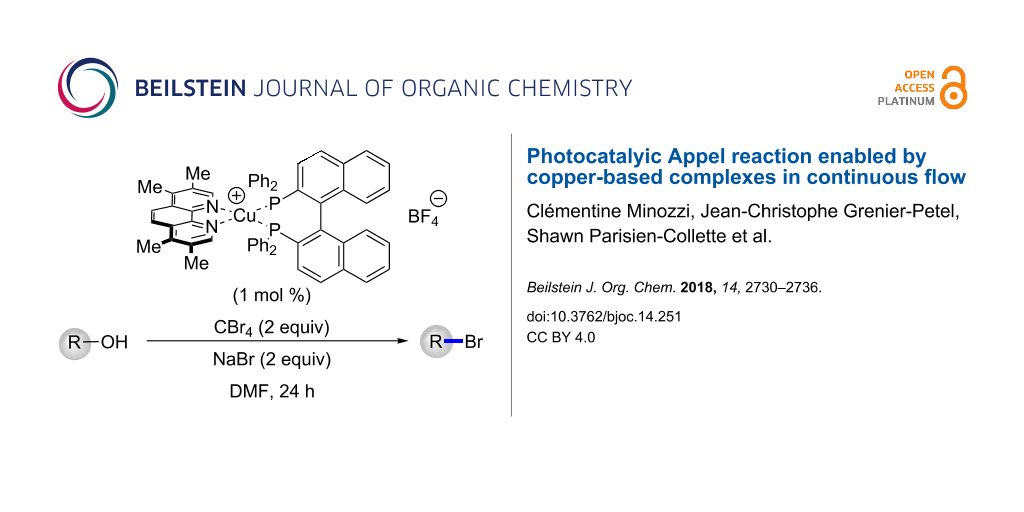
Abstract
A copper-based photocatalyst, Cu(tmp)(BINAP)BF4, was found to be active in a photoredox Appel-type conversion of alcohols to bromides. The catalyst was identified from a screening of 50 complexes and promoted the transformation of primary and secondary alcohols to their corresponding bromides and carboxylic acids to their anhydrides. The protocol was also amendable and optimized under continuous flow conditions.

Graphical Abstract
Introduction
Synthetic photochemistry and photocatalysis continues to influence molecular synthesis [1-4]. In exploring photochemical reactivity manifolds, there exists the potential to discover new methods to construct important molecular fragments, as well as revamp traditional chemical transformations. One such process is the Appel reaction [5], which employs PPh3 and an electrophilic halogen source to promote the formation of an organic halide from the corresponding alcohol (Figure 1) [6,7]. The Appel reaction is representative of a host of transformations that require stoichiometric reagents to effect a functional group change of an alcohol. In 2011, Stephenson and co-workers reported that photocatalysis could be used to promote the alcohol→halide conversion using low catalyst loadings of a ruthenium-based catalyst (Ru(bpy)3Cl2, 1 mol %) in the absence of PPh3 as a reductant (Figure 1) [8]. The method possesses numerous advantages (wide functional group tolerance, no formation of oxidized phosphine byproducts [9-14], mild reaction conditions and visible-light irradiation), which should be easily embraced by the synthetic community. To further develop the photochemical alcohol→halide transformation, the use of alternative photocatalysts based upon more abundant metals was envisioned [15-18]. Specifically, our group has demonstrated that heteroleptic Cu(I) complexes [19-21] have significant potential as photocatalysts that can promote a variety of mechanistically distinct photochemical transformations including single electron transfer (SET), energy transfer (ET), and proton-coupled electron transfer (PCET) reactions [22-26]. Herein, the evaluation of Cu(I)-complexes for photocatalytic Appel reactions and demonstration in continuous flow is described.
![[1860-5397-14-251-1]](/bjoc/content/figures/1860-5397-14-251-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Alcohol→bromide functional group transformations.
Figure 1: Alcohol→bromide functional group transformations.
Results and Discussion
The first step in identifying a heteroleptic diamine/bisphosphine Cu(I)-based photocatalyst for the conversion of an alcohol to bromide involved screening a wide variety of structurally varied complexes. Our group has previously demonstrated that the nature of each ligand influences the physical and photophysical properties as well as catalytic activity of the resulting catalyst (Figure 2) [27].
