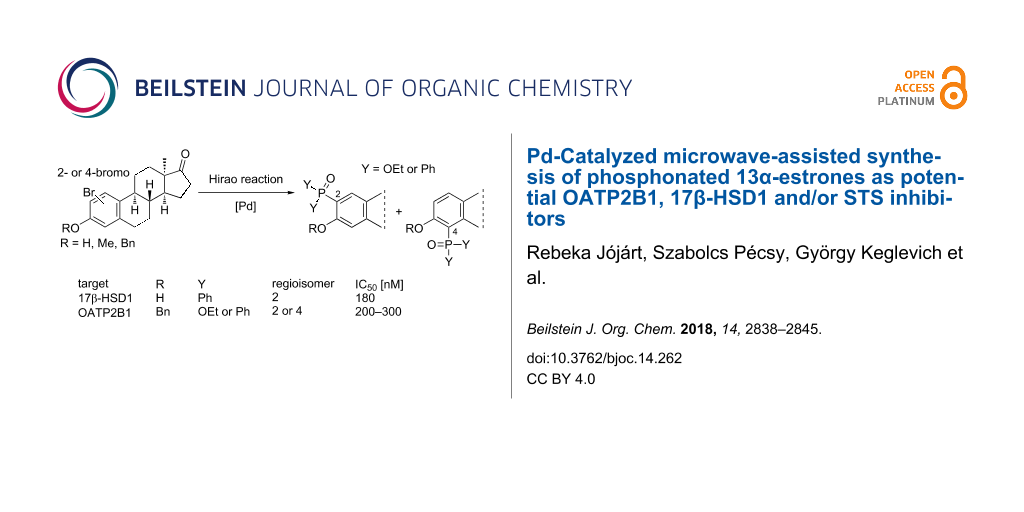
Abstract
Novel 2- or 4-phosphonated 13α-estrone derivatives were synthesized via the Hirao reaction. Bromo regioisomers (2- or 4-) of 13α-estrone and its 3-benzyl or 3-methyl ether were reacted with diethyl phosphite or diphenylphosphine oxide using Pd(PPh3)4 as catalyst under microwave irradiation. The influence of the new compounds on the transport function of the organic anion transporting polypeptide OATP2B1 was investigated by measuring Cascade Blue uptake. Derivatives bearing a 3-benzyl ether function displayed substantial submicromolar OATP2B1 inhibitory activity. The inhibitory effects of the compounds on human placental steroid sulfatase (STS) and 17β-hydroxysteroid dehydrogenase type 1 isozyme (17β-HSD1) were investigated by in vitro radiosubstrate incubation methods. None of the test compounds inhibited the STS markedly. The structure–activity relationship evaluation revealed that 2-substituted 3-hydroxy derivatives are able to inhibit the 17β-HSD1 enzyme with submicromolar IC50 values. Dual OATP2B1 and 17β-HSD1 inhibitors have been identified.

Graphical Abstract
Introduction
The biosynthesis of estrogens occurs via various enzymatic routes. Cytochrome P450 aromatase catalyzes the conversion of nonaromatic steroids to estrogens [1]. Moreover, hydrolysis of estrone 3-sulfate, existing as a large circulatory reservoir catalyzed by steroid sulfatase (STS), is one of the major alternative sources of prehormone estrone for the local supply of estrogens [2]. The availability of free estrone on the cellular level depends on the expression of STS and/or SULT, which catalyze the sulfation of the phenolic hydroxy function. On the other hand, estrone sulfate is not able to cross the cell membrane passively; therefore, a carrier is needed to mediate its transport across the lipid bilayer. These carriers are certain representatives of the solute carrier superfamily (SLC), including members of the organic anion transporting polypeptides (OATP) protein family [3,4]. Human OATP2B1 is one of the OATPs transporting estrone 3-sulfate, expressed in the intestine, blood–brain barrier, liver and placenta [5-9]. Moreover, OATP2B1 is overexpressed in certain malignancies, including breast cancer [10]. OATP2B1 transports its substrates, including estrone-sulfate in a sodium- and ATP-independent manner [11,12]. After entering the cells, estrone-3-sulfate is subjected to enzymatic conversions involving different cytosolic enzymes. The desulfation catalyzed by STS is followed by stereospecific reduction of the 17-oxo function leading to 17β-estradiol. This last step of the estrogen biosynthesis is catalyzed by the 17β-hydroxy steroid dehydrogenase type 1 isoenzyme (17β-HSD1) [13]. Thus STS and 17β-HSD1 became important drug targets in estrogen-dependent diseases [1,14]. Their inhibition could be a powerful strategy for the suppression of local estrogen production. Additionally, the entry of estrone-sulfate into the cells by inhibition of OATPs can be a good alternative [15,16]. Prior suppression of cytosolic enzymes involved in the estradiol biosynthesis, the transport of conjugated estrone derivatives, including estrone-3-sulfate could be blocked. Inhibitors based on the estrane core could have multiple inhibitory properties concerning the two enzymatic steps of the sulfatase pathway and OATP2B1-mediated membrane transport of estrone-sulfate.
