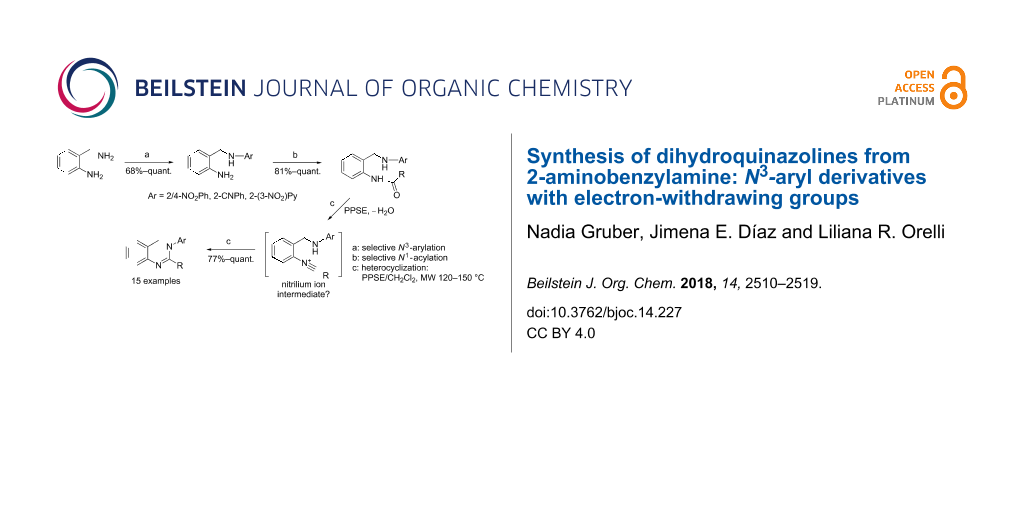
Abstract
The sequential N-functionalization of 2-aminobenzylamine (2-ABA) followed by cyclodehydration allowed for a straightforward and efficient synthesis of 3,4-dihydroquinazolines with N-aryl substituents bearing electron-withdrawing groups. The sequence involves an initial SNAr displacement, N-acylation and MW-assisted ring closure. Remarkably, the uncatalyzed N-arylation of 2-ABA led to the monosubstitution product using equimolar amounts of both reagents. The individual steps were optimized achieving good to excellent overall yields of the desired heterocycles, avoiding additional protection and deprotection steps. A mechanistic interpretation for the cyclodehydration reaction promoted by trimethylsilyl polyphosphate (PPSE) is also proposed on the basis of literature data and our experimental observations.

Graphical Abstract
Introduction
Dihydroquinazolines (DHQs) represent heterocyclic cores of pharmacological interest. For example, vasicine is a 3,4-dihydroquinazoline alkaloid isolated from natural sources with antitumor activity [1]. Vasicine and deoxyvasicine are also potent butyrylcholinesterase (BChE) inhibitors [2]. Some synthetic derivatives containing the dihydroquinazoline scaffold have shown antimicrobial [3] and antifungal properties [4]. In addition, antiparasitic activity has been studied for some members of this family as inhibitors of trypanothione reductase [5], an essential enzyme of the kinetoplastid Trypanosoma brucei. Their activity as selective T-type calcium channel blockers [6-11], as tumor suppressors [12] and as neuroprotective agents has also been reported [13]. Some related 2-amino-DHQs were studied as blood platelet aggregation inhibitors [14], antihypertensive agents [15] or inhibitors of β-secretase, an important target for Alzheimer’s disease [16]. Additionally, some ruthenium complexes bearing a 3,4-dihydroquinazoline ligand have been studied as hydrogenation-transfer catalysts, showing good to excellent activity for the reduction of ketones [17]. In the context of our research on heterocyclic amidine N-oxides [18-22], we recently prepared some suitably 2-substituted N-aryl-3,4-DHQs with electron-withdrawing groups (EWGs) in their aryl moiety (Figure 1) as synthetic intermediates for novel tetracyclic nitrones, to be studied as spin traps and antimicrobials.
![[1860-5397-14-227-1]](/bjoc/content/figures/1860-5397-14-227-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: N-Aryl-3,4-dihydroquinazolines 1.
Figure 1: N-Aryl-3,4-dihydroquinazolines 1.
The literature describes several methods for the synthesis of 3,4-DHQs. Some of them involve oxidation or reduction of related heterocycles (quinazolinones, quinazolines or tetrahydroquinazolines) [23-28]. Dihydroquinazolines can also be synthesized by heterocyclization of 2-aminobenzylamines (2-ABAs) with different C2 donors [3,16,17,29-36]. An alternative method is the ring closure of N-acyl-2-ABAs, but this strategy generally requires high temperatures and/or long reaction times [5,37-40].
Recently, our group described efficient strategies to prepare 3,4- and 1,4-DHQs with different substitution patterns from 2-ABA [41]. However, N-aryl-3,4-DHQs were not accessible through these routes. There are very few reported strategies for the synthesis of C4-unsubstituted N-aryl-3,4-DHQs, and all of them show limitations. The classical method involves the reaction between p-substituted anilines and paraformaldehyde in mineral [42-44] or formic [45] acid to yield symmetrically substituted 3,4-DHQs, usually in low yields [45] and accompanied by several by-products [46,47]. In addition, this strategy is limited to the preparation of 3,6-disubstituted compounds, because a p-substituted aniline is required to block this condensation position [48-50]. An exception to this is the recently reported synthesis of 3-(m-nitrophenyl)-5-nitro-3,4-dihydroquinazoline from m-nitroaniline and 1,3-dioxolane in the presence of strong protic acids [51]. In a related methodology, the ring closure of N-aryl-2-ABA is promoted by formic acid or other sources of C2 like ethyl orthoformate [52], diarylformamidines [53] or 1,1-dimethoxy-N,N-dimethylmethanamine [17]. Using an alternative approach, the corresponding 2-ABA was treated with acetic anhydride in concentrated sulfuric acid affording 2-methyl-6-nitro-3-(p-nitrophenyl)-3,4-DHQ [54]. o-Nitrobenzylanilines were also employed as starting materials, although this methodology involves a reduction step to form the functionalized 2-ABA and suffers from byproduct formation [55-57]. A different strategy involves the oxidation of N-aryl-1,2,3,4-tetrahydroquinazolines [45,58]. A more recent method starts from an aromatic amine and formaldehyde in the presence of ionic liquids and a controlled amount of 1-methyl-3-(2-(sulfoxy)ethyl)-1H-imidazol-3-ium chloride as the catalyst. The reaction affords the corresponding 1,4-dihydroquinzolin-3-ium tetrafluoroborates, which upon treatment with NaOH/EtOH evolve to the symmetrically substituted 3,4-DHQs [59]. Other authors report the synthesis of N-phenyl-3,4-DHQ by alkylation of sodium formanilide with 2-nitrobenzyl chloride followed by cyclization under reductive conditions [60], or from 2-ABA and CO2 as starting materials in catalytic reductive conditions [61].
In conclusion, the already described methods for the synthesis of 4-unsustituted N-aryl-3,4-DHQs are either limited to symmetrical substitution patterns, lead to byproducts and afford low yields, require the use of complex catalysts and ionic liquids or involve reductive conditions that are incompatible with nitro or cyano groups. In this context, and as part of our ongoing research on heterocyclic amidines (vide infra) and amidine N-oxides we report herein the first method for the synthesis of 2-alkyl/aryl-N-aryl-3,4-DHQs 1 bearing electron-withdrawing groups in the aryl moiety.
Our approach involves two selective functionalizations of 2-ABA followed by a ring-closure step. Some selective N-acylations and N-alkylations of this precursor had been described in the literature, involving in many cases additional protection and deprotection steps or affording modest yields [40,62-67]. N-Arylations of this substrate have been attempted, although with poor results [68]. Alternative strategies leading to N-functionalized 2-ABAs start from isatoic anhydride, 2-nitrobenzonitrile, 2-nitrobenzaldehyde, 2-aminobenzophenone or 2-nitrobenzyl halides, among others, and involve a reduction step which would be incompatible with the presence of nitro or cyano groups [5,13,14,39,68-71].
The synthetic sequence towards compounds 1 requires the chemoselective arylation of the benzylic amino group of the precursor with active haloaryl derivatives. SNAr is a widely employed reaction in organic synthesis [72]. However, the possibility of achieving selective N-substitution of diamines in equimolar conditions with high yields still represents a challenge. Selective SNAr reactions were reported for polyhaloaryl substrates [73,74], while alkanediamines require in most cases either a large excess of the nucleophile [75-77] or Boc protection [78,79] in order to obtain the monoarylated product in acceptable yields. There are some reports on selective SNAr reactions of piperazine [80] but, to the best of our knowledge, uncatalyzed selective N-arylations of unprotected primary diamines have not been systematically investigated yet.