![[1860-5397-14-251-2]](/bjoc/content/figures/1860-5397-14-251-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Ligands used in the library generation of heteroleptic copper(I)-based complexes for photocatalysis.
Figure 2: Ligands used in the library generation of heteroleptic copper(I)-based complexes for photocatalysis....
A library of 50 different catalysts was evaluated in the conversion of alcohol 1 to bromide 2 (Figure 2 and Figure 3). Several homoleptic complexes were not evaluated due to problematic oxidation or low solubility. Reactions were irradiated at either 394 nm (purple LEDs) or 450 nm (blue LEDs), depending on the UV–vis absorption characteristics of the photocatalysts [28]. Stephenson and co-workers had previously reported that a primary alcohol structurally similar to 1 underwent conversion to the corresponding bromide in 96% yield upon irradiation in the presence of Ru(bpy)3Cl2 (1 mol %). The screening for a suitable copper-based catalyst was performed under identical reaction conditions whereby the Ru-based photocatalyst was substituted for the Cu-based complex. Control reactions performed in the absence of light or in the absence of catalyst at either 394 or 450 nm revealed no conversion to the bromide. From the results, none of the homoleptic complexes promoted the alcohol-to-halide conversion (1→2, see the light blue entries in the front row of Figure 3). While many of the heteroleptic complexes promoted the reaction, some trends were apparent. In general, amongst the phosphines the dppf-based complexes were poor catalysts, while when considering the diamine ligands the dq and bathophenthroline catalysts provided poor to modest yields. Also, BINAP and Xantphos-based catalysts tended to afford higher yields of 2, while amongst the diamines, the triazole-based complexes were almost all efficient at providing 2 (54–87% yield, not including dppf-based complexes). Interestingly, the best catalyst for the transformation (Cu(tmp)(BINAP)BF4, 99% of 2) was a poor catalyst for a previously reported photoredox reaction [27]. It should be noted that Cu(tmp)(BINAP)+ possesses an excited state reduction potential of −1.93 V vs. SCE, much greater than that of Ru(bpy)3+2 (−0.81 V vs SCE), albeit the copper complex has a much shorter excited state lifetime (≈4 ns vs ≈1100 ns for Ru(bpy)3+2). The excited state reduction potential should match favorably with CBr4 (E½ = 0.30 V vs SCE) in DMF [29]. Note that many of the corresponding homoleptic copper-based sensitizers were ineffective at promoting the Appel-type reaction.
![[1860-5397-14-251-3]](/bjoc/content/figures/1860-5397-14-251-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Evaluation of the library of copper-based complexes in photocatalytic alcohol→bromide conversion. Reactions irradiated with 394 nm light (pink) or 450 nm (blue). Front entries without an indicated phosphine ligand pertain to homoleptic Cu(diamine)2BF4 complexes and are colored in lighter blue. Entries without a color indicate reactions which could not be performed due to solubility or overoxidation of the complex.
Figure 3: Evaluation of the library of copper-based complexes in photocatalytic alcohol→bromide conversion. R...
With conditions in hand for the formation of the bromides, different alcohols were converted to their corresponding halides (Table 1). As shown previously, a benzyl-protected alcohol 1 could be transformed to the corresponding bromide 2 in 99% yield, respectively (Table 1, entry 1). The corresponding bromide of citronellol (4) was also formed in high yield (91%, Table 1, entry 2). A long chain methyl ester 5 was also tolerated under the reaction conditions (98% of the bromide, Table 1, entry 3). The corresponding dibromide could be formed from 1,9-nonadiol (6) in quantitative yield (99%, Table 1, entry 4). A sulfur-containing alcohol 7 was smoothly converted to its bromide in 99% yield (Table 1, entry 5). An allylic alcohol 8 having a cis-olefin underwent alcohol-to-halide conversion in 89% yield and was isolated as a 1:1 mixture of cis and trans isomers (Table 1, entry 6). Finally, a racemic secondary alcohol 9 was easily transformed to the corresponding racemic bromide (99%, Table 1, entry 7).
