 1a,1b(NO3)2. Additionally, the protons of naphthacenedione in 3a and naphthaquinone in 3b were not considerably shifted (Figure 1 and Supporting Information File 1, Figure S14). The appearance of a singlet peak in the 31P NMR evidenced the formation of a single product and the fact that the phosphorus moieties are in the same chemical environment (Supporting Information File 1, Figures S15 and S16). The same observation is recorded for the 195Pt NMR analyses of all the heterometallic cages (Supporting Information File 1, Figures S17 and S18).
![[1860-5397-14-199-1]](/bjoc/content/figures/1860-5397-14-199-1.png?scale=2.0&max-width=1024&background=FFFFFF)
The DOSY NMR experiments also confirmed the formation of a single product in all the cages with the hydrodynamic radii (rH) of the heterometallic prismatic cages calculated from the Stokes–Einstein equation using the diffusion coefficients (D) obtained from the DOSY NMR experiments. The obtained values of D from the experiment are −9.631 log (m2 s−1) for 3a and −9.567 log (m2 s−1) for 3b, respectively. The calculated hydrodynamic radii (rH) of 3a and 3b are 15.57 Å, and 13.43 Å, respectively (Supporting Information File 1, Figures S19 and S20). The 1H,1H COSY NMR spectra also showed the correlation between the protons of the arene–ruthenium(II) clips as well as the correlation between the protons within the metalloligand (Supporting Information File 1, Figures S19 and S20).
The vibrational symmetrical stretching frequency of the coordinated carbonyl groups (νC–O) in the dinuclear arene–ruthenium(II) clips 1a and 1b was found at 1536.16 cm−1 for 3a and 1528.93 cm−1 for 3b in the infrared spectra of the heterometallic cages while the vibrational symmetrical stretching frequency bands of =C–Haromatic showed strong stretching bands at 3074.07 cm−1 for 3a and 3064.78 cm−1 for 3b, respectively. Additionally, the stretching bands at 543.67 cm−1 for 3a and 545.44 cm−1 for 3b correspond to the νRu–O symmetrical stretching frequency (Supporting Information File 1, Figure S21).
The UV–vis absorption spectra recorded in methanol at room temperature show intense bands at λmax = 544, 514, 334, 290, 205 nm for 3a and λmax = 698, 644, 339, 204 nm for 3b. The intense bands at 335 nm and 291 nm for 2 correspond to charge-transfer transitions, which shift slightly to shorter wavelengths in the spectra of the heterometallic prismatic cages. The peaks in the ranges of 514–698 nm and 204–339 nm can be assigned to intramolecular and intermolecular π–π* transitions and metal-to-ligand charge transfer (MLCT) transitions associated with capped p-cymene ruthenium cap, respectively. A hypochromic shift (decrease in absorption intensity) was also observed in the spectra of the heterometallic prismatic cages as compared to the metalloligand probably as a result of coordination of electron-rich metalloligand to the electron-deficient ruthenium center (Figure 2).
![[1860-5397-14-199-2]](/bjoc/content/figures/1860-5397-14-199-2.png?scale=1.3&max-width=1024&background=FFFFFF)
Figure 2: UV–vis spectra of the metalloligand 2 and heterometallic prismatic cages 3a and 3b in methanol (1.0 × 10−5 M) at 298 K.
Figure 2: UV–vis spectra of the metalloligand 2 and heterometallic prismatic cages 3a and 3b in methanol (1.0...
Mass spectrometry experiments also established the formation of the heterometallic prismatic cages in which all the cages maintain good stability. The ESIMS analysis of the [3 + 2] self-assembled heterometallic cages showed multiply charged fragmented ions for 3a at m/z = 1473.83 [3a(NO3−)2]4+, 1166.86 [3a(NO3−)]5+, 961.88 [3a]6+; 3b at m/z = 1398.79 [3b(NO3−)2]4+, 1106.64 [3b(NO3−)]5+, 911.87 [3b]6+ and all peaks are well-resolved isotopically and matched with the theoretical isotopic distribution patterns (Figure 3 and Supporting Information File 1, Figure S22).
![[1860-5397-14-199-3]](/bjoc/content/figures/1860-5397-14-199-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ESIMS spectrum of 3a in methanol. Inset: experimentally observed isotopic distribution patterns of the charged fragments.
Figure 3: ESIMS spectrum of 3a in methanol. Inset: experimentally observed isotopic distribution patterns of ...
Geometry optimization of the heterometallic cages
All efforts to obtain single crystals of the prismatic cages were unsuccessful so far. Thus the structures of 3a and 3b were optimized to get insights into their structural features. The tritopic platinum(II) metalloligand was optimized using the B3LYP method while the heterometallic cage structures 3a and 3b were optimized with the semiempirical method using the PM6 basis set. The energy-minimized structures showed that cage 3a has a dimension of 8.414 Å × 26.321 Å × 26.755 Å while cage 3b has a dimension of 8.231 Å × 26.227 Å × 26.598 Å. The phenyl cores of the two triplatinum metalloligands are separated by a distance of 11.376 Å in 3a and 11.456 Å in 3b. The phenyl cores are slightly out-of-plane with regards to the pyridyl groups possibly as a result of steric influence upon metal–ligand coordination (Figure 4 and Supporting Information File 1, Figure S23).
![[1860-5397-14-199-4]](/bjoc/content/figures/1860-5397-14-199-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Energy-minimized structure of heterometallic trigonal prismatic cage 3a. Hydrogen atoms are omitted for the sake of clarity [Ru: green, Pt: pink, O: red, N: blue, P: orange, C: grey].
Figure 4: Energy-minimized structure of heterometallic trigonal prismatic cage 3a. Hydrogen atoms are omitted...
Conclusion
The metalloligand synthetic approach has been utilized to synthesize heterobimetallic trigonal prismatic coordination cages 3a and 3b through the one-pot coordination-driven self-assembly of dinuclear arene–ruthenium(II) clips and a tritopic platinum(II) metalloligand. The formations of the [3 + 2] trigonal prismatic Ru–Pt cages are confirmed by multinuclear NMR and ESIMS studies. The optimized structures of cages 3a and 3b showed large prismatic cages composed of twelve metal centers comprising six ruthenium(II) and six platinum(II) metal centers thus showcasing how two different metal components can be incorporated to form a single framework architecture through a one-pot self-assembly strategy.