Regarding the last step of the sequence leading to compounds 1, our group has worked extensively on ring-closure reactions leading to nitrogen-containing heterocycles such as 5–8-membered cyclic amidines [81-83], N-aryl-2-iminoazacycloalkanes [84] and 2-oxazolines or their higher homologues [85], using polyphosphoric acid esters PPE (ethyl polyphosphate) [86] and PPSE (trimethylsilyl polyphosphate) [87] under microwave irradiation. PPE and PPSE are aprotic irreversible dehydrating agents of the Lewis acid-type, which are able at the same time, to activate nitrogen and oxygen functionalities towards nucleophilic attack. This dual behavior makes them valuable reagents, capable of promoting certain cyclodehydrations that cannot be achieved by using classical Lewis acids [84,85]. Additional advantages of PPA esters are their low cost, operational simplicity and minimum environmental impact. Besides, their use together with microwave irradiation brings about shorter reaction times, cleaner crude products and consequently higher yields. On the basis of our previous work on DHQs [41], we explore herein the use of PPA esters for the optimization of the heterocyclization reaction leading to compounds 1.
Results and Discussion
The synthetic strategy leading to DHQs 1 is depicted in Scheme 1.
![[1860-5397-14-227-i1]](/bjoc/content/inline/1860-5397-14-227-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathway leading to N-aryl-3,4-dihydroquinazolines 1.
Scheme 1: Synthetic pathway leading to N-aryl-3,4-dihydroquinazolines 1.
The key step of the sequence involves an N-arylation of 2-ABA with active haloaromatics (Scheme 2). As previously mentioned, SNAr displacements in 1,n-diamines usually require a large excess of the nucleophile in order to achieve acceptable results. As 2-ABA has two nucleophilic nitrogen centers of different reactivity, the challenge consisted in achieving complete selectivity together with high yields using equimolar amounts of both reactants.
![[1860-5397-14-227-i2]](/bjoc/content/inline/1860-5397-14-227-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The reaction between 2-ABA and 2-nitrohalobenzenes was chosen for the initial optimization of the experimental conditions, whose results are collected in Table 1. No reaction was observed with 2-nitrochlorobenzene after 8 h either in solvent-free conditions at 125 °C or using THF at reflux, even with a two-fold excess of the amine (Table 1, entries 1–3). Better results were achieved with DME as the solvent and adding K2CO3 as proton acceptor (Table 1, entry 4). The replacement of 2-nitrochlorobenzene by the more reactive fluoro derivative resulted in a significant increase of the reaction yield (Table 1, entry 5). The best results were achieved using water as the solvent (Table 1, entry 6). Under such conditions, the conversion using equimolar amounts of the reagents was almost quantitative, with complete selectivity towards the N-monoarylation product 2a.
Table 1: Reaction conditions screening for the synthesis of compounds 2.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-227-i5.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | 2 | R1 | R2 | Y | X | Solvent; mL/mmol ArX | Temperature (°C) | Time (h) | Yield (%)a |
|---|---|---|---|---|---|---|---|---|---|
| 1b | a | NO2 | H | CH | Cl | – | 125 | 8 | NR |
| 2b | a | NO2 | H | CH | Cl | THF; 0.5 | reflux | 8 | NR |
| 3b,c | a | NO2 | H | CH | Cl | THF; 0.5 | reflux | 8 | NR |
| 4 | a | NO2 | H | CH | Cl | DME; 0.5 | reflux | 24 | 42 |
| 5 | a | NO2 | H | CH | F | DME; 0.5 | reflux | 4 | 90 |
| 6 | a | NO2 | H | CH | F | H2O; 1 | 80 | 4 | 96 |
| 7 | b | H | NO2 | CH | F | H2O; 1 | 80 | 4 | 52 |
| 8 | b | H | NO2 | CH | F | H2O; 1 | 80 | 16 | 58 |
| 9 | b | H | NO2 | CH | F | H2O; 1 | reflux | 4 | 71 |
| 10 | b | H | NO2 | CH | F | DMSO; 0.25 | 125 | 4 | 96 |
| 11 | c | NO2 | H | N | Cl | H2O; 1 | 80 | 8 | 20 |
| 12 | c | NO2 | H | N | Cl | H2O; 1 | 50 | 8 | 55 |
| 13 | c | NO2 | H | N | Cl | DMSO; 0.5 | rt | 8 | quant. |
| 14 | d | CN | H | CH | F | DMSO; 0.25 | 125 | 8 | 66 |
| 15 | d | CN | H | CH | F | DMSO; 0.25 | 135 | 8 | 68 |
| 16 | d | CN | H | CH | F | DMSO; 0.25 | 150 | 8 | 53 |
aYields correspond to pure compounds. bThe reaction was carried out without K2CO3. cThe reaction was carried out with a two-fold excess of 2-ABA.
In the same experimental conditions, the reaction of 2-ABA with 4-nitrofluorobenzene afforded acceptable yields (Table 1, entry 7), although complete conversion was not achieved even working at a higher temperature or with longer reaction times (Table 1, entries 8 and 9). DMF was then tested as the solvent in order to improve the yield by raising the temperature, but the reaction performed at 125 °C afforded N,N-dimethyl-4-nitroaniline as the main product together with a low percentage (<10%) of the desired compound 2b. DMSO gave the best results due to its higher boiling point, stability and a better solubility of the reactants, leading to nearly quantitative yields of 2b with complete chemoselectivity (Table 1, entry 10).
The different reactivity of o- and p-nitrofluorobenzenes in SNAr displacements had previously been reported, and is attributed to a differential stabilization of the reaction intermediate in the former. Such effect would result from an intramolecular hydrogen bond between the ammonium and nitro groups in the reaction intermediate, also known as “built-in solvation” [71] (Figure 2).
![[1860-5397-14-227-2]](/bjoc/content/figures/1860-5397-14-227-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reaction intermediate in the synthesis of compound 2a.
Figure 2: Reaction intermediate in the synthesis of compound 2a.
When 2-ABA was reacted with 2-chloro-3-nitropyridine (CNP) in analogous experimental conditions as 2a, compound 2c was isolated in low yield (Table 1, entry 11). A second product was obtained, which was identified as the corresponding N,N’-diaryl derivative. This behavior was related to the enhanced reactivity of CNP due to the presence of two strong EWGs in the aromatic ring. Lowering the reaction temperature had the expected effect on chemoselectivity, and the desired product was isolated in comparatively better yields (Table 1, entry 12). In order to improve the solubility of the reagents in the reaction mixture, water was replaced by small amounts of DMSO, resulting in a quantitative conversion with complete selectivity toward diamine 2c (Table 1, entry 13).
Analogous conditions as for 2b, although doubling the reaction time, were then tested for the arylation of 2-ABA with the less reactive 2-fluorobenzonitrile (Table 1, entry 14). The best results were attained working at a slightly higher temperature, affording 2d in good yield (Table 1, entry 15). A further increase in the reaction temperature, however, was unfavorable due to decomposition of the substrate (Table 1, entry 16).
Next, N-aryl-2-aminobenzylamines 2 were selectively acylated to yield compounds 3, using acid chlorides or anhydrides. No N,N’-diacylation products were observed due to the low nucleophilicity of the secondary amino group, bearing a deactivating aromatic ring and all desired compounds were obtained with excellent yields (Table 2).
Table 2: Selective N-acylation of N-aryl-2-ABAs 2.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-227-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | 3 | Ar | R | Yield (%)a |
|---|---|---|---|---|
| 1 | a | 2-NO2C6H4 | CH2CH3 | 93 |
| 2 | b | 2-NO2C6H4 | CH(CH3)2 | 92 |
| 3 | c | 2-NO2C6H4 | C6H5 | 89 |
| 4 | d | 2-NO2C6H4 | 4-ClC6H4 | quant. |
| 5 | e | 2-NO2C6H4 | CH2C6H5 | quant. |
| 6 | f | 2-NO2C6H4 | 4-ClC6H4CH2 | quant. |
| 7 | g | 4-NO2C6H4 | CH3 | 92 |
| 8 | h | 4-NO2C6H4 | CH2CH3 | quant. |
| 9 | i | 4-NO2C6H4 | CH(CH3)2 | 95 |
| 10 | j | 4-NO2C6H4 | C(CH3)3 | 86 |
| 11 | k | 4-NO2C6H4 | C6H5 | quant. |
| 12 | l | 2-(3-NO2)C5H3N | CH2CH3 | 97 |
| 13 | m | 2-(3-NO2)C5H3N | CH(CH3)2 | 87 |
| 14 | n | 2-CNC6H4 | CH3 | 93 |
| 15 | o | 2-CNC6H4 | CH(CH3)2 | 81 |
aYields correspond to pure compounds.
Then, the microwave-assisted cyclization reaction leading to 3,4-dihydroquinazolines 1 was initially tested with compound 3b using PPE/Cl3CH as the dehydrating agent, on the basis of our previous results [41]. After disappearance of the starting material, the resulting yield of 1b estimated from the crude product was disappointingly low (<20%). This behavior was unexpected and can be attributed to the low basicity of the dihydroquinazoline, which is not extracted in the acidic aqueous phase using the classical work-up procedure for PPE [41].