Table 1: Photocatalytic conversion of alcohols to bromides in batch.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-251-i2.svg?max-width=637&scale=1.0)
|
|||
| entry | alcohol | yield (%)a | |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-251-i3.svg?max-width=637&scale=1.0)
|
1 | 99 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-251-i4.svg?max-width=637&scale=1.0)
|
4 | 91 |
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-251-i5.svg?max-width=637&scale=1.0)
|
5 | 98 |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-251-i6.svg?max-width=637&scale=1.0)
|
6 | 99 |
| 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-251-i7.svg?max-width=637&scale=1.0)
|
7 | 99 |
| 6 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-251-i8.svg?max-width=637&scale=1.0)
|
8 | 89b |
| 7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-251-i9.svg?max-width=637&scale=1.0)
|
9 | 99 |
aYield determined by isolation via chromatography. bIsolated as a 1:1 mixture of cis and trans isomers.
Following the optimization of the catalyst structure and exploration of scope, the batch reaction conditions were then transferred to continuous flow (Table 2). Initially, an experimental set-up using a previously reported reactor for purple LEDs was selected for the reaction [30,31]. Following injection of the reaction mixture with a target residence time of 60 min, only traces of the desired bromide 2 were observed. Extending the residence time to 120 or 240 min increased the yield to 32–53%, but significant quantities of the starting alcohol 1 and the corresponding formate ester 3 were observed. Using tetra-n-butylammonium bromide (TBAB) as the halide source did not improve the yield, but resulted in larger amount of the formylated product 3 (Table 2, entry 4). A possible explanation for the increased yield of 3 when using TBAB could be due to slower displacement of leaving group by the “bulkier” source of bromide. In attempting to extend the residence time, the flow rate of the reaction mixture was decreased. Knowing that faster flow rates can improve mixing and reaction rates [32], an additional reactor was placed in line and the residence time of 240 min was repeated but with an increased flow rate (110 μL/min, Figure 4 and Table 2 entry 5). Gratifyingly, the desired bromide 2 was isolated in 91% yield.
Table 2: Photocatalytic conversion of alcohol 1 to bromide 2 in continuous flow.
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-251-i10.svg?max-width=637&scale=1.0)
|
||||||
| entry | halide source | Tres (min) | flow rate (μL/min) | recovered 1 (%)a | yield 2 (%)a | yield 3 (%)a |
|---|---|---|---|---|---|---|
| 1 | NaBr | 60 | 216 | 89 | <5 | <5 |
| 2 | NaBr | 120 | 110 | 47 | 53 | – |
| 3 | NaBr | 240 | 54.2 | 45 | 32 | 23 |
| 4 | TBAB | 240 | 110 | – | 21 | 63 |
| 5 | NaBr | 240 | 110 | – | 91(83)b | – |
aYield determined by analysis of 1H NMR. bYield determined by isolation via chromatography.
![[1860-5397-14-251-4]](/bjoc/content/figures/1860-5397-14-251-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Experimental set-up for the photocatalytic conversion of alcohols to bromides. PFA tubing is wrapped around purple LEDs (394 nm) and fans are placed underneath reactors to maintain cooling.
Figure 4: Experimental set-up for the photocatalytic conversion of alcohols to bromides. PFA tubing is wrappe...
With optimized flow conditions in hand for the formation of bromides in continuous flow, five different alcohols were converted to their corresponding halides (Table 3). The benzyl-protected alcohol 1 could be transformed to the bromide in 83% yield (Table 3, entry 1), as was citronellol (4, 83% yield, Table 3, entry 2). A methyl ester 5, allylic alcohol 8 and racemic secondary alcohol 9 could all undergo conversion to their corresponding bromides in 240 min using the continuous flow protocol (Table 3, entries 3 to 5).
Table 3: Photocatalytic conversion of alcohols to bromides in continuous flow.
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-251-i11.svg?max-width=637&scale=1.0)
|
|||
| entry | alcohol | yield (%)a | |
| 1 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-251-i12.svg?max-width=637&scale=1.0)
|
1 | 83 |
| 2 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-251-i13.svg?max-width=637&scale=1.0)
|
4 | 83 |
| 3 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-251-i14.svg?max-width=637&scale=1.0)
|
5 | 85 |
| 4 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-251-i15.svg?max-width=637&scale=1.0)
|
8 | 86 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-251-i16.svg?max-width=637&scale=1.0)
|
9 | 86 |
aYield determined by isolation via chromatography.
The continuous flow protocol was also applicable to the synthesis of anhydrides, which has also been previously reported by Stephenson and co-workers [33]. The carboxylic acid 10 was submitted to a flow protocol using the optimized Cu(tmp)(BINAP)BF4 catalyst, CBr4 (1 equiv) and 2,6-lutidine as base with a residence time of 20 min (Scheme 1). The anhydride derived from p-methoxybenzoic acid was isolated in 90% yield.