Supporting Information
| Supporting Information File 1: Experimental procedures, multinuclear NMR spectra data, ESIMS data and infrared spectra of the hexanuclear trigonal prismatic cages. | ||
| Format: PDF | Size: 978.9 KB | Download |
References
-
Lehn, J.-M. Angew. Chem., Int. Ed. Engl. 1990, 29, 1304–1319. doi:10.1002/anie.199013041
Return to citation in text: [1] -
Lehn, J.-M. Science 1993, 260, 1762–1763. doi:10.1126/science.8511582
Return to citation in text: [1] -
Harris, K.; Fujita, D.; Fujita, M. Chem. Commun. 2013, 49, 6703–6712. doi:10.1039/c3cc43191f
Return to citation in text: [1] -
Saha, M. L.; Neogi, S.; Schmittel, M. Dalton Trans. 2014, 43, 3815–3834. doi:10.1039/C3DT53570C
Return to citation in text: [1] -
Leininger, S.; Olenyuk, B.; Stang, P. J. Chem. Rev. 2000, 100, 853–908. doi:10.1021/cr9601324
Return to citation in text: [1] -
Nehete, U. N.; Anantharaman, G.; Chandrasekhar, V.; Murugavel, R.; Walawalkar, M. G.; Roesky, H. W.; Vidovic, D.; Magull, J.; Samwer, K.; Sass, B. Angew. Chem., Int. Ed. 2004, 43, 3832–3835. doi:10.1002/anie.200453740
Return to citation in text: [1] -
Lifschitz, A. M.; Rosen, M. S.; McGuirk, C. M.; Mirkin, C. A. J. Am. Chem. Soc. 2015, 137, 7252–7261. doi:10.1021/jacs.5b01054
Return to citation in text: [1] -
Smulders, M. M. J.; Riddell, I. A.; Browne, C.; Nitschke, J. R. Chem. Soc. Rev. 2013, 42, 1728–1754. doi:10.1039/C2CS35254K
Return to citation in text: [1] -
Newkome, G. R.; Moorefield, C. N. Chem. Soc. Rev. 2015, 44, 3954–3967. doi:10.1039/C4CS00234B
Return to citation in text: [1] -
Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784–5814. doi:10.1021/cr100357h
Return to citation in text: [1] -
Brown, C. J.; Toste, F. D.; Bergman, R. G.; Raymond, K. N. Chem. Rev. 2015, 115, 3012–3035. doi:10.1021/cr4001226
Return to citation in text: [1] [2] -
Northrop, B. H.; Chercka, D.; Stang, P. J. Tetrahedron 2008, 64, 11495–11503. doi:10.1016/j.tet.2008.08.062
Return to citation in text: [1] -
Guillerm, V.; Kim, D.; Eubank, J. F.; Luebke, R.; Liu, X.; Adil, K.; Lah, M. S.; Eddaoudi, M. Chem. Soc. Rev. 2014, 43, 6141–6172. doi:10.1039/C4CS00135D
Return to citation in text: [1] -
Ward, M. D.; Raithby, P. R. Chem. Soc. Rev. 2013, 42, 1619–1636. doi:10.1039/C2CS35123D
Return to citation in text: [1] -
Amouri, H.; Desmarets, C.; Moussa, J. Chem. Rev. 2012, 112, 2015–2041. doi:10.1021/cr200345v
Return to citation in text: [1] -
Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571–3579. doi:10.1021/ar500335a
Return to citation in text: [1] -
Stock, N.; Biswas, S. Chem. Rev. 2012, 112, 933–969. doi:10.1021/cr200304e
Return to citation in text: [1] -
Wang, W.; Wang, Y.-X.; Yang, H.-B. Chem. Soc. Rev. 2016, 45, 2656–2693. doi:10.1039/C5CS00301F
Return to citation in text: [1] -
Liu, Y.; Perez, L.; Mettry, M.; Easley, C. J.; Hooley, R. J.; Zhong, W. J. Am. Chem. Soc. 2016, 138, 10746–10749. doi:10.1021/jacs.6b05897
Return to citation in text: [1] -
Chen, L.-J.; Chen, S.; Qin, Y.; Xu, L.; Yin, G.-Q.; Zhu, J.-L.; Zhu, F.-F.; Zheng, W.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2018, 140, 5049–5052. doi:10.1021/jacs.8b02386
Return to citation in text: [1] -
Huang, C.-B.; Xu, L.; Zhu, J.-L.; Wang, Y.-X.; Sun, B.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2017, 139, 9459–9462. doi:10.1021/jacs.7b04659
Return to citation in text: [1] -
Jiang, B.; Chen, L.-J.; Zhang, Y.; Tan, H.-W.; Xu, L.; Yang, H.-B. Chin. Chem. Lett. 2016, 27, 607–612. doi:10.1016/j.cclet.2016.03.017
Return to citation in text: [1] -
Fu, T.-F.; Ao, L.; Gao, Z.-C.; Zhang, X.-L.; Wang, F. Chin. Chem. Lett. 2016, 27, 1147–1154. doi:10.1016/j.cclet.2016.06.054
Return to citation in text: [1] -
Vajpayee, V.; Kim, H.; Mishra, A.; Mukherjee, P. S.; Stang, P. J.; Lee, M. H.; Kim, H. K.; Chi, K.-W. Dalton Trans. 2011, 40, 3112–3115. doi:10.1039/c0dt01481h
Return to citation in text: [1] [2] [3] -
Mal, P.; Breiner, B.; Rissanen, K.; Nitschke, J. R. Science 2009, 324, 1697–1699. doi:10.1126/science.1175313
Return to citation in text: [1] -
Riddell, I. A.; Smulders, M. M. J.; Clegg, J. K.; Nitschke, J. R. Chem. Commun. 2011, 47, 457–459. doi:10.1039/C0CC02573A
Return to citation in text: [1] -
Yan, X.; Wang, H.; Hauke, C. E.; Cook, T. R.; Wang, M.; Saha, M. L.; Zhou, Z.; Zhang, M.; Li, X.; Huang, F.; Stang, P. J. J. Am. Chem. Soc. 2015, 137, 15276–15286. doi:10.1021/jacs.5b10130
Return to citation in text: [1] -
Kumar, A.; Sun, S.-S.; Lees, A. J. Coord. Chem. Rev. 2008, 252, 922–939. doi:10.1016/j.ccr.2007.07.023
Return to citation in text: [1] -
Chen, L.-J.; Ren, Y.-Y.; Wu, N.-W.; Sun, B.; Ma, J.-Q.; Zhang, L.; Tan, H.; Liu, M.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2015, 137, 11725–11735. doi:10.1021/jacs.5b06565