It had previously been reported that PPSE is a more efficient cyclization agent than PPE, allowing to perform some conversions that cannot be achieved by the latter [85,88]. Additionally, the classical work-up procedure of PPSE-promoted reactions involves basic conditions [85]. In fact, treatment of 3b with PPSE/Cl2CH2 under microwave irradiation afforded the desired product 1b with 90% yield (Table 3). In a control experiment, 3b was reacted with PPE/Cl3CH and the product was isolated under basic conditions, affording compound 1b in 81% yield after a cumbersome purification. Using this protocol, cyclization of precursor 3a afforded 85% of the corresponding dihydroquinazoline 1a vs 92% for the reaction with PPSE, which was thus chosen as the dehydrating agent. Employing the optimized reaction conditions, a series of novel N-aryl-3,4-dihydroquinazolines 1 was synthesized in high to excellent yields (Table 3). The reactions were performed, with the exception of 1j, at 120 °C. Lower temperatures hindered the complete dissolution of the substrates thus increasing the reaction times. The irradiation times were individually adjusted according to the reactivity of the substrates.
Table 3: Synthesis of N-aryl-3,4-dihydroquinazolines 1.
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-227-i7.svg?max-width=637&scale=1.0)
|
||||||
| Entry | 1 | Ar | R | Time (min) | Yield (%)a | Yield 2-ABA → 1 (%) |
|---|---|---|---|---|---|---|
| 1 | a | 2-NO2C6H4 | CH2CH3 | 15 | 92 | 82 |
| 2 | b | 2-NO2C6H4 | CH(CH3)2 | 25 | 90 | 79 |
| 3 | c | 2-NO2C6H4 | C6H5 | 90 | 87 | 74 |
| 4 | d | 2-NO2C6H4 | 4-ClC6H4 | 85 | 83 | 80 |
| 5 | e | 2-NO2C6H4 | CH2C6H5 | 40 | 87 | 84 |
| 6 | f | 2-NO2C6H4 | 4-ClC6H4CH2 | 45 | 88 | 84 |
| 7 | g | 4-NO2C6H4 | CH3 | 5 | quant. | 88 |
| 8 | h | 4-NO2C6H4 | CH2CH3 | 12 | 93 | 89 |
| 9 | i | 4-NO2C6H4 | CH(CH3)2 | 15 | 89 | 81 |
| 10b | j | 4-NO2C6H4 | C(CH3)3 | 90 | 87 | 72 |
| 11 | k | 4-NO2C6H4 | C6H5 | 45 | 92 | 88 |
| 12 | l | 2-(3-NO2)C5H3N | CH2CH3 | 15 | 83 | 81 |
| 13 | m | 2-(3-NO2)C5H3N | CH(CH3)2 | 25 | 80 | 70 |
| 14 | n | 2-CNC6H4 | CH3 | 5 | 91 | 58 |
| 15 | o | 2-CNC6H4 | CH(CH3)2 | 30 | 77 | 42 |
aYields correspond to pure compounds. bThe reaction was carried out at 150 °C.
Interestingly, the 1H NMR spectra of some ortho-substituted derivatives show nonequivalent hydrogens within the benzylic methylenes, which appear as AB spin systems (compounds 1a,b,e,f) or as broadened signals (compounds 1c,d,n,o). These spectral features would arise from restricted rotation around the N3-aryl bond, which entails the existence of conformational enantiomers [89].
Except for 2-cyano derivatives 1n,o, the whole sequence afforded compounds 1 in remarkably high (>70%) to excellent (>80%) overall yields from 2-ABA in three steps (Table 3, last column).
Regarding the ring-closure step, Table 3 shows a clear dependence of the reaction times with steric hindrance of the amide group (R) in the substrate, displaying the order R = CH3 < CH2CH3 < CH(CH3)2 << C(CH3)3. In fact, the conversion of the tert-butyl derivative 3j at 120 °C was slow, affording the desired product 1 in low yield (<10%) after 60 min. In this case, harsher reaction conditions were required for complete consumption of the starting material. In turn, the less reactive benzamides 3c,d,k required relatively longer reaction times than alkaneamides to achieve complete conversion at 120 °C.
On the other hand, the reaction times were almost insensitive to the electronic features of the N-aryl group. This was quite unexpected in the context of the accepted mechanism for this heterocyclization, which involves a nucleophilic attack of the arylamino group on the amide carbonyl followed by H2O elimination (Scheme 3) [90]. In such processes, PPSE would play a dual role, activating the amide towards the nucleophilic attack and acting as water scavenger. However, deactivated arylamino groups such as 2/4-nitrophenylanilines within compounds 3 are poor nucleophiles, and the N-2-(3-nitropyridyl) group is even less reactive. This is reflected by the fact that, as previously mentioned, compounds 3 do not react further with acylating agents to give the N,N’-diacylation products.
![[1860-5397-14-227-i3]](/bjoc/content/inline/1860-5397-14-227-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Addition–elimination mechanism for the heterocyclization.
Scheme 3: Addition–elimination mechanism for the heterocyclization.
On the basis of some literature data, an alternative mechanism involving nitrilium ions as intermediates can be proposed (Scheme 4). Nitrilium ions [91] are known to mediate different organic reactions such as the Beckmann rearrangement and the Bischler–Napieralski, von Braun and Ritter reactions, among others [92,93]. Stable nitrilium salts can be generated by Lewis acid-promoted halide abstraction from imidoyl chlorides or by alkylation of nitriles. A nucleophilic attack on such species followed by cyclization provides access to a variety of heterocycles. Intramolecular reactions of in situ generated nitrilium ions leading to heterocyclic compounds have also been reported [91]. These transient intermediates have been characterized spectroscopically and, in some cases, were isolated from the reaction medium [94-97]. In relation to our research, nitrilium ions are also known to mediate the synthesis of acyclic amidines by reaction with primary and secondary amines [98]. On the other hand, it is known that N-unsubstituted amides react with PPSE affording nitriles [99]. In this context, the cyclodehydration of N-monosubstituted amides 3 could in principle involve an initial PPSE-mediated elimination generating a reactive nitrilium ion in situ (Scheme 4). This transient species is a powerful internal electrophile which would readily undergo intramolecular attack even by poor nucleophiles like the deactivated arylamino groups present in compounds 3.
![[1860-5397-14-227-i4]](/bjoc/content/inline/1860-5397-14-227-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism involving an intermediate nitrilium ion.
Scheme 4: Proposed mechanism involving an intermediate nitrilium ion.
Conclusion
Our synthetic approach represents the first method for the preparation of 2-substituted-3-aryl-3,4-dihydroquinazolines with EWGs in the aryl moiety. The sequence involves a minimum number of steps, is operationally simple and requires easily available and inexpensive reagents. A careful optimization of the individual steps allowed for the selective functionalization of 2-ABA, avoiding the use of protecting groups, and affording the desired heterocycles in good to excellent overall yields.
A suitable balance between reactivity and selectivity was achieved in the SNAr reaction which, to our knowledge, is the first report on a successful uncatalyzed N-monoarylation of the unprotected diamine performed under equimolar conditions. These results are of particular relevance due to the wide use of the SNAr reaction in synthetic organic chemistry.