![[1860-5397-14-251-i1]](/bjoc/content/inline/1860-5397-14-251-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Copper-based photocatalysis for photocatalytic synthesis of an anhydride.
Scheme 1: Copper-based photocatalysis for photocatalytic synthesis of an anhydride.
Conclusion
In summary, a heteroleptic copper-based photocatalyst Cu(tmp)(BINAP)BF4 was discovered for the photochemical Appel-type conversion of alcohols to bromides, as well as carboxylic acids to their anhydrides. The protocol was highly efficient and could be adapted to continuous flow using purple LED reactors. The batch and continuous flow processes were all made possible due to the ability to screen highly modular copper-based complexes for photocatalysis.
Supporting Information
| Supporting Information File 1: Experimental details and compound characterization. | ||
| Format: PDF | Size: 450.2 KB | Download |
Acknowledgements
The authors acknowledge the Natural Sciences and Engineering Research Council of Canada (NSERC), the NSERC CREATE program in Continuous Flow Science, the Canadian Foundation for Innovation for financial support for continuous flow infrastructure and the Centre in Green Chemistry and Catalysis (CGCC) for funding. Ms. Vanessa Kairouz is thanked for assistance with the continuous flow infrastructure.
References
-
Stephenson, C. R. J.; Yoon, T. P.; MacMillan, D. W. C., Eds. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH: Hoboken, New Jersey, 2018.
Return to citation in text: [1] -
Albini, A.; Fagnoni, M., Eds. Handbook of Synthetic Photochemistry; Wiley-VCH: Germany, 2010.
Return to citation in text: [1] -
Rackl, D.; Kreitmeier, P.; Reiser, O. Green Chem. 2016, 18, 214–219. doi:10.1039/C5GC01792K
Return to citation in text: [1] -
Lima, C. G. S.; de M. Lima, T.; Duarte, M.; Jurberg, I. D.; Paixão, M. W. ACS Catal. 2016, 6, 1389–1407. doi:10.1021/acscatal.5b02386
Return to citation in text: [1] -
Appel, R. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. doi:10.1002/anie.197508011
Return to citation in text: [1] -
Weiss, R. G.; Snyder, E. I. Chem. Commun. 1968, 1358–1359. doi:10.1039/C19680001358
Return to citation in text: [1] -
Weiss, R. G.; Snyder, E. I. J. Org. Chem. 1971, 36, 403–406. doi:10.1021/jo00802a009
Return to citation in text: [1] -
Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949
Return to citation in text: [1] -
Larock, R. C. Comprehensive Organic Transformations, 2nd ed.; John Wiley & Sons, 1999; p 689.
Return to citation in text: [1] -
Fujisawa, T.; Iida, S.; Sato, T. Chem. Lett. 1984, 13, 1173–1174. doi:10.1246/cl.1984.1173
Return to citation in text: [1] -
Mukaiyama, T.; Shoda, S.-i.; Watanabe, Y. Chem. Lett. 1977, 6, 383–386. doi:10.1246/cl.1977.383
Return to citation in text: [1] -
Benazza, T.; Uzan, R.; Beaupère, D.; Demailly, G. Tetrahedron Lett. 1992, 33, 4901–4904. doi:10.1016/S0040-4039(00)61228-5
Return to citation in text: [1] -
Benazza, T.; Uzan, R.; Beaupère, D.; Demailly, G. Tetrahedron Lett. 1992, 33, 3129–3132. doi:10.1016/S0040-4039(00)79831-5
Return to citation in text: [1] -
Kelly, B. D.; Lambert, T. H. J. Am. Chem. Soc. 2009, 131, 13930–13931. doi:10.1021/ja906520p
Return to citation in text: [1] -
Zhao, Y.; Antonietti, M. ChemPhotoChem 2018, 2, 720–724. doi:10.1002/cptc.201800084
See for an alternative route to convert alcohols to iodides in the absence of catalyst.