Return to citation in text: [1] -
Shanmugaraju, S.; Bar, A. K.; Chi, K.-W.; Mukherjee, P. S. Organometallics 2010, 29, 2971–2980. doi:10.1021/om100202c
Return to citation in text: [1] -
Kang, S. O.; Llinares, J. M.; Day, V. W.; Bowman-James, K. Chem. Soc. Rev. 2010, 39, 3980–4003. doi:10.1039/c0cs00083c
Return to citation in text: [1] -
Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373
Return to citation in text: [1] -
Samanta, D.; Mukherjee, P. S. Chem. Commun. 2013, 49, 4307–4309. doi:10.1039/c2cc37377g
Return to citation in text: [1] -
Biros, S. M.; Bergman, R. G.; Raymond, K. N. J. Am. Chem. Soc. 2007, 129, 12094–12095. doi:10.1021/ja075236i
Return to citation in text: [1] -
Samanta, D.; Mukherjee, S.; Patil, Y. P.; Mukherjee, P. S. Chem. – Eur. J. 2012, 18, 12322–12329. doi:10.1002/chem.201201679
Return to citation in text: [1] -
Jans, A. C. H.; Gómez-Suárez, A.; Nolan, S. P.; Reek, J. N. H. Chem. – Eur. J. 2016, 22, 14836–14839. doi:10.1002/chem.201603162
Return to citation in text: [1] -
Schneider, M. W.; Oppel, I. M.; Mastalerz, M. Chem. – Eur. J. 2012, 18, 4156–4160. doi:10.1002/chem.201200032
Return to citation in text: [1] -
Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340
Return to citation in text: [1] -
Howlader, P.; Das, P.; Zangrando, E.; Mukherjee, P. S. J. Am. Chem. Soc. 2016, 138, 1668–1676. doi:10.1021/jacs.5b12237
Return to citation in text: [1] -
Murase, T.; Nishijima, Y.; Fujita, M. J. Am. Chem. Soc. 2012, 134, 162–164. doi:10.1021/ja210068f
Return to citation in text: [1] -
Salles, A. G., Jr.; Zarra, S.; Turner, R. M.; Nitschke, J. R. J. Am. Chem. Soc. 2013, 135, 19143–19146. doi:10.1021/ja412235e
Return to citation in text: [1] -
Zhao, C.; Toste, F. D.; Raymond, K. N.; Bergman, R. G. J. Am. Chem. Soc. 2014, 136, 14409–14412. doi:10.1021/ja508799p
Return to citation in text: [1] -
Kaphan, D. M.; Levin, M. D.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. Science 2015, 350, 1235–1238. doi:10.1126/science.aad3087
Return to citation in text: [1] -
Neelakandan, P. P.; Jiménez, A.; Thoburn, J. D.; Nitschke, J. R. Angew. Chem., Int. Ed. 2015, 54, 14378–14382. doi:10.1002/anie.201507045
Return to citation in text: [1] -
Hasegawa, S.; Horike, S.; Matsuda, R.; Furukawa, S.; Mochizuki, K.; Kinoshita, Y.; Kitagawa, S. J. Am. Chem. Soc. 2007, 129, 2607–2614. doi:10.1021/ja067374y
Return to citation in text: [1] -
Shultz, A. M.; Farha, O. K.; Hupp, J. T.; Nguyen, S. T. J. Am. Chem. Soc. 2009, 131, 4204–4205. doi:10.1021/ja900203f
Return to citation in text: [1] -
Therrien, B.; Süss-Fink, G.; Govindaswamy, P.; Renfrew, A. K.; Dyson, P. J. Angew. Chem., Int. Ed. 2008, 47, 3773–3776. doi:10.1002/anie.200800186
Return to citation in text: [1] -
Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/C2SC00899H
Return to citation in text: [1] -
Samanta, S. K.; Moncelet, D.; Briken, V.; Isaacs, L. J. Am. Chem. Soc. 2016, 138, 14488–14496. doi:10.1021/jacs.6b09504
Return to citation in text: [1] -
Nguyen, T. D.; Liu, Y.; Saha, S.; Leung, K. C.-F.; Stoddart, J. F.; Zink, J. I. J. Am. Chem. Soc. 2007, 129, 626–634. doi:10.1021/ja065485r
Return to citation in text: [1] -
Ma, Z.; Moulton, B. Coord. Chem. Rev. 2011, 255, 1623–1641. doi:10.1016/j.ccr.2011.01.031
Return to citation in text: [1] -
Singh, N.; Jo, J.-H.; Song, Y. H.; Kim, H.; Kim, D.; Lah, M. S.; Chi, K.-W. Chem. Commun. 2015, 51, 4492–4495. doi:10.1039/C4CC09494H
Return to citation in text: [1] -
Schmitt, F.; Freudenreich, J.; Barry, N. P. E.; Juillerat-Jeanneret, L.; Süss-Fink, G.; Therrien, B. J. Am. Chem. Soc. 2012, 134, 754–757. doi:10.1021/ja207784t
Return to citation in text: [1] -
Lewis, J. E. M.; Elliott, A. B. S.; McAdam, C. J.; Gordon, K. C.; Crowley, J. D. Chem. Sci. 2014, 5, 1833–1843. doi:10.1039/C4SC00434E
Return to citation in text: [1] -
Li, H.; Han, Y.-F.; Lin, Y.-J.; Guo, Z.-W.; Jin, G.-X. J. Am. Chem. Soc. 2014, 136, 2982–2985. doi:10.1021/ja412667t
Return to citation in text: [1] [2] -
Barry, N. P. E.; Therrien, B. Eur. J. Inorg. Chem. 2009, 4695–4700. doi:10.1002/ejic.200900649
Return to citation in text: [1] -
Rowan, A. E.; Nolte, R. J. M. Angew. Chem., Int. Ed. 1998, 37, 63–68. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<63::AID-ANIE63>3.0.CO;2-4
Return to citation in text: [1] -
Saha, M. L.; Pramanik, S.; Schmittel, M. Chem. Commun. 2012, 48, 9459–9461. doi:10.1039/c2cc35036j
Return to citation in text: [1] -
Zheng, Y.-R.; Zhao, Z.; Wang, M.; Ghosh, K.; Pollock, J. B.; Cook, T. R.; Stang, P. J. J. Am. Chem. Soc. 2010, 132, 16873–16882. doi:10.1021/ja106251f
Return to citation in text: [1] -
Neogi, S.; Lorenz, Y.; Engeser, M.; Samanta, D.; Schmittel, M. Inorg. Chem. 2013, 52, 6975–6984. doi:10.1021/ic400328d