The cyclization of functionalized 2-ABAs was performed efficiently under MW irradiation using PPSE as the dehydrating agent. This approach avoids the use of strong protic acids, which may be incompatible with sensitive substrates. Two alternative mechanisms can be proposed for the reaction. An initial elimination yielding a transient nitrilium ion intermediate followed by a nucleophilic attack seems more likely on the basis of literature data and our experimental observations.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization of new compounds. | ||
| Format: PDF | Size: 5.1 MB | Download |
References
-
Joshi, B. S.; Bai, Y.; Puar, M. S.; Dubose, K. K.; Pelletier, S. W. J. Nat. Prod. 1994, 57, 953–962. doi:10.1021/np50109a012
Return to citation in text: [1] -
Yang, Y.; Cheng, X.; Liu, W.; Chou, G.; Wang, Z.; Wang, C. J. Ethnopharmacol. 2015, 168, 279–286. doi:10.1016/j.jep.2015.03.070
Return to citation in text: [1] -
Los, R.; Wesołowska-Trojanowska, M.; Malm, A.; Karpińska, M. M.; Matysiak, J.; Niewiadomy, A.; Głaszcz, U. Heteroat. Chem. 2012, 23, 265–275. doi:10.1002/hc.21012
Return to citation in text: [1] [2] -
Li, W.-J.; Li, Q.; Liu, D.-L.; Ding, M.-W. J. Agric. Food Chem. 2013, 61, 1419–1426. doi:10.1021/jf305355u
Return to citation in text: [1] -
Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v
Return to citation in text: [1] [2] [3] -
Lee, Y. S.; Lee, B. H.; Park, S. J.; Kang, S. B.; Rhim, H.; Park, J.-Y.; Lee, J.-H.; Jeong, S.-W.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2004, 14, 3379–3384. doi:10.1016/j.bmcl.2004.04.090
Return to citation in text: [1] -
Choe, Y. J.; Seo, H. N.; Jung, S. Y.; Rhim, H.; Kim, J.; Choo, D. J.; Lee, J. Y. Arch. Pharm. 2008, 341, 661–664. doi:10.1002/ardp.200800079
Return to citation in text: [1] -
Park, S. J.; Park, S. J.; Lee, M. J.; Rhim, H.; Kim, Y.; Lee, J.-H.; Chung, B. Y.; Lee, J. Y. Bioorg. Med. Chem. 2006, 14, 3502–3511. doi:10.1016/j.bmc.2006.01.005
Return to citation in text: [1] -
Rhim, H.; Lee, Y. S.; Park, S. J.; Chung, B. Y.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2005, 15, 283–286. doi:10.1016/j.bmcl.2004.10.078
Return to citation in text: [1] -
Heo, J. H.; Seo, H. N.; Choe, Y. J.; Kim, S.; Oh, C. R.; Kim, Y. D.; Rhim, H.; Choo, D. J.; Kim, J.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2008, 18, 3899–3901. doi:10.1016/j.bmcl.2008.06.034
Return to citation in text: [1] -
Byun, J. S.; Sohn, J. M.; Leem, D. G.; Park, B.; Nam, J. H.; Shin, D. H.; Shin, J. S.; Kim, H. J.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 1073–1079. doi:10.1016/j.bmcl.2015.12.010
Return to citation in text: [1] -
Jung, S. Y.; Lee, S. H.; Kang, H. B.; Park, H. A.; Chang, S. K.; Kim, J.; Choo, D. J.; Oh, C. R.; Kim, Y. D.; Seo, J. H.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2010, 20, 6633–6636. doi:10.1016/j.bmcl.2010.09.020
Return to citation in text: [1] -
Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424
Return to citation in text: [1] [2] -
Ishikawa, F.; Watanabe, Y.; Saegusa, J. Chem. Pharm. Bull. 1980, 28, 1357–1364. doi:10.1248/cpb.28.1357
Return to citation in text: [1] [2] -
Danielewicz, J. C.; Snarey, M.; Thomas, G. N. (2-Imidazolin-2-ylamino) substituted quinolines, -quinoxalines and –quinazolines as antihypertensive agents. U.S. Patent 3,890,319, June 17, 1975.
Return to citation in text: [1] -
Ghosh, A. K.; Pandey, S.; Gangarajula, S.; Kulkarni, S.; Xu, X.; Rao, K. V.; Huang, X.; Tang, J. Bioorg. Med. Chem. Lett. 2012, 22, 5460–5465. doi:10.1016/j.bmcl.2012.07.043
Return to citation in text: [1] [2] -
Mercan, D.; Çetinkaya, E.; Şahin, E. Inorg. Chim. Acta 2013, 400, 74–81. doi:10.1016/j.ica.2013.02.005
Return to citation in text: [1] [2] [3] -
García, M. B.; Orelli, L. R.; Magri, M. L.; Perillo, I. A. Synthesis 2002, 18, 2687–2690. doi:10.1055/s-2002-35980
Return to citation in text: [1] -
Lavaggi, M. L.; Aguirre, G.; Boiani, L.; Orelli, L.; García, B.; Cerecetto, H.; González, M. Eur. J. Med. Chem. 2008, 43, 1737–1741. doi:10.1016/j.ejmech.2007.10.031
Return to citation in text: [1] -
Díaz, J. E.; Vanthuyne, N.; Rispaud, H.; Roussel, C.; Vega, D.; Orelli, L. R. J. Org. Chem. 2015, 80, 1689–1695. doi:10.1021/jo502626f
Return to citation in text: [1] -
Gruber, N.; Piehl, L. L.; Rubin de Celis, E.; Díaz, J. E.; García, M. B.; Stipa, P.; Orelli, L. R. RSC Adv. 2015, 5, 2724–2731. doi:10.1039/c4ra14335c
Return to citation in text: [1] -
Gruber, N.; Orelli, L. R.; Cipolletti, R.; Stipa, P. Org. Biomol. Chem. 2017, 15, 7685–7695. doi:10.1039/c7ob01387f
Return to citation in text: [1] -
Decker, M. Eur. J. Med. Chem. 2005, 40, 305–313. doi:10.1016/j.ejmech.2004.12.003
Return to citation in text: [1] -
Jaén, J. C.; Gregor, V. E.; Lee, C.; Davis, R.; Emmerling, M. Bioorg. Med. Chem. Lett. 1996, 6, 737–742. doi:10.1016/0960-894x(96)00102-3
Return to citation in text: [1] -
Armarego, W. L. F. J. Chem. Soc. 1961, 2697–2701. doi:10.1039/jr9610002697
Return to citation in text: [1] -
Armarego, W. L. F. Adv. Heterocycl. Chem. 1963, 11, 253–309. doi:10.1016/s0065-2725(08)60527-9
Return to citation in text: [1] -
Richers, M. T.; Zhao, C.; Seidel, D. Beilstein J. Org. Chem. 2013, 9, 1194–1201. doi:10.3762/bjoc.9.135
Return to citation in text: [1] -
He, K.-H.; Tan, F.-F.; Zhou, C.-Z.; Zhou, G.-J.; Yang, X.-L.; Li, Y. Angew. Chem., Int. Ed. 2017, 56, 3080–3084. doi:10.1002/anie.201612486
Return to citation in text: [1] -
Kumar, R. A.; Saidulu, G.; Sridhar, B.; Liu, S. T.; Reddy, K. R. J. Org. Chem. 2013, 78, 10240–10250. doi:10.1021/jo401622r
Return to citation in text: [1] -
Hati, S.; Sen, S. Synthesis 2016, 48, 1389–1398. doi:10.1055/s-0035-1560416
Return to citation in text: [1] -
Li, C.; An, S.; Zhu, Y.; Zhang, J.; Kang, Y.; Liu, P.; Wang, Y.; Li, J. RSC Adv. 2014, 4, 49888–49891. doi:10.1039/c4ra09240f
Return to citation in text: [1] -
Hsu, H.-Y.; Tseng, C.-C.; Matii, B.; Sun, C.-M. Mol. Diversity 2012, 16, 241–249. doi:10.1007/s11030-011-9350-1
Return to citation in text: [1] -
Baxter, E. W.; Conway, K. A.; Kennis, L.; Bischoff, F.; Mercken, M. H.; De Winter, H. L.; Reynolds, C. H.; Tounge, B. A.; Luo, C.; Scott, M. K.; Huang, Y.; Braeken, M.; Pieters, S. M. A.; Berthelot, D. J. C.; Masure, S.; Bruinzeel, W. D.; Jordan, A. D.; Parker, M. H.; Boyd, R. E.; Qu, J.; Alexander, R. S.; Brenneman, D. E.; Reitz, A. B. J. Med. Chem. 2007, 50, 4261–4264. doi:10.1021/jm0705408
Return to citation in text: [1] -
Grasso, S.; Micale, N.; Monforte, A.-M.; Monforte, P.; Polimeni, S.; Zappalà, M. Eur. J. Med. Chem. 2000, 35, 1115–1119. doi:10.1016/s0223-5234(00)01195-8
Return to citation in text: [1] -
Papadopoulos, E. P.; George, B. J. Org. Chem. 1977, 42, 2530–2532. doi:10.1021/jo00434a049
Return to citation in text: [1] -
Burdick, B. A.; Benkovic, P. A.; Benkovic, S. J. J. Am. Chem. Soc. 1977, 99, 5716–5725. doi:10.1021/ja00459a032
Return to citation in text: [1] -
Zhong, Y.; Wang, L.; Ding, M.-W. Tetrahedron 2011, 67, 3714–3723. doi:10.1016/j.tet.2011.03.056
Return to citation in text: [1] -
He, P.; Wu, J.; Nie, Y.-B.; Ding, M.-W. Eur. J. Org. Chem. 2010, 1088–1095. doi:10.1002/ejoc.200901287
Return to citation in text: [1] -
Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/s0040-4039(01)01842-1
Return to citation in text: [1] [2] -
Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108
Return to citation in text: [1] [2] -
Díaz, J. E.; Ranieri, S.; Gruber, N.; Orelli, L. R. Beilstein J. Org. Chem. 2017, 13, 1470–1477. doi:10.3762/bjoc.13.145
Return to citation in text: [1] [2] [3] [4] -
Wagner, E. C.; Eisner, A. J. Am. Chem. Soc. 1937, 59, 879–883. doi:10.1021/ja01284a033
Return to citation in text: [1] -
Wagner, E. C. J. Org. Chem. 1954, 19, 1862–1881. doi:10.1021/jo01377a002
And references therein.