Return to citation in text: [1] -
Reiser, O.; Kachkovskyi, G.; Kais, V.; Kohls, P.; Paria, S.; Pirtsch, M.; Rackl, D.; Seo, H. Chem. Photocatal. 2013, 139–150. doi:10.1515/9783110269246.139
Return to citation in text: [1] -
Higgins, R. F.; Fatur, S. M.; Shepard, S. G.; Stevenson, S. M.; Boston, D. J.; Ferreira, E. M.; Damrauer, N. H.; Rappé, A. K.; Shores, M. P. J. Am. Chem. Soc. 2016, 138, 5451–5464. doi:10.1021/jacs.6b02723
Return to citation in text: [1] -
Ruhl, K. E.; Rovis, T. J. Am. Chem. Soc. 2016, 138, 15527–15530. doi:10.1021/jacs.6b08792
Return to citation in text: [1] -
Hernandez-Perez, A. C.; Collins, S. K. Acc. Chem. Res. 2016, 49, 1557–1565. doi:10.1021/acs.accounts.6b00250
Return to citation in text: [1] -
Hernandez-Perez, A. C.; Collins, S. K. Angew. Chem., Int. Ed. 2013, 52, 12696–12700. doi:10.1002/anie.201306920
Return to citation in text: [1] -
Knorn, M.; Rawner, T.; Czerwieniec, R.; Reiser, O. ACS Catal. 2015, 5, 5186–5193. doi:10.1021/acscatal.5b01071
Return to citation in text: [1] -
Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474–1484. doi:10.1021/acs.accounts.5b00068
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449
Return to citation in text: [1] -
Arias-Rotondo, D. M.; McCusker, J. K. Chem. Soc. Rev. 2016, 45, 5803–5820. doi:10.1039/C6CS00526H
See for energy transfer.
Return to citation in text: [1] -
Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546–1556. doi:10.1021/acs.accounts.6b00272
See for proton-coupled electron transfer.
Return to citation in text: [1] -
Minozzi, C.; Caron, A.; Grenier-Petel, J.-C.; Santandrea, J.; Collins, S. K. Angew. Chem., Int. Ed. 2018, 57, 5477–5481. doi:10.1002/anie.201800144
Return to citation in text: [1] [2] -
See Supporting Information File 1 for tabular data on reaction yields.
Return to citation in text: [1] -
Fukui, K.; Morokuma, K.; Kato, H.; Yonezawa, T. Bull. Chem. Soc. Jpn. 1963, 36, 217–222. doi:10.1246/bcsj.36.217
Return to citation in text: [1] -
Parisien-Collette, S.; Cruché, C.; Abel-Snape, X.; Collins, S. K. Green Chem. 2017, 19, 4798–4803. doi:10.1039/C7GC02261A
Return to citation in text: [1] -
Parisien-Collette, S.; Collins, S. K. ChemPhotoChem 2018, 2, 855–859. doi:10.1002/cptc.201800096
Return to citation in text: [1] -
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1] -
Konieczynska, M. D.; Dai, C.; Stephenson, C. R. J. Org. Biomol. Chem. 2012, 10, 4509–4511. doi:10.1039/c2ob25463h
Return to citation in text: [1]
| 1. | Stephenson, C. R. J.; Yoon, T. P.; MacMillan, D. W. C., Eds. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH: Hoboken, New Jersey, 2018. |
| 2. | Albini, A.; Fagnoni, M., Eds. Handbook of Synthetic Photochemistry; Wiley-VCH: Germany, 2010. |
| 3. | Rackl, D.; Kreitmeier, P.; Reiser, O. Green Chem. 2016, 18, 214–219. doi:10.1039/C5GC01792K |
| 4. | Lima, C. G. S.; de M. Lima, T.; Duarte, M.; Jurberg, I. D.; Paixão, M. W. ACS Catal. 2016, 6, 1389–1407. doi:10.1021/acscatal.5b02386 |
| 9. | Larock, R. C. Comprehensive Organic Transformations, 2nd ed.; John Wiley & Sons, 1999; p 689. |
| 10. | Fujisawa, T.; Iida, S.; Sato, T. Chem. Lett. 1984, 13, 1173–1174. doi:10.1246/cl.1984.1173 |
| 11. | Mukaiyama, T.; Shoda, S.-i.; Watanabe, Y. Chem. Lett. 1977, 6, 383–386. doi:10.1246/cl.1977.383 |