Return to citation in text: [1] -
Stephenson, A.; Ward, M. D. Dalton Trans. 2011, 40, 10360–10369. doi:10.1039/c1dt10263j
Return to citation in text: [1] -
Park, Y. J.; Ryu, J. Y.; Begum, H.; Lee, M. H.; Stang, P. J.; Lee, J. J. Am. Chem. Soc. 2015, 137, 5863–5866. doi:10.1021/jacs.5b01253
Return to citation in text: [1] -
Govender, P.; Lemmerhirt, H.; Hutton, A. T.; Therrien, B.; Bednarski, P. J.; Smith, G. S. Organometallics 2014, 33, 5535–5545. doi:10.1021/om500809g
Return to citation in text: [1] -
Shen, X.-Y.; Zhang, Y.-Y.; Zhang, L.; Lin, Y.-J.; Lin, G.-X. Chem. – Eur. J. 2015, 21, 16975–16981. doi:10.1002/chem.201502387
Return to citation in text: [1] -
Singh, N.; Jang, S.; Jo, J.-H.; Kim, D. H.; Park, D. W.; Kim, I.; Kim, H.; Kang, S. C.; Chi, K.-W. Chem. – Eur. J. 2016, 22, 16157–16164. doi:10.1002/chem.201603521
Return to citation in text: [1] -
Sepehrpour, H.; Saha, M. L.; Stang, P. J. J. Am. Chem. Soc. 2017, 139, 2553–2556. doi:10.1021/jacs.6b11860
Return to citation in text: [1] -
Wise, M. D.; Holstein, J. J.; Pattison, P.; Besnard, C.; Solari, E.; Scopelliti, R.; Bricogne, G.; Severin, K. Chem. Sci. 2015, 6, 1004–1010. doi:10.1039/C4SC03046J
Return to citation in text: [1] -
Li, K.; Zhang, L.-Y.; Yan, C.; Wei, S.-C.; Pan, M.; Zhang, L.; Su, C.-Y. J. Am. Chem. Soc. 2014, 136, 4456–4459. doi:10.1021/ja410044r
Return to citation in text: [1] -
Lu, X.; Li, X.; Guo, K.; Xie, T.-Z.; Moorefield, C. N.; Wesdemiotis, C.; Newkome, G. R. J. Am. Chem. Soc. 2014, 136, 18149–18155. doi:10.1021/ja511341z
Return to citation in text: [1] -
Kobayashi, A.; Suzuki, Y.; Ohba, T.; Noro, S.-i.; Chang, H.-C.; Kato, M. Inorg. Chem. 2011, 50, 2061–2063. doi:10.1021/ic102361d
Return to citation in text: [1] -
Kent, C. A.; Mehl, B. P.; Ma, L.; Papanikolas, J. M.; Meyer, T. J.; Lin, W. J. Am. Chem. Soc. 2010, 132, 12767–12769. doi:10.1021/ja102804s
Return to citation in text: [1] -
Clemente-León, M.; Coronado, E.; Gómez-García, C. J.; Soriano-Portillo, A. Inorg. Chem. 2006, 45, 5653–5660. doi:10.1021/ic060442x
Return to citation in text: [1] -
Kumar, G.; Gupta, R. Chem. Soc. Rev. 2013, 42, 9403–9453. doi:10.1039/c3cs60255a
Return to citation in text: [1] -
Halper, S. R.; Do, L.; Stork, J. R.; Cohen, S. M. J. Am. Chem. Soc. 2006, 128, 15255–15268. doi:10.1021/ja0645483
Return to citation in text: [1] -
Wei, P.; Yan, X.; Huang, F. Chem. Soc. Rev. 2015, 44, 815–832. doi:10.1039/C4CS00327F
Return to citation in text: [1] -
Chandler, B. D.; Cramb, D. T.; Shimizu, G. K. H. J. Am. Chem. Soc. 2006, 128, 10403–10412. doi:10.1021/ja060666e
Return to citation in text: [1] -
Kitaura, R.; Onoyama, G.; Sakamoto, H.; Matsuda, R.; Noro, S.-i.; Kitagawa, S. Angew. Chem., Int. Ed. 2004, 43, 2684–2687. doi:10.1002/anie.200352596
Return to citation in text: [1] -
Kent, C. A.; Liu, D.; Meyer, T. J.; Lin, W. J. Am. Chem. Soc. 2012, 134, 3991–3994. doi:10.1021/ja211271m
Return to citation in text: [1] -
Xu, L.; Wang, Y.-X.; Chen, L.-J.; Yang, H.-B. Chem. Soc. Rev. 2015, 44, 2148–2167. doi:10.1039/C5CS00022J
Return to citation in text: [1] -
Alvarado, L.; Brewer, G.; Carpenter, E. E.; Viragh, C.; Zavalij, P. Y. Inorg. Chim. Acta 2010, 363, 817–822. doi:10.1016/j.ica.2009.12.005
Return to citation in text: [1] -
Li, Q.-J.; Zhao, G.-Z.; Chen, L.-J.; Tan, H.; Wang, C.-H.; Wang, D.-X.; Lehman, D. A.; Muddiman, D. C.; Yang, H.-B. Organometallics 2012, 31, 7241–7247. doi:10.1021/om3007932
Return to citation in text: [1] -
Reichel, F.; Clegg, J. K.; Gloe, K.; Gloe, K.; Weigand, J. J.; Reynolds, J. K.; Li, C.-G.; Aldrich-Wright, J. R.; Kepert, C. J.; Lindoy, L. F.; Yao, H.-C.; Li, F. Inorg. Chem. 2014, 53, 688–690. doi:10.1021/ic402686s
Return to citation in text: [1] -
Ou-Yang, J.-K.; Chen, L.-J.; Xu, L.; Wang, C.-H.; Yang, H.-B. Chin. Chem. Lett. 2013, 24, 471–474. doi:10.1016/j.cclet.2013.03.055
Return to citation in text: [1] -
Chen, B.; Liang, C.; Yang, J.; Contreras, D. S.; Clancy, Y. L.; Lobkovsky, E. B.; Yaghi, O. M.; Dai, S. Angew. Chem. 2006, 118, 1418–1421. doi:10.1002/ange.200502844
Return to citation in text: [1] -
Farrusseng, D.; Aguado, S.; Pinel, C. Angew. Chem., Int. Ed. 2009, 48, 7502–7513. doi:10.1002/anie.200806063
Return to citation in text: [1] -
Meek, S. T.; Greathouse, J. A.; Allendorf, M. D. Adv. Mater. 2011, 23, 249–267. doi:10.1002/adma.201002854
Return to citation in text: [1] -
Farha, O. K.; Hupp, J. T. Acc. Chem. Res. 2010, 43, 1166–1175. doi:10.1021/ar1000617
Return to citation in text: [1] -
Wu, C.-D.; Hu, A.; Zhang, L.; Lin, W. J. Am. Chem. Soc. 2005, 127, 8940–8941. doi:10.1021/ja052431t
Return to citation in text: [1] -
Corma, A.; García, H.; Llabrés i Xamena, F. X. Chem. Rev. 2010, 110, 4606–4655. doi:10.1021/cr9003924