Return to citation in text: [1] -
Simons, J. K. J. Am. Chem. Soc. 1937, 59, 518–523. doi:10.1021/ja01282a026
Return to citation in text: [1] -
Fales, H. M. J. Am. Chem. Soc. 1955, 77, 5118–5121. doi:10.1021/ja01624a050
Return to citation in text: [1] [2] [3] -
Eisner, A.; Wagner, E. C. J. Am. Chem. Soc. 1934, 56, 1938–1943. doi:10.1021/ja01324a033
Return to citation in text: [1] -
Cairncross, S. E.; Bogert, M. T. Collect. Czech. Chem. Commun. 1935, 7, 548–554. doi:10.1135/cccc19350548
Return to citation in text: [1] -
Borkowski, W. L.; Wagner, E. C. J. Org. Chem. 1952, 17, 1128–1140. doi:10.1021/jo50008a012
Return to citation in text: [1] -
Clarke, H. T.; Gillespie, H. B.; Weisshaus, S. Z. J. Am. Chem. Soc. 1933, 55, 4571–4587. doi:10.1021/ja01338a041
Return to citation in text: [1] -
Emerson, W. S.; Neumann, F. W.; Moundres, T. P. J. Am. Chem. Soc. 1941, 63, 972–974. doi:10.1021/ja01849a023
Return to citation in text: [1] -
Yunnikova, L. P.; Esenbaeva, V. V. Russ. J. Gen. Chem. 2016, 86, 1769–1771. doi:10.1134/s1070363216070392
Return to citation in text: [1] -
von Walther, R.; Bamberg, R. J. Prakt. Chem. 1906, 73, 209–228. doi:10.1002/prac.19060730111
Return to citation in text: [1] -
Wagner, E. C. J. Org. Chem. 1940, 5, 133–141. doi:10.1021/jo01208a007
And references therein.
Return to citation in text: [1] -
Stillich, O. Ber. Dtsch. Chem. Ges. 1903, 36, 3115–3121. doi:10.1002/cber.19030360387
Return to citation in text: [1] -
Paal, C.; Krecke, F. Ber. Dtsch. Chem. Ges. 1890, 23, 2634–2641. doi:10.1002/cber.189002302160
Return to citation in text: [1] -
Paal, C.; Krecke, F. Ber. Dtsch. Chem. Ges. 1891, 24, 3049–3058. doi:10.1002/cber.189102402141
Return to citation in text: [1] -
Widman, O. J. Prakt. Chem. 1893, 47, 343–366. doi:10.1002/prac.18930470127
Return to citation in text: [1] -
McLaughlin, P. J.; Wagner, E. C. J. Am. Chem. Soc. 1944, 66, 251–254. doi:10.1021/ja01230a028
Return to citation in text: [1] -
Wan, Y.; Yuan, R.; Zhang, W.-c.; Shi, Y.-h.; Lin, W.; Yin, W.; Bo, R.-c.; Shi, J.-j.; Wu, H. Tetrahedron 2010, 66, 3405–3409. doi:10.1016/j.tet.2010.03.057
Return to citation in text: [1] -
Denney, D. B.; Rosen, J. Army Ordnance Contract No: DA-30-069-CRD-1689, 1956.
Return to citation in text: [1] -
Jacquet, O.; Das Neves Gomes, C.; Ephritikhine, M.; Cantat, T. ChemCatChem 2013, 5, 117–120. doi:10.1002/cctc.201200732
Return to citation in text: [1] -
Charton, J.; Girault-Mizzi, S.; Debreu-Fontaine, M.-A.; Foufelle, F.; Hainault, I.; Bizot-Espiard, J.-G.; Caignard, D.-H.; Sergheraert, C. Bioorg. Med. Chem. 2006, 14, 4490–4518. doi:10.1016/j.bmc.2006.02.028
Return to citation in text: [1] -
Hoang, L. T. M.; Ngo, L. H.; Nguyen, H. L.; Nguyen, H. T. H.; Nguyen, C. K.; Nguyen, B. T.; Ton, Q. T.; Nguyen, H. K. D.; Cordova, K. E.; Truong, T. Chem. Commun. 2015, 51, 17132–17135. doi:10.1039/c5cc05985b
Return to citation in text: [1] -
Yadav, D. K. T.; Bhanage, B. M. Synlett 2015, 26, 1862–1866. doi:10.1055/s-0034-1380811
Return to citation in text: [1] -
Pelagalli, R.; Chiarotto, I.; Feroci, M.; Vecchio, S. Green Chem. 2012, 14, 2251–2255. doi:10.1039/c2gc35485c
Return to citation in text: [1] -
Apfel, C.; Banner, D. W.; Bur, D.; Dietz, M.; Hubschwerlen, C.; Locher, H.; Marlin, F.; Masciadri, R.; Pirson, W.; Stalder, H. J. Med. Chem. 2001, 44, 1847–1852. doi:10.1021/jm000352g
Return to citation in text: [1] -
Bar-Haim, G.; Kol, M. Tetrahedron Lett. 1998, 39, 2643–2644. doi:10.1016/s0040-4039(98)00227-5
Return to citation in text: [1] -
Rzasa, R. M.; Kaller, M. R.; Liu, G.; Magal, E.; Nguyen, T. T.; Osslund, T. D.; Powers, D.; Santora, V. J.; Viswanadhan, V. N.; Wang, H.-L.; Xiong, X.; Zhong, W.; Norman, M. H. Bioorg. Med. Chem. 2007, 15, 6574–6595. doi:10.1016/j.bmc.2007.07.005
Return to citation in text: [1] [2] -
Coyne, W. E.; Cusic, J. W. J. Med. Chem. 1968, 11, 1208–1213. doi:10.1021/jm00312a022
Return to citation in text: [1] -
Camacho, M. E.; Chayah, M.; García, M. E.; Fernández-Sáez, N.; Arias, F.; Gallo, M. A.; Carrión, M. D. Arch. Pharm. 2016, 349, 638–650. doi:10.1002/ardp.201600020
Return to citation in text: [1] -
Carabateas, P. M.; Schodack, N. Y. 2-Tertiary aminomethyl-N-acylanilines. U.S. Patent 3360562A, Dec 26, 1967.
Return to citation in text: [1] [2] -
Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527656141
Return to citation in text: [1] -
Golf, H. R. A.; Reissig, H.-U.; Wiehe, A. Eur. J. Org. Chem. 2015, 1548–1568. doi:10.1002/ejoc.201403503
Return to citation in text: [1] -
Zhu, L.; Zhang, M. J. Org. Chem. 2004, 69, 7371–7374. doi:10.1021/jo049056s
Return to citation in text: [1] -
Perillo, I.; Fernández, B.; Lamdan, S. J. Chem. Soc., Perkin Trans. 2 1977, 2068–2072. doi:10.1039/p29770002068
Return to citation in text: [1] -
Martin, G. E.; Elgin, R. J.; Mathiasen, J. R.; Davis, C. B.; Kesslick, J. M.; Baldy, W. J.; Shank, R. P.; DiStefano, D. L.; Fedde, C. L.; Scott, M. K. J. Med. Chem. 1989, 32, 1052–1056. doi:10.1021/jm00125a020
Return to citation in text: [1] -
Lagu, B.; Tian, D.; Nagarathnam, D.; Marzabadi, M. R.; Wong, W. C.; Miao, S. W.; Zhang, F.; Sun, W.; Chiu, G.; Fang, J.; Forray, C.; Chang, R. S. L.; Ransom, R. W.; Chen, T. B.; O'Malley, S.; Zhang, K.; Vyas, K. P.; Gluchowski, C. J. Med. Chem. 1999, 42, 4794–4803. doi:10.1021/jm990202+
Return to citation in text: [1] -
Taliani, S.; Simorini, F.; Sergianni, V.; La Motta, C.; Da Settimo, F.; Cosimelli, B.; Abignente, E.; Greco, G.; Novellino, E.; Rossi, L.; Gremigni, V.; Spinetti, F.; Chelli, B.; Martini, C. J. Med. Chem. 2007, 50, 404–407. doi:10.1021/jm061137o
Return to citation in text: [1] -
Neuville, L.; Zhu, J. Tetrahedron Lett. 1997, 38, 4091–4094. doi:10.1016/s0040-4039(97)00831-9
Return to citation in text: [1] -
Eckert, J.; Chan, T.-M.; Osterman, R. M.; Lambert, J. B.; Gala, D. Tetrahedron Lett. 1999, 40, 5661–5665. doi:10.1016/s0040-4039(99)01104-1
And references therein.
Return to citation in text: [1] -
García, M. B.; Torres, R. A.; Orelli, L. R. Tetrahedron Lett. 2006, 47, 4857–4859. doi:10.1016/j.tetlet.2006.05.042
Return to citation in text: [1] -
Díaz, J. E.; Bisceglia, J. Á.; Mollo, M. C.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 1895–1897. doi:10.1016/j.tetlet.2011.02.042
Return to citation in text: [1] -
Díaz, J. E.; Gruber, N.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 6443–6445. doi:10.1016/j.tetlet.2011.09.097
Return to citation in text: [1] -
Díaz, J. E.; Mollo, M. C.; Orelli, L. R. Beilstein J. Org. Chem. 2016, 12, 2026–2031. doi:10.3762/bjoc.12.190
Return to citation in text: [1] [2] -
Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122
Return to citation in text: [1] [2] [3] [4] -
Dixon, L. A. Polyphosphate Ester. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2001. doi:10.1002/047084289x.rp185
Return to citation in text: [1] -
Imamoto, T. Cerium(III) Iodide. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2001. doi:10.1002/047084289x.rc043
Return to citation in text: [1] -
Li, W.; Fuchs, P. L. Org. Lett. 2003, 5, 4061–4064. doi:10.1021/ol035425f
Return to citation in text: [1] -
Mislow, K.; Raban, M. Stereoisomeric Relationships of Groups in Molecules. In Topics in Stereochemistry; Allinger, N. L.; Eliel, E. L., Eds.; Wiley-Blackwell: New York City, NY, U.S.A., 1967; Vol. 1, pp 1–38. doi:10.1002/9780470147108.ch1
Return to citation in text: [1] -
Perillo, I. A.; Caterina, M. C.; Salerno, A. ARKIVOC 2018, No. 1, 288–318. doi:10.24820/ark.5550190.p010.359
Return to citation in text: [1] -
van Dijk, T.; Chris Slootweg, J.; Lammertsma, K. Org. Biomol. Chem. 2017, 15, 10134–10144. doi:10.1039/c7ob02533e
See for a recent review.