| 12. | Benazza, T.; Uzan, R.; Beaupère, D.; Demailly, G. Tetrahedron Lett. 1992, 33, 4901–4904. doi:10.1016/S0040-4039(00)61228-5 |
| 13. | Benazza, T.; Uzan, R.; Beaupère, D.; Demailly, G. Tetrahedron Lett. 1992, 33, 3129–3132. doi:10.1016/S0040-4039(00)79831-5 |
| 14. | Kelly, B. D.; Lambert, T. H. J. Am. Chem. Soc. 2009, 131, 13930–13931. doi:10.1021/ja906520p |
| 33. | Konieczynska, M. D.; Dai, C.; Stephenson, C. R. J. Org. Biomol. Chem. 2012, 10, 4509–4511. doi:10.1039/c2ob25463h |
| 8. | Dai, C.; Narayanam, J. M. R.; Stephenson, C. R. J. Nat. Chem. 2011, 3, 140–145. doi:10.1038/nchem.949 |
| 6. | Weiss, R. G.; Snyder, E. I. Chem. Commun. 1968, 1358–1359. doi:10.1039/C19680001358 |
| 7. | Weiss, R. G.; Snyder, E. I. J. Org. Chem. 1971, 36, 403–406. doi:10.1021/jo00802a009 |
| 30. | Parisien-Collette, S.; Cruché, C.; Abel-Snape, X.; Collins, S. K. Green Chem. 2017, 19, 4798–4803. doi:10.1039/C7GC02261A |
| 31. | Parisien-Collette, S.; Collins, S. K. ChemPhotoChem 2018, 2, 855–859. doi:10.1002/cptc.201800096 |
| 5. | Appel, R. Angew. Chem., Int. Ed. Engl. 1975, 14, 801–811. doi:10.1002/anie.197508011 |
| 32. | Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707 |
| 27. | Minozzi, C.; Caron, A.; Grenier-Petel, J.-C.; Santandrea, J.; Collins, S. K. Angew. Chem., Int. Ed. 2018, 57, 5477–5481. doi:10.1002/anie.201800144 |
| 27. | Minozzi, C.; Caron, A.; Grenier-Petel, J.-C.; Santandrea, J.; Collins, S. K. Angew. Chem., Int. Ed. 2018, 57, 5477–5481. doi:10.1002/anie.201800144 |
| 22. | Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474–1484. doi:10.1021/acs.accounts.5b00068 |
| 23. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 24. | Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449 |
| 25. |
Arias-Rotondo, D. M.; McCusker, J. K. Chem. Soc. Rev. 2016, 45, 5803–5820. doi:10.1039/C6CS00526H
See for energy transfer. |
| 26. |
Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546–1556. doi:10.1021/acs.accounts.6b00272
See for proton-coupled electron transfer. |
| 29. | Fukui, K.; Morokuma, K.; Kato, H.; Yonezawa, T. Bull. Chem. Soc. Jpn. 1963, 36, 217–222. doi:10.1246/bcsj.36.217 |
| 19. | Hernandez-Perez, A. C.; Collins, S. K. Acc. Chem. Res. 2016, 49, 1557–1565. doi:10.1021/acs.accounts.6b00250 |
| 20. | Hernandez-Perez, A. C.; Collins, S. K. Angew. Chem., Int. Ed. 2013, 52, 12696–12700. doi:10.1002/anie.201306920 |
| 21. | Knorn, M.; Rawner, T.; Czerwieniec, R.; Reiser, O. ACS Catal. 2015, 5, 5186–5193. doi:10.1021/acscatal.5b01071 |
| 15. |
Zhao, Y.; Antonietti, M. ChemPhotoChem 2018, 2, 720–724. doi:10.1002/cptc.201800084
See for an alternative route to convert alcohols to iodides in the absence of catalyst. |
| 16. | Reiser, O.; Kachkovskyi, G.; Kais, V.; Kohls, P.; Paria, S.; Pirtsch, M.; Rackl, D.; Seo, H. Chem. Photocatal. 2013, 139–150. doi:10.1515/9783110269246.139 |
| 17. | Higgins, R. F.; Fatur, S. M.; Shepard, S. G.; Stevenson, S. M.; Boston, D. J.; Ferreira, E. M.; Damrauer, N. H.; Rappé, A. K.; Shores, M. P. J. Am. Chem. Soc. 2016, 138, 5451–5464. doi:10.1021/jacs.6b02723 |
| 18. | Ruhl, K. E.; Rovis, T. J. Am. Chem. Soc. 2016, 138, 15527–15530. doi:10.1021/jacs.6b08792 |
© 2018 Minozzi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)