Return to citation in text: [1] -
Dhakshinamoorthy, A.; Alvaro, M.; Corma, A.; Garcia, H. Dalton Trans. 2011, 40, 6344–6360. doi:10.1039/c1dt10354g
Return to citation in text: [1] -
Rowsell, J. L. C.; Yaghi, O. M. Angew. Chem., Int. Ed. 2005, 44, 4670–4679. doi:10.1002/anie.200462786
Return to citation in text: [1] -
Li, J.-R.; Sculley, J.; Zhou, H.-C. Chem. Rev. 2012, 112, 869–932. doi:10.1021/cr200190s
Return to citation in text: [1] -
Vajpayee, V.; Song, Y. H.; Lee, M. H.; Kim, H.; Wang, M.; Stang, P. J.; Chi, K.-W. Chem. – Eur. J. 2011, 17, 7837–7844. doi:10.1002/chem.201100242
Return to citation in text: [1] [2] -
Schmittel, M.; Mahata, K. Inorg. Chem. 2009, 48, 822–824. doi:10.1021/ic8021084
Return to citation in text: [1] -
Jiang, B.; Zhang, J.; Ma, J.-Q.; Zheng, W.; Chen, L.-J.; Sun, B.; Li, C.; Hu, B.-W.; Tan, H.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2016, 138, 738–741. doi:10.1021/jacs.5b11409
Return to citation in text: [1] -
Wang, L.; Chen, L.-J.; Ma, J.-Q.; Wang, C.-H.; Tan, H.; Huang, J.; Xiao, F.; Xu, L. J. Organomet. Chem. 2016, 823, 1–7. doi:10.1016/j.jorganchem.2016.09.001
Return to citation in text: [1] -
Adeyemo, A. A.; Shettar, A.; Bhat, I. A.; Kondaiah, P.; Mukherjee, P. S. Dalton Trans. 2018, 47, 8466–8475. doi:10.1039/C8DT00962G
Return to citation in text: [1] -
Gupta, G.; Das, A.; Ghate, N. B.; Kim, T.; Ryu, J. Y.; Lee, J.; Mandal, N.; Lee, C. Y. Chem. Commun. 2016, 52, 4274–4277. doi:10.1039/C6CC00046K
Return to citation in text: [1] -
Shanmugaraju, S.; Bar, A. K.; Mukherjee, P. S. Inorg. Chem. 2010, 49, 10235–10237. doi:10.1021/ic101823s
Return to citation in text: [1] -
Shanmugaraju, S.; Samanta, D.; Mukherjee, P. S. Beilstein J. Org. Chem. 2012, 8, 313–322. doi:10.3762/bjoc.8.34
Return to citation in text: [1] [2] -
Mirtschin, S.; Slabon-Turski, A.; Scopelliti, R.; Velders, A. H.; Severin, K. J. Am. Chem. Soc. 2010, 132, 14004–14005. doi:10.1021/ja1063789
Return to citation in text: [1] -
Vajpayee, V.; Song, Y. H.; Yang, Y. J.; Kang, S. C.; Cook, T. R.; Kim, D. W.; Lah, M. S.; Kim, I. S.; Wang, M.; Stang, P. J.; Chi, K.-W. Organometallics 2011, 30, 6482–6489. doi:10.1021/om200908c
Return to citation in text: [1] [2] -
Mishra, A.; Dubey, A.; Min, J. W.; Kim, H.; Stang, P. J.; Chi, K.-W. Chem. Commun. 2014, 50, 7542–7544. doi:10.1039/C4CC01991A
Return to citation in text: [1] [2] -
Barry, N. P. E.; Furrer, J.; Therrien, B. Helv. Chim. Acta 2010, 93, 1313–1328. doi:10.1002/hlca.200900422
Return to citation in text: [1] -
Yan, H.; Suss-Fink, G.; Neels, A.; Stoeckli-Evans, H. J. Chem. Soc., Dalton Trans. 1997, 4345–4350. doi:10.1039/A704658H
Return to citation in text: [1] -
Samanta, D.; Shanmugaraju, S.; Adeyemo, A. A.; Mukherjee, P. S. J. Organomet. Chem. 2014, 751, 703–710. doi:10.1016/j.jorganchem.2013.09.037
Return to citation in text: [1] -
Adeyemo, A. A.; Shanmugaraju, S.; Samanta, D.; Mukherjee, P. S. Inorg. Chim. Acta 2016, 440, 62–68. doi:10.1016/j.ica.2015.10.029
Return to citation in text: [1] -
Adeyemo, A. A.; Shettar, A.; Bhat, I. A.; Kondaiah, P.; Mukherjee, P. S. Inorg. Chem. 2017, 56, 608–617. doi:10.1021/acs.inorgchem.6b02488
Return to citation in text: [1]
| 1. | Lehn, J.-M. Angew. Chem., Int. Ed. Engl. 1990, 29, 1304–1319. doi:10.1002/anie.199013041 |
| 2. | Lehn, J.-M. Science 1993, 260, 1762–1763. doi:10.1126/science.8511582 |
| 3. | Harris, K.; Fujita, D.; Fujita, M. Chem. Commun. 2013, 49, 6703–6712. doi:10.1039/c3cc43191f |
| 4. | Saha, M. L.; Neogi, S.; Schmittel, M. Dalton Trans. 2014, 43, 3815–3834. doi:10.1039/C3DT53570C |
| 5. | Leininger, S.; Olenyuk, B.; Stang, P. J. Chem. Rev. 2000, 100, 853–908. doi:10.1021/cr9601324 |
| 6. | Nehete, U. N.; Anantharaman, G.; Chandrasekhar, V.; Murugavel, R.; Walawalkar, M. G.; Roesky, H. W.; Vidovic, D.; Magull, J.; Samwer, K.; Sass, B. Angew. Chem., Int. Ed. 2004, 43, 3832–3835. doi:10.1002/anie.200453740 |
| 7. | Lifschitz, A. M.; Rosen, M. S.; McGuirk, C. M.; Mirkin, C. A. J. Am. Chem. Soc. 2015, 137, 7252–7261. doi:10.1021/jacs.5b01054 |
| 8. | Smulders, M. M. J.; Riddell, I. A.; Browne, C.; Nitschke, J. R. Chem. Soc. Rev. 2013, 42, 1728–1754. doi:10.1039/C2CS35254K |
| 9. | Newkome, G. R.; Moorefield, C. N. Chem. Soc. Rev. 2015, 44, 3954–3967. doi:10.1039/C4CS00234B |
| 10. | Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784–5814. doi:10.1021/cr100357h |
| 11. | Brown, C. J.; Toste, F. D.; Bergman, R. G.; Raymond, K. N. Chem. Rev. 2015, 115, 3012–3035. doi:10.1021/cr4001226 |
| 12. | Northrop, B. H.; Chercka, D.; Stang, P. J. Tetrahedron 2008, 64, 11495–11503. doi:10.1016/j.tet.2008.08.062 |
| 13. | Guillerm, V.; Kim, D.; Eubank, J. F.; Luebke, R.; Liu, X.; Adil, K.; Lah, M. S.; Eddaoudi, M. Chem. Soc. Rev. 2014, 43, 6141–6172. doi:10.1039/C4CS00135D |