Return to citation in text: [1] [2] -
Truce, W. E. Org. React. 1957, 9, 37–58. doi:10.1002/0471264180.or009.02
Return to citation in text: [1] -
Houben, J. Ber. Dtsch. Chem. Ges. 1926, 59, 2878–2891. doi:10.1002/cber.19260591135
Return to citation in text: [1] -
Marziano, N. C.; Ronchin, L.; Tortato, C.; Tonon, O.; Bertani, R. Int. J. Chem. Kinet. 2004, 36, 417–426. doi:10.1002/kin.20011
Return to citation in text: [1] -
Olah, G. A.; Kiovsky, T. E. J. Am. Chem. Soc. 1968, 90, 4666–4672. doi:10.1021/ja01019a028
Return to citation in text: [1] -
van Dijk, T.; Burck, S.; Rong, M. K.; Rosenthal, A. J.; Nieger, M.; Slootweg, J. C.; Lammertsma, K. Angew. Chem., Int. Ed. 2014, 53, 9068–9071. doi:10.1002/anie.201405027
Return to citation in text: [1] -
Doherty, S.; Hogarth, G.; Waugh, M.; Scanlan, T. H.; Clegg, W.; Elsegood, M. R. J. Organometallics 1999, 18, 3178–3186. doi:10.1021/om9901630
Return to citation in text: [1] -
Hegarty, A. Acc. Chem. Res. 1980, 13, 448–454. doi:10.1021/ar50156a003
Return to citation in text: [1] -
Yokoyama, M.; Yoshida, S.; Imamoto, T. Synthesis 1982, 591–592. doi:10.1055/s-1982-29874
Return to citation in text: [1]
| 55. | Paal, C.; Krecke, F. Ber. Dtsch. Chem. Ges. 1890, 23, 2634–2641. doi:10.1002/cber.189002302160 |
| 56. | Paal, C.; Krecke, F. Ber. Dtsch. Chem. Ges. 1891, 24, 3049–3058. doi:10.1002/cber.189102402141 |
| 57. | Widman, O. J. Prakt. Chem. 1893, 47, 343–366. doi:10.1002/prac.18930470127 |
| 45. | Fales, H. M. J. Am. Chem. Soc. 1955, 77, 5118–5121. doi:10.1021/ja01624a050 |
| 58. | McLaughlin, P. J.; Wagner, E. C. J. Am. Chem. Soc. 1944, 66, 251–254. doi:10.1021/ja01230a028 |
| 59. | Wan, Y.; Yuan, R.; Zhang, W.-c.; Shi, Y.-h.; Lin, W.; Yin, W.; Bo, R.-c.; Shi, J.-j.; Wu, H. Tetrahedron 2010, 66, 3405–3409. doi:10.1016/j.tet.2010.03.057 |
| 73. | Golf, H. R. A.; Reissig, H.-U.; Wiehe, A. Eur. J. Org. Chem. 2015, 1548–1568. doi:10.1002/ejoc.201403503 |
| 74. | Zhu, L.; Zhang, M. J. Org. Chem. 2004, 69, 7371–7374. doi:10.1021/jo049056s |
| 75. | Perillo, I.; Fernández, B.; Lamdan, S. J. Chem. Soc., Perkin Trans. 2 1977, 2068–2072. doi:10.1039/p29770002068 |
| 76. | Martin, G. E.; Elgin, R. J.; Mathiasen, J. R.; Davis, C. B.; Kesslick, J. M.; Baldy, W. J.; Shank, R. P.; DiStefano, D. L.; Fedde, C. L.; Scott, M. K. J. Med. Chem. 1989, 32, 1052–1056. doi:10.1021/jm00125a020 |
| 77. | Lagu, B.; Tian, D.; Nagarathnam, D.; Marzabadi, M. R.; Wong, W. C.; Miao, S. W.; Zhang, F.; Sun, W.; Chiu, G.; Fang, J.; Forray, C.; Chang, R. S. L.; Ransom, R. W.; Chen, T. B.; O'Malley, S.; Zhang, K.; Vyas, K. P.; Gluchowski, C. J. Med. Chem. 1999, 42, 4794–4803. doi:10.1021/jm990202+ |
| 5. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 13. | Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424 |
| 14. | Ishikawa, F.; Watanabe, Y.; Saegusa, J. Chem. Pharm. Bull. 1980, 28, 1357–1364. doi:10.1248/cpb.28.1357 |
| 39. | Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/s0040-4039(01)01842-1 |
| 68. | Rzasa, R. M.; Kaller, M. R.; Liu, G.; Magal, E.; Nguyen, T. T.; Osslund, T. D.; Powers, D.; Santora, V. J.; Viswanadhan, V. N.; Wang, H.-L.; Xiong, X.; Zhong, W.; Norman, M. H. Bioorg. Med. Chem. 2007, 15, 6574–6595. doi:10.1016/j.bmc.2007.07.005 |
| 69. | Coyne, W. E.; Cusic, J. W. J. Med. Chem. 1968, 11, 1208–1213. doi:10.1021/jm00312a022 |
| 70. | Camacho, M. E.; Chayah, M.; García, M. E.; Fernández-Sáez, N.; Arias, F.; Gallo, M. A.; Carrión, M. D. Arch. Pharm. 2016, 349, 638–650. doi:10.1002/ardp.201600020 |
| 71. | Carabateas, P. M.; Schodack, N. Y. 2-Tertiary aminomethyl-N-acylanilines. U.S. Patent 3360562A, Dec 26, 1967. |
| 72. | Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527656141 |
| 40. | Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108 |
| 62. | Charton, J.; Girault-Mizzi, S.; Debreu-Fontaine, M.-A.; Foufelle, F.; Hainault, I.; Bizot-Espiard, J.-G.; Caignard, D.-H.; Sergheraert, C. Bioorg. Med. Chem. 2006, 14, 4490–4518. doi:10.1016/j.bmc.2006.02.028 |
| 63. | Hoang, L. T. M.; Ngo, L. H.; Nguyen, H. L.; Nguyen, H. T. H.; Nguyen, C. K.; Nguyen, B. T.; Ton, Q. T.; Nguyen, H. K. D.; Cordova, K. E.; Truong, T. Chem. Commun. 2015, 51, 17132–17135. doi:10.1039/c5cc05985b |
| 64. | Yadav, D. K. T.; Bhanage, B. M. Synlett 2015, 26, 1862–1866. doi:10.1055/s-0034-1380811 |
| 65. | Pelagalli, R.; Chiarotto, I.; Feroci, M.; Vecchio, S. Green Chem. 2012, 14, 2251–2255. doi:10.1039/c2gc35485c |
| 66. | Apfel, C.; Banner, D. W.; Bur, D.; Dietz, M.; Hubschwerlen, C.; Locher, H.; Marlin, F.; Masciadri, R.; Pirson, W.; Stalder, H. J. Med. Chem. 2001, 44, 1847–1852. doi:10.1021/jm000352g |
| 67. | Bar-Haim, G.; Kol, M. Tetrahedron Lett. 1998, 39, 2643–2644. doi:10.1016/s0040-4039(98)00227-5 |
| 68. | Rzasa, R. M.; Kaller, M. R.; Liu, G.; Magal, E.; Nguyen, T. T.; Osslund, T. D.; Powers, D.; Santora, V. J.; Viswanadhan, V. N.; Wang, H.-L.; Xiong, X.; Zhong, W.; Norman, M. H. Bioorg. Med. Chem. 2007, 15, 6574–6595. doi:10.1016/j.bmc.2007.07.005 |
| 60. | Denney, D. B.; Rosen, J. Army Ordnance Contract No: DA-30-069-CRD-1689, 1956. |
| 61. | Jacquet, O.; Das Neves Gomes, C.; Ephritikhine, M.; Cantat, T. ChemCatChem 2013, 5, 117–120. doi:10.1002/cctc.201200732 |
| 78. | Taliani, S.; Simorini, F.; Sergianni, V.; La Motta, C.; Da Settimo, F.; Cosimelli, B.; Abignente, E.; Greco, G.; Novellino, E.; Rossi, L.; Gremigni, V.; Spinetti, F.; Chelli, B.; Martini, C. J. Med. Chem. 2007, 50, 404–407. doi:10.1021/jm061137o |
| 79. | Neuville, L.; Zhu, J. Tetrahedron Lett. 1997, 38, 4091–4094. doi:10.1016/s0040-4039(97)00831-9 |
| 80. |
Eckert, J.; Chan, T.-M.; Osterman, R. M.; Lambert, J. B.; Gala, D. Tetrahedron Lett. 1999, 40, 5661–5665. doi:10.1016/s0040-4039(99)01104-1
And references therein. |
| 81. | García, M. B.; Torres, R. A.; Orelli, L. R. Tetrahedron Lett. 2006, 47, 4857–4859. doi:10.1016/j.tetlet.2006.05.042 |
| 82. | Díaz, J. E.; Bisceglia, J. Á.; Mollo, M. C.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 1895–1897. doi:10.1016/j.tetlet.2011.02.042 |
| 83. | Díaz, J. E.; Gruber, N.; Orelli, L. R. Tetrahedron Lett. 2011, 52, 6443–6445. doi:10.1016/j.tetlet.2011.09.097 |
| 71. | Carabateas, P. M.; Schodack, N. Y. 2-Tertiary aminomethyl-N-acylanilines. U.S. Patent 3360562A, Dec 26, 1967. |
| 41. | Díaz, J. E.; Ranieri, S.; Gruber, N.; Orelli, L. R. Beilstein J. Org. Chem. 2017, 13, 1470–1477. doi:10.3762/bjoc.13.145 |
| 84. | Díaz, J. E.; Mollo, M. C.; Orelli, L. R. Beilstein J. Org. Chem. 2016, 12, 2026–2031. doi:10.3762/bjoc.12.190 |
| 85. | Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122 |
| 41. | Díaz, J. E.; Ranieri, S.; Gruber, N.; Orelli, L. R. Beilstein J. Org. Chem. 2017, 13, 1470–1477. doi:10.3762/bjoc.13.145 |
| 86. | Dixon, L. A. Polyphosphate Ester. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2001. doi:10.1002/047084289x.rp185 |
| 87. | Imamoto, T. Cerium(III) Iodide. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2001. doi:10.1002/047084289x.rc043 |
| 84. | Díaz, J. E.; Mollo, M. C.; Orelli, L. R. Beilstein J. Org. Chem. 2016, 12, 2026–2031. doi:10.3762/bjoc.12.190 |
| 85. | Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122 |
| 85. | Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122 |
| 88. | Li, W.; Fuchs, P. L. Org. Lett. 2003, 5, 4061–4064. doi:10.1021/ol035425f |
| 85. | Mollo, M. C.; Orelli, L. R. Org. Lett. 2016, 18, 6116–6119. doi:10.1021/acs.orglett.6b03122 |
| 41. | Díaz, J. E.; Ranieri, S.; Gruber, N.; Orelli, L. R. Beilstein J. Org. Chem. 2017, 13, 1470–1477. doi:10.3762/bjoc.13.145 |
| 1. | Joshi, B. S.; Bai, Y.; Puar, M. S.; Dubose, K. K.; Pelletier, S. W. J. Nat. Prod. 1994, 57, 953–962. doi:10.1021/np50109a012 |
| 5. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 3. | Los, R.; Wesołowska-Trojanowska, M.; Malm, A.; Karpińska, M. M.; Matysiak, J.; Niewiadomy, A.; Głaszcz, U. Heteroat. Chem. 2012, 23, 265–275. doi:10.1002/hc.21012 |
| 16. | Ghosh, A. K.; Pandey, S.; Gangarajula, S.; Kulkarni, S.; Xu, X.; Rao, K. V.; Huang, X.; Tang, J. Bioorg. Med. Chem. Lett. 2012, 22, 5460–5465. doi:10.1016/j.bmcl.2012.07.043 |
| 17. | Mercan, D.; Çetinkaya, E.; Şahin, E. Inorg. Chim. Acta 2013, 400, 74–81. doi:10.1016/j.ica.2013.02.005 |
| 29. | Kumar, R. A.; Saidulu, G.; Sridhar, B.; Liu, S. T.; Reddy, K. R. J. Org. Chem. 2013, 78, 10240–10250. doi:10.1021/jo401622r |
| 30. | Hati, S.; Sen, S. Synthesis 2016, 48, 1389–1398. doi:10.1055/s-0035-1560416 |
| 31. | Li, C.; An, S.; Zhu, Y.; Zhang, J.; Kang, Y.; Liu, P.; Wang, Y.; Li, J. RSC Adv. 2014, 4, 49888–49891. doi:10.1039/c4ra09240f |
| 32. | Hsu, H.-Y.; Tseng, C.-C.; Matii, B.; Sun, C.-M. Mol. Diversity 2012, 16, 241–249. doi:10.1007/s11030-011-9350-1 |
| 33. | Baxter, E. W.; Conway, K. A.; Kennis, L.; Bischoff, F.; Mercken, M. H.; De Winter, H. L.; Reynolds, C. H.; Tounge, B. A.; Luo, C.; Scott, M. K.; Huang, Y.; Braeken, M.; Pieters, S. M. A.; Berthelot, D. J. C.; Masure, S.; Bruinzeel, W. D.; Jordan, A. D.; Parker, M. H.; Boyd, R. E.; Qu, J.; Alexander, R. S.; Brenneman, D. E.; Reitz, A. B. J. Med. Chem. 2007, 50, 4261–4264. doi:10.1021/jm0705408 |
| 34. | Grasso, S.; Micale, N.; Monforte, A.-M.; Monforte, P.; Polimeni, S.; Zappalà, M. Eur. J. Med. Chem. 2000, 35, 1115–1119. doi:10.1016/s0223-5234(00)01195-8 |
| 35. | Papadopoulos, E. P.; George, B. J. Org. Chem. 1977, 42, 2530–2532. doi:10.1021/jo00434a049 |
| 36. | Burdick, B. A.; Benkovic, P. A.; Benkovic, S. J. J. Am. Chem. Soc. 1977, 99, 5716–5725. doi:10.1021/ja00459a032 |
| 4. | Li, W.-J.; Li, Q.; Liu, D.-L.; Ding, M.-W. J. Agric. Food Chem. 2013, 61, 1419–1426. doi:10.1021/jf305355u |
| 5. | Patterson, S.; Alphey, M. S.; Jones, D. C.; Shanks, E. J.; Street, I. P.; Frearson, J. A.; Wyatt, P. G.; Gilbert, I. H.; Fairlamb, A. H. J. Med. Chem. 2011, 54, 6514–6530. doi:10.1021/jm200312v |
| 37. | Zhong, Y.; Wang, L.; Ding, M.-W. Tetrahedron 2011, 67, 3714–3723. doi:10.1016/j.tet.2011.03.056 |
| 38. | He, P.; Wu, J.; Nie, Y.-B.; Ding, M.-W. Eur. J. Org. Chem. 2010, 1088–1095. doi:10.1002/ejoc.200901287 |
| 39. | Zhang, J.; Barker, J.; Lou, B.; Saneii, H. Tetrahedron Lett. 2001, 42, 8405–8408. doi:10.1016/s0040-4039(01)01842-1 |
| 40. | Dietrich, J.; Kaiser, C.; Meurice, N.; Hulme, C. Tetrahedron Lett. 2010, 51, 3951–3955. doi:10.1016/j.tetlet.2010.05.108 |
| 3. | Los, R.; Wesołowska-Trojanowska, M.; Malm, A.; Karpińska, M. M.; Matysiak, J.; Niewiadomy, A.; Głaszcz, U. Heteroat. Chem. 2012, 23, 265–275. doi:10.1002/hc.21012 |
| 18. | García, M. B.; Orelli, L. R.; Magri, M. L.; Perillo, I. A. Synthesis 2002, 18, 2687–2690. doi:10.1055/s-2002-35980 |
| 19. | Lavaggi, M. L.; Aguirre, G.; Boiani, L.; Orelli, L.; García, B.; Cerecetto, H.; González, M. Eur. J. Med. Chem. 2008, 43, 1737–1741. doi:10.1016/j.ejmech.2007.10.031 |
| 20. | Díaz, J. E.; Vanthuyne, N.; Rispaud, H.; Roussel, C.; Vega, D.; Orelli, L. R. J. Org. Chem. 2015, 80, 1689–1695. doi:10.1021/jo502626f |
| 21. | Gruber, N.; Piehl, L. L.; Rubin de Celis, E.; Díaz, J. E.; García, M. B.; Stipa, P.; Orelli, L. R. RSC Adv. 2015, 5, 2724–2731. doi:10.1039/c4ra14335c |
| 22. | Gruber, N.; Orelli, L. R.; Cipolletti, R.; Stipa, P. Org. Biomol. Chem. 2017, 15, 7685–7695. doi:10.1039/c7ob01387f |
| 91. |
van Dijk, T.; Chris Slootweg, J.; Lammertsma, K. Org. Biomol. Chem. 2017, 15, 10134–10144. doi:10.1039/c7ob02533e
See for a recent review. |
| 2. | Yang, Y.; Cheng, X.; Liu, W.; Chou, G.; Wang, Z.; Wang, C. J. Ethnopharmacol. 2015, 168, 279–286. doi:10.1016/j.jep.2015.03.070 |