| 14. | Ward, M. D.; Raithby, P. R. Chem. Soc. Rev. 2013, 42, 1619–1636. doi:10.1039/C2CS35123D |
| 15. | Amouri, H.; Desmarets, C.; Moussa, J. Chem. Rev. 2012, 112, 2015–2041. doi:10.1021/cr200345v |
| 16. | Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571–3579. doi:10.1021/ar500335a |
| 17. | Stock, N.; Biswas, S. Chem. Rev. 2012, 112, 933–969. doi:10.1021/cr200304e |
| 18. | Wang, W.; Wang, Y.-X.; Yang, H.-B. Chem. Soc. Rev. 2016, 45, 2656–2693. doi:10.1039/C5CS00301F |
| 19. | Liu, Y.; Perez, L.; Mettry, M.; Easley, C. J.; Hooley, R. J.; Zhong, W. J. Am. Chem. Soc. 2016, 138, 10746–10749. doi:10.1021/jacs.6b05897 |
| 20. | Chen, L.-J.; Chen, S.; Qin, Y.; Xu, L.; Yin, G.-Q.; Zhu, J.-L.; Zhu, F.-F.; Zheng, W.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2018, 140, 5049–5052. doi:10.1021/jacs.8b02386 |
| 21. | Huang, C.-B.; Xu, L.; Zhu, J.-L.; Wang, Y.-X.; Sun, B.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2017, 139, 9459–9462. doi:10.1021/jacs.7b04659 |
| 22. | Jiang, B.; Chen, L.-J.; Zhang, Y.; Tan, H.-W.; Xu, L.; Yang, H.-B. Chin. Chem. Lett. 2016, 27, 607–612. doi:10.1016/j.cclet.2016.03.017 |
| 23. | Fu, T.-F.; Ao, L.; Gao, Z.-C.; Zhang, X.-L.; Wang, F. Chin. Chem. Lett. 2016, 27, 1147–1154. doi:10.1016/j.cclet.2016.06.054 |
| 52. | Singh, N.; Jo, J.-H.; Song, Y. H.; Kim, H.; Kim, D.; Lah, M. S.; Chi, K.-W. Chem. Commun. 2015, 51, 4492–4495. doi:10.1039/C4CC09494H |
| 53. | Schmitt, F.; Freudenreich, J.; Barry, N. P. E.; Juillerat-Jeanneret, L.; Süss-Fink, G.; Therrien, B. J. Am. Chem. Soc. 2012, 134, 754–757. doi:10.1021/ja207784t |
| 54. | Lewis, J. E. M.; Elliott, A. B. S.; McAdam, C. J.; Gordon, K. C.; Crowley, J. D. Chem. Sci. 2014, 5, 1833–1843. doi:10.1039/C4SC00434E |
| 55. | Li, H.; Han, Y.-F.; Lin, Y.-J.; Guo, Z.-W.; Jin, G.-X. J. Am. Chem. Soc. 2014, 136, 2982–2985. doi:10.1021/ja412667t |
| 56. | Barry, N. P. E.; Therrien, B. Eur. J. Inorg. Chem. 2009, 4695–4700. doi:10.1002/ejic.200900649 |
| 47. | Therrien, B.; Süss-Fink, G.; Govindaswamy, P.; Renfrew, A. K.; Dyson, P. J. Angew. Chem., Int. Ed. 2008, 47, 3773–3776. doi:10.1002/anie.200800186 |
| 48. | Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/C2SC00899H |
| 49. | Samanta, S. K.; Moncelet, D.; Briken, V.; Isaacs, L. J. Am. Chem. Soc. 2016, 138, 14488–14496. doi:10.1021/jacs.6b09504 |
| 50. | Nguyen, T. D.; Liu, Y.; Saha, S.; Leung, K. C.-F.; Stoddart, J. F.; Zink, J. I. J. Am. Chem. Soc. 2007, 129, 626–634. doi:10.1021/ja065485r |
| 51. | Ma, Z.; Moulton, B. Coord. Chem. Rev. 2011, 255, 1623–1641. doi:10.1016/j.ccr.2011.01.031 |
| 11. | Brown, C. J.; Toste, F. D.; Bergman, R. G.; Raymond, K. N. Chem. Rev. 2015, 115, 3012–3035. doi:10.1021/cr4001226 |
| 36. | Jans, A. C. H.; Gómez-Suárez, A.; Nolan, S. P.; Reek, J. N. H. Chem. – Eur. J. 2016, 22, 14836–14839. doi:10.1002/chem.201603162 |
| 37. | Schneider, M. W.; Oppel, I. M.; Mastalerz, M. Chem. – Eur. J. 2012, 18, 4156–4160. doi:10.1002/chem.201200032 |
| 38. | Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340 |
| 39. | Howlader, P.; Das, P.; Zangrando, E.; Mukherjee, P. S. J. Am. Chem. Soc. 2016, 138, 1668–1676. doi:10.1021/jacs.5b12237 |
| 40. | Murase, T.; Nishijima, Y.; Fujita, M. J. Am. Chem. Soc. 2012, 134, 162–164. doi:10.1021/ja210068f |
| 41. | Salles, A. G., Jr.; Zarra, S.; Turner, R. M.; Nitschke, J. R. J. Am. Chem. Soc. 2013, 135, 19143–19146. doi:10.1021/ja412235e |
| 42. | Zhao, C.; Toste, F. D.; Raymond, K. N.; Bergman, R. G. J. Am. Chem. Soc. 2014, 136, 14409–14412. doi:10.1021/ja508799p |
| 43. | Kaphan, D. M.; Levin, M. D.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. Science 2015, 350, 1235–1238. doi:10.1126/science.aad3087 |
| 44. | Neelakandan, P. P.; Jiménez, A.; Thoburn, J. D.; Nitschke, J. R. Angew. Chem., Int. Ed. 2015, 54, 14378–14382. doi:10.1002/anie.201507045 |
| 45. | Hasegawa, S.; Horike, S.; Matsuda, R.; Furukawa, S.; Mochizuki, K.; Kinoshita, Y.; Kitagawa, S. J. Am. Chem. Soc. 2007, 129, 2607–2614. doi:10.1021/ja067374y |
| 46. | Shultz, A. M.; Farha, O. K.; Hupp, J. T.; Nguyen, S. T. J. Am. Chem. Soc. 2009, 131, 4204–4205. doi:10.1021/ja900203f |
| 24. | Vajpayee, V.; Kim, H.; Mishra, A.; Mukherjee, P. S.; Stang, P. J.; Lee, M. H.; Kim, H. K.; Chi, K.-W. Dalton Trans. 2011, 40, 3112–3115. doi:10.1039/c0dt01481h |
| 93. | Vajpayee, V.; Song, Y. H.; Lee, M. H.; Kim, H.; Wang, M.; Stang, P. J.; Chi, K.-W. Chem. – Eur. J. 2011, 17, 7837–7844. doi:10.1002/chem.201100242 |
| 98. | Gupta, G.; Das, A.; Ghate, N. B.; Kim, T.; Ryu, J. Y.; Lee, J.; Mandal, N.; Lee, C. Y. Chem. Commun. 2016, 52, 4274–4277. doi:10.1039/C6CC00046K |
| 99. | Shanmugaraju, S.; Bar, A. K.; Mukherjee, P. S. Inorg. Chem. 2010, 49, 10235–10237. doi:10.1021/ic101823s |
| 100. | Shanmugaraju, S.; Samanta, D.; Mukherjee, P. S. Beilstein J. Org. Chem. 2012, 8, 313–322. doi:10.3762/bjoc.8.34 |
| 101. | Mirtschin, S.; Slabon-Turski, A.; Scopelliti, R.; Velders, A. H.; Severin, K. J. Am. Chem. Soc. 2010, 132, 14004–14005. doi:10.1021/ja1063789 |
| 102. | Vajpayee, V.; Song, Y. H.; Yang, Y. J.; Kang, S. C.; Cook, T. R.; Kim, D. W.; Lah, M. S.; Kim, I. S.; Wang, M.; Stang, P. J.; Chi, K.-W. Organometallics 2011, 30, 6482–6489. doi:10.1021/om200908c |
| 103. | Mishra, A.; Dubey, A.; Min, J. W.; Kim, H.; Stang, P. J.; Chi, K.-W. Chem. Commun. 2014, 50, 7542–7544. doi:10.1039/C4CC01991A |
| 24. | Vajpayee, V.; Kim, H.; Mishra, A.; Mukherjee, P. S.; Stang, P. J.; Lee, M. H.; Kim, H. K.; Chi, K.-W. Dalton Trans. 2011, 40, 3112–3115. doi:10.1039/c0dt01481h |
| 25. | Mal, P.; Breiner, B.; Rissanen, K.; Nitschke, J. R. Science 2009, 324, 1697–1699. doi:10.1126/science.1175313 |
| 26. | Riddell, I. A.; Smulders, M. M. J.; Clegg, J. K.; Nitschke, J. R. Chem. Commun. 2011, 47, 457–459. doi:10.1039/C0CC02573A |
| 27. | Yan, X.; Wang, H.; Hauke, C. E.; Cook, T. R.; Wang, M.; Saha, M. L.; Zhou, Z.; Zhang, M.; Li, X.; Huang, F.; Stang, P. J. J. Am. Chem. Soc. 2015, 137, 15276–15286. doi:10.1021/jacs.5b10130 |
| 28. | Kumar, A.; Sun, S.-S.; Lees, A. J. Coord. Chem. Rev. 2008, 252, 922–939. doi:10.1016/j.ccr.2007.07.023 |
| 29. | Chen, L.-J.; Ren, Y.-Y.; Wu, N.-W.; Sun, B.; Ma, J.-Q.; Zhang, L.; Tan, H.; Liu, M.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2015, 137, 11725–11735. doi:10.1021/jacs.5b06565 |
| 30. | Shanmugaraju, S.; Bar, A. K.; Chi, K.-W.; Mukherjee, P. S. Organometallics 2010, 29, 2971–2980. doi:10.1021/om100202c |
| 31. | Kang, S. O.; Llinares, J. M.; Day, V. W.; Bowman-James, K. Chem. Soc. Rev. 2010, 39, 3980–4003. doi:10.1039/c0cs00083c |
| 32. | Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373 |
| 33. | Samanta, D.; Mukherjee, P. S. Chem. Commun. 2013, 49, 4307–4309. doi:10.1039/c2cc37377g |
| 34. | Biros, S. M.; Bergman, R. G.; Raymond, K. N. J. Am. Chem. Soc. 2007, 129, 12094–12095. doi:10.1021/ja075236i |
| 35. | Samanta, D.; Mukherjee, S.; Patil, Y. P.; Mukherjee, P. S. Chem. – Eur. J. 2012, 18, 12322–12329. doi:10.1002/chem.201201679 |
| 100. | Shanmugaraju, S.; Samanta, D.; Mukherjee, P. S. Beilstein J. Org. Chem. 2012, 8, 313–322. doi:10.3762/bjoc.8.34 |
| 102. | Vajpayee, V.; Song, Y. H.; Yang, Y. J.; Kang, S. C.; Cook, T. R.; Kim, D. W.; Lah, M. S.; Kim, I. S.; Wang, M.; Stang, P. J.; Chi, K.-W. Organometallics 2011, 30, 6482–6489. doi:10.1021/om200908c |
| 103. | Mishra, A.; Dubey, A.; Min, J. W.; Kim, H.; Stang, P. J.; Chi, K.-W. Chem. Commun. 2014, 50, 7542–7544. doi:10.1039/C4CC01991A |
| 104. | Barry, N. P. E.; Furrer, J.; Therrien, B. Helv. Chim. Acta 2010, 93, 1313–1328. doi:10.1002/hlca.200900422 |
| 105. | Yan, H.; Suss-Fink, G.; Neels, A.; Stoeckli-Evans, H. J. Chem. Soc., Dalton Trans. 1997, 4345–4350. doi:10.1039/A704658H |
| 106. | Samanta, D.; Shanmugaraju, S.; Adeyemo, A. A.; Mukherjee, P. S. J. Organomet. Chem. 2014, 751, 703–710. doi:10.1016/j.jorganchem.2013.09.037 |
| 107. | Adeyemo, A. A.; Shanmugaraju, S.; Samanta, D.; Mukherjee, P. S. Inorg. Chim. Acta 2016, 440, 62–68. doi:10.1016/j.ica.2015.10.029 |
| 108. | Adeyemo, A. A.; Shettar, A.; Bhat, I. A.; Kondaiah, P.; Mukherjee, P. S. Inorg. Chem. 2017, 56, 608–617. doi:10.1021/acs.inorgchem.6b02488 |
| 79. | Xu, L.; Wang, Y.-X.; Chen, L.-J.; Yang, H.-B. Chem. Soc. Rev. 2015, 44, 2148–2167. doi:10.1039/C5CS00022J |
| 80. | Alvarado, L.; Brewer, G.; Carpenter, E. E.; Viragh, C.; Zavalij, P. Y. Inorg. Chim. Acta 2010, 363, 817–822. doi:10.1016/j.ica.2009.12.005 |
| 81. | Li, Q.-J.; Zhao, G.-Z.; Chen, L.-J.; Tan, H.; Wang, C.-H.; Wang, D.-X.; Lehman, D. A.; Muddiman, D. C.; Yang, H.-B. Organometallics 2012, 31, 7241–7247. doi:10.1021/om3007932 |
| 82. | Reichel, F.; Clegg, J. K.; Gloe, K.; Gloe, K.; Weigand, J. J.; Reynolds, J. K.; Li, C.-G.; Aldrich-Wright, J. R.; Kepert, C. J.; Lindoy, L. F.; Yao, H.-C.; Li, F. Inorg. Chem. 2014, 53, 688–690. doi:10.1021/ic402686s |
| 83. | Ou-Yang, J.-K.; Chen, L.-J.; Xu, L.; Wang, C.-H.; Yang, H.-B. Chin. Chem. Lett. 2013, 24, 471–474. doi:10.1016/j.cclet.2013.03.055 |
| 24. | Vajpayee, V.; Kim, H.; Mishra, A.; Mukherjee, P. S.; Stang, P. J.; Lee, M. H.; Kim, H. K.; Chi, K.-W. Dalton Trans. 2011, 40, 3112–3115. doi:10.1039/c0dt01481h |
| 93. | Vajpayee, V.; Song, Y. H.; Lee, M. H.; Kim, H.; Wang, M.; Stang, P. J.; Chi, K.-W. Chem. – Eur. J. 2011, 17, 7837–7844. doi:10.1002/chem.201100242 |
| 94. | Schmittel, M.; Mahata, K. Inorg. Chem. 2009, 48, 822–824. doi:10.1021/ic8021084 |