| 23. | Decker, M. Eur. J. Med. Chem. 2005, 40, 305–313. doi:10.1016/j.ejmech.2004.12.003 |
| 24. | Jaén, J. C.; Gregor, V. E.; Lee, C.; Davis, R.; Emmerling, M. Bioorg. Med. Chem. Lett. 1996, 6, 737–742. doi:10.1016/0960-894x(96)00102-3 |
| 25. | Armarego, W. L. F. J. Chem. Soc. 1961, 2697–2701. doi:10.1039/jr9610002697 |
| 26. | Armarego, W. L. F. Adv. Heterocycl. Chem. 1963, 11, 253–309. doi:10.1016/s0065-2725(08)60527-9 |
| 27. | Richers, M. T.; Zhao, C.; Seidel, D. Beilstein J. Org. Chem. 2013, 9, 1194–1201. doi:10.3762/bjoc.9.135 |
| 28. | He, K.-H.; Tan, F.-F.; Zhou, C.-Z.; Zhou, G.-J.; Yang, X.-L.; Li, Y. Angew. Chem., Int. Ed. 2017, 56, 3080–3084. doi:10.1002/anie.201612486 |
| 94. | Marziano, N. C.; Ronchin, L.; Tortato, C.; Tonon, O.; Bertani, R. Int. J. Chem. Kinet. 2004, 36, 417–426. doi:10.1002/kin.20011 |
| 95. | Olah, G. A.; Kiovsky, T. E. J. Am. Chem. Soc. 1968, 90, 4666–4672. doi:10.1021/ja01019a028 |
| 96. | van Dijk, T.; Burck, S.; Rong, M. K.; Rosenthal, A. J.; Nieger, M.; Slootweg, J. C.; Lammertsma, K. Angew. Chem., Int. Ed. 2014, 53, 9068–9071. doi:10.1002/anie.201405027 |
| 97. | Doherty, S.; Hogarth, G.; Waugh, M.; Scanlan, T. H.; Clegg, W.; Elsegood, M. R. J. Organometallics 1999, 18, 3178–3186. doi:10.1021/om9901630 |
| 14. | Ishikawa, F.; Watanabe, Y.; Saegusa, J. Chem. Pharm. Bull. 1980, 28, 1357–1364. doi:10.1248/cpb.28.1357 |
| 16. | Ghosh, A. K.; Pandey, S.; Gangarajula, S.; Kulkarni, S.; Xu, X.; Rao, K. V.; Huang, X.; Tang, J. Bioorg. Med. Chem. Lett. 2012, 22, 5460–5465. doi:10.1016/j.bmcl.2012.07.043 |
| 91. |
van Dijk, T.; Chris Slootweg, J.; Lammertsma, K. Org. Biomol. Chem. 2017, 15, 10134–10144. doi:10.1039/c7ob02533e
See for a recent review. |
| 13. | Tian, Y.; Ma, C.; Feng, L.; Zhang, L.; Hao, F.; Pan, L.; Cheng, M. Arch. Pharm. 2012, 345, 423–430. doi:10.1002/ardp.201100424 |
| 17. | Mercan, D.; Çetinkaya, E.; Şahin, E. Inorg. Chim. Acta 2013, 400, 74–81. doi:10.1016/j.ica.2013.02.005 |
| 92. | Truce, W. E. Org. React. 1957, 9, 37–58. doi:10.1002/0471264180.or009.02 |
| 93. | Houben, J. Ber. Dtsch. Chem. Ges. 1926, 59, 2878–2891. doi:10.1002/cber.19260591135 |
| 12. | Jung, S. Y.; Lee, S. H.; Kang, H. B.; Park, H. A.; Chang, S. K.; Kim, J.; Choo, D. J.; Oh, C. R.; Kim, Y. D.; Seo, J. H.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2010, 20, 6633–6636. doi:10.1016/j.bmcl.2010.09.020 |
| 89. | Mislow, K.; Raban, M. Stereoisomeric Relationships of Groups in Molecules. In Topics in Stereochemistry; Allinger, N. L.; Eliel, E. L., Eds.; Wiley-Blackwell: New York City, NY, U.S.A., 1967; Vol. 1, pp 1–38. doi:10.1002/9780470147108.ch1 |
| 6. | Lee, Y. S.; Lee, B. H.; Park, S. J.; Kang, S. B.; Rhim, H.; Park, J.-Y.; Lee, J.-H.; Jeong, S.-W.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2004, 14, 3379–3384. doi:10.1016/j.bmcl.2004.04.090 |
| 7. | Choe, Y. J.; Seo, H. N.; Jung, S. Y.; Rhim, H.; Kim, J.; Choo, D. J.; Lee, J. Y. Arch. Pharm. 2008, 341, 661–664. doi:10.1002/ardp.200800079 |
| 8. | Park, S. J.; Park, S. J.; Lee, M. J.; Rhim, H.; Kim, Y.; Lee, J.-H.; Chung, B. Y.; Lee, J. Y. Bioorg. Med. Chem. 2006, 14, 3502–3511. doi:10.1016/j.bmc.2006.01.005 |
| 9. | Rhim, H.; Lee, Y. S.; Park, S. J.; Chung, B. Y.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2005, 15, 283–286. doi:10.1016/j.bmcl.2004.10.078 |
| 10. | Heo, J. H.; Seo, H. N.; Choe, Y. J.; Kim, S.; Oh, C. R.; Kim, Y. D.; Rhim, H.; Choo, D. J.; Kim, J.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2008, 18, 3899–3901. doi:10.1016/j.bmcl.2008.06.034 |
| 11. | Byun, J. S.; Sohn, J. M.; Leem, D. G.; Park, B.; Nam, J. H.; Shin, D. H.; Shin, J. S.; Kim, H. J.; Lee, K.-T.; Lee, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 1073–1079. doi:10.1016/j.bmcl.2015.12.010 |
| 15. | Danielewicz, J. C.; Snarey, M.; Thomas, G. N. (2-Imidazolin-2-ylamino) substituted quinolines, -quinoxalines and –quinazolines as antihypertensive agents. U.S. Patent 3,890,319, June 17, 1975. |
| 90. | Perillo, I. A.; Caterina, M. C.; Salerno, A. ARKIVOC 2018, No. 1, 288–318. doi:10.24820/ark.5550190.p010.359 |
| 41. | Díaz, J. E.; Ranieri, S.; Gruber, N.; Orelli, L. R. Beilstein J. Org. Chem. 2017, 13, 1470–1477. doi:10.3762/bjoc.13.145 |
| 42. | Wagner, E. C.; Eisner, A. J. Am. Chem. Soc. 1937, 59, 879–883. doi:10.1021/ja01284a033 |
| 43. |
Wagner, E. C. J. Org. Chem. 1954, 19, 1862–1881. doi:10.1021/jo01377a002
And references therein. |
| 44. | Simons, J. K. J. Am. Chem. Soc. 1937, 59, 518–523. doi:10.1021/ja01282a026 |
| 99. | Yokoyama, M.; Yoshida, S.; Imamoto, T. Synthesis 1982, 591–592. doi:10.1055/s-1982-29874 |
| 17. | Mercan, D.; Çetinkaya, E.; Şahin, E. Inorg. Chim. Acta 2013, 400, 74–81. doi:10.1016/j.ica.2013.02.005 |
| 54. | Stillich, O. Ber. Dtsch. Chem. Ges. 1903, 36, 3115–3121. doi:10.1002/cber.19030360387 |
| 52. | von Walther, R.; Bamberg, R. J. Prakt. Chem. 1906, 73, 209–228. doi:10.1002/prac.19060730111 |
| 53. |
Wagner, E. C. J. Org. Chem. 1940, 5, 133–141. doi:10.1021/jo01208a007
And references therein. |
| 48. | Borkowski, W. L.; Wagner, E. C. J. Org. Chem. 1952, 17, 1128–1140. doi:10.1021/jo50008a012 |
| 49. | Clarke, H. T.; Gillespie, H. B.; Weisshaus, S. Z. J. Am. Chem. Soc. 1933, 55, 4571–4587. doi:10.1021/ja01338a041 |
| 50. | Emerson, W. S.; Neumann, F. W.; Moundres, T. P. J. Am. Chem. Soc. 1941, 63, 972–974. doi:10.1021/ja01849a023 |
| 51. | Yunnikova, L. P.; Esenbaeva, V. V. Russ. J. Gen. Chem. 2016, 86, 1769–1771. doi:10.1134/s1070363216070392 |
| 46. | Eisner, A.; Wagner, E. C. J. Am. Chem. Soc. 1934, 56, 1938–1943. doi:10.1021/ja01324a033 |
| 47. | Cairncross, S. E.; Bogert, M. T. Collect. Czech. Chem. Commun. 1935, 7, 548–554. doi:10.1135/cccc19350548 |
© 2018 Gruber et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)