| 71. | Kent, C. A.; Mehl, B. P.; Ma, L.; Papanikolas, J. M.; Meyer, T. J.; Lin, W. J. Am. Chem. Soc. 2010, 132, 12767–12769. doi:10.1021/ja102804s |
| 72. | Clemente-León, M.; Coronado, E.; Gómez-García, C. J.; Soriano-Portillo, A. Inorg. Chem. 2006, 45, 5653–5660. doi:10.1021/ic060442x |
| 73. | Kumar, G.; Gupta, R. Chem. Soc. Rev. 2013, 42, 9403–9453. doi:10.1039/c3cs60255a |
| 74. | Halper, S. R.; Do, L.; Stork, J. R.; Cohen, S. M. J. Am. Chem. Soc. 2006, 128, 15255–15268. doi:10.1021/ja0645483 |
| 75. | Wei, P.; Yan, X.; Huang, F. Chem. Soc. Rev. 2015, 44, 815–832. doi:10.1039/C4CS00327F |
| 76. | Chandler, B. D.; Cramb, D. T.; Shimizu, G. K. H. J. Am. Chem. Soc. 2006, 128, 10403–10412. doi:10.1021/ja060666e |
| 77. | Kitaura, R.; Onoyama, G.; Sakamoto, H.; Matsuda, R.; Noro, S.-i.; Kitagawa, S. Angew. Chem., Int. Ed. 2004, 43, 2684–2687. doi:10.1002/anie.200352596 |
| 78. | Kent, C. A.; Liu, D.; Meyer, T. J.; Lin, W. J. Am. Chem. Soc. 2012, 134, 3991–3994. doi:10.1021/ja211271m |
| 95. | Jiang, B.; Zhang, J.; Ma, J.-Q.; Zheng, W.; Chen, L.-J.; Sun, B.; Li, C.; Hu, B.-W.; Tan, H.; Li, X.; Yang, H.-B. J. Am. Chem. Soc. 2016, 138, 738–741. doi:10.1021/jacs.5b11409 |
| 96. | Wang, L.; Chen, L.-J.; Ma, J.-Q.; Wang, C.-H.; Tan, H.; Huang, J.; Xiao, F.; Xu, L. J. Organomet. Chem. 2016, 823, 1–7. doi:10.1016/j.jorganchem.2016.09.001 |
| 97. | Adeyemo, A. A.; Shettar, A.; Bhat, I. A.; Kondaiah, P.; Mukherjee, P. S. Dalton Trans. 2018, 47, 8466–8475. doi:10.1039/C8DT00962G |
| 55. | Li, H.; Han, Y.-F.; Lin, Y.-J.; Guo, Z.-W.; Jin, G.-X. J. Am. Chem. Soc. 2014, 136, 2982–2985. doi:10.1021/ja412667t |
| 62. | Park, Y. J.; Ryu, J. Y.; Begum, H.; Lee, M. H.; Stang, P. J.; Lee, J. J. Am. Chem. Soc. 2015, 137, 5863–5866. doi:10.1021/jacs.5b01253 |
| 63. | Govender, P.; Lemmerhirt, H.; Hutton, A. T.; Therrien, B.; Bednarski, P. J.; Smith, G. S. Organometallics 2014, 33, 5535–5545. doi:10.1021/om500809g |
| 64. | Shen, X.-Y.; Zhang, Y.-Y.; Zhang, L.; Lin, Y.-J.; Lin, G.-X. Chem. – Eur. J. 2015, 21, 16975–16981. doi:10.1002/chem.201502387 |
| 65. | Singh, N.; Jang, S.; Jo, J.-H.; Kim, D. H.; Park, D. W.; Kim, I.; Kim, H.; Kang, S. C.; Chi, K.-W. Chem. – Eur. J. 2016, 22, 16157–16164. doi:10.1002/chem.201603521 |
| 66. | Sepehrpour, H.; Saha, M. L.; Stang, P. J. J. Am. Chem. Soc. 2017, 139, 2553–2556. doi:10.1021/jacs.6b11860 |
| 67. | Wise, M. D.; Holstein, J. J.; Pattison, P.; Besnard, C.; Solari, E.; Scopelliti, R.; Bricogne, G.; Severin, K. Chem. Sci. 2015, 6, 1004–1010. doi:10.1039/C4SC03046J |
| 68. | Li, K.; Zhang, L.-Y.; Yan, C.; Wei, S.-C.; Pan, M.; Zhang, L.; Su, C.-Y. J. Am. Chem. Soc. 2014, 136, 4456–4459. doi:10.1021/ja410044r |
| 69. | Lu, X.; Li, X.; Guo, K.; Xie, T.-Z.; Moorefield, C. N.; Wesdemiotis, C.; Newkome, G. R. J. Am. Chem. Soc. 2014, 136, 18149–18155. doi:10.1021/ja511341z |
| 70. | Kobayashi, A.; Suzuki, Y.; Ohba, T.; Noro, S.-i.; Chang, H.-C.; Kato, M. Inorg. Chem. 2011, 50, 2061–2063. doi:10.1021/ic102361d |
| 57. | Rowan, A. E.; Nolte, R. J. M. Angew. Chem., Int. Ed. 1998, 37, 63–68. doi:10.1002/(SICI)1521-3773(19980202)37:1/2<63::AID-ANIE63>3.0.CO;2-4 |
| 58. | Saha, M. L.; Pramanik, S.; Schmittel, M. Chem. Commun. 2012, 48, 9459–9461. doi:10.1039/c2cc35036j |
| 59. | Zheng, Y.-R.; Zhao, Z.; Wang, M.; Ghosh, K.; Pollock, J. B.; Cook, T. R.; Stang, P. J. J. Am. Chem. Soc. 2010, 132, 16873–16882. doi:10.1021/ja106251f |
| 60. | Neogi, S.; Lorenz, Y.; Engeser, M.; Samanta, D.; Schmittel, M. Inorg. Chem. 2013, 52, 6975–6984. doi:10.1021/ic400328d |
| 61. | Stephenson, A.; Ward, M. D. Dalton Trans. 2011, 40, 10360–10369. doi:10.1039/c1dt10263j |
| 84. | Chen, B.; Liang, C.; Yang, J.; Contreras, D. S.; Clancy, Y. L.; Lobkovsky, E. B.; Yaghi, O. M.; Dai, S. Angew. Chem. 2006, 118, 1418–1421. doi:10.1002/ange.200502844 |
| 85. | Farrusseng, D.; Aguado, S.; Pinel, C. Angew. Chem., Int. Ed. 2009, 48, 7502–7513. doi:10.1002/anie.200806063 |
| 86. | Meek, S. T.; Greathouse, J. A.; Allendorf, M. D. Adv. Mater. 2011, 23, 249–267. doi:10.1002/adma.201002854 |
| 87. | Farha, O. K.; Hupp, J. T. Acc. Chem. Res. 2010, 43, 1166–1175. doi:10.1021/ar1000617 |
| 88. | Wu, C.-D.; Hu, A.; Zhang, L.; Lin, W. J. Am. Chem. Soc. 2005, 127, 8940–8941. doi:10.1021/ja052431t |
| 89. | Corma, A.; García, H.; Llabrés i Xamena, F. X. Chem. Rev. 2010, 110, 4606–4655. doi:10.1021/cr9003924 |
| 90. | Dhakshinamoorthy, A.; Alvaro, M.; Corma, A.; Garcia, H. Dalton Trans. 2011, 40, 6344–6360. doi:10.1039/c1dt10354g |
| 91. | Rowsell, J. L. C.; Yaghi, O. M. Angew. Chem., Int. Ed. 2005, 44, 4670–4679. doi:10.1002/anie.200462786 |
| 92. | Li, J.-R.; Sculley, J.; Zhou, H.-C. Chem. Rev. 2012, 112, 869–932. doi:10.1021/cr200190s |
© 2018 Adeyemo and Mukherjee; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)