The inhibitor design is usually based on the substrate of the target protein. The literature reveals diverse synthetic estrone derivatives as STS or 17β-HSD1 inhibitors [1,14], but to the best of our knowledge, there are no reports on estrone-based OATP2B1 inhibitors. Note that estrone itself has been shown to slightly inhibit estrone-3-sulfate uptake by OATP2B1 [6]. None of the steroidal STS or 17β-HSD1 inhibitors reached the clinical trial, which is mainly due to their retained estrogenic activity. This side-effect could be eliminated by the inhibitor design based on the 13-epimer of natural estrone (13α-estrone, 13αE1OH) [17,18]. Poirier et al. proved that 13α-derivatives of 3,17-estradiols have reduced binding affinity for estrogen receptor alpha and display no uterotropic activity [19]. The conformational change resulting from the inversion at C-13 leads to an epimer unable to bind to its nuclear receptors. We have recently established that 13α-estrone, irrespective of the substantial structural changes, displays an affinity to 17β-HSD1 similar to that of the natural substrate estrone [20]. This finding led us to design novel 17β-HSD1 inhibitors based on the hormonally inactive 13α-estrane core. Ring A of 13α-estrone was transformed in order to get 2-substituted derivatives [21-23]. On the basis of the crystal structure of 17β-HSD1 in its complex with 17β-estradiol, Möller et al. described, that there is an unoccupied lipophilic tunnel located near the C-2 atom of the steroid [24]. Their strategy for improving the inhibitory activity of natural estrone derivatives was the introduction of large halogens and phenethynyl or phenethyl functions onto C-2 of estrone. Their most potent compounds displayed nanomolar inhibitory activities. Our related previous work included the synthesis and in vitro 17β-HSD1 determination of similar derivatives of the hormonally inactive 13α-estrane core [21-23]. We found that 2- and/or 4-substitution with large halogens (Br or I) lead to submicromolar inhibitors irrespective of the position and number of the introduced halogens. In the case of phenethynyl- or phenethyl-13α-estrones, only 2-regioisomers displayed substantial inhibitory action, which did not depend on the hybrid state of carbon attached to C-2 [22]. Phan et al. described that halogenation of ring A of estrone is a powerful strategy in the synthesis of effective STS inhibitors [25]. Certain 4-halogenated estrone derivatives displayed higher affinity to the enzyme than that of their parent compound estrone. Based on these findings, we tested our halogenated 13α-estrone derivatives against STS, too [23]. Both the nature and the position of the introduced halogen influenced the STS inhibitory potential markedly. The presence of large substituents in 4-iodo and 2,4-diiodo compounds proved to be advantageous, since these compounds inhibited the enzyme with IC50 values in the low micromolar range. It can be stated that introduction of large lipophilic groups onto C-2 or C-4 of 13α-estrone might improve their affinity to 17β-HSD1 and STS, respectively. In order to get more structure–activity data concerning the effect of the nature and the position of the introduced functional group onto inhibitory activity, the synthesis and testing of novel ring A substituted 13α-estrone derivatives would be of particular interest. Taking into consideration that OATP2B1 is expressed in certain human malignancies and it transports estrone-3-sulfate effectively, it would be worth designing, synthesizing and investigating potential 13α-estrone-based inhibitors against this transporter protein, too. Ring A substituents differing in position, size and polarity are expected to have remarkable influence on the affinity for the target proteins. Investigation of the influence of the nature of the atom attached directly to the aromatic ring A of 13α-estrone and the stereochemistry of the introduced moiety would also be of high interest.
Here we disclose the synthesis of novel 2- or 4-substituted 13α-estrone derivatives 8–13 via the Hirao reaction (Scheme 1). Diethyl phosphite (7a) or diphenylphosphine oxide (7b) were chosen as >P(O)H reagents and C–P couplings were planned under microwave irradiation using Pd-based catalysis. Investigation of inhibitory activities of the newly-synthesized compounds 8–13 against the OATP2B1 transporter protein and 17β-HSD1 or STS cytosolic enzymes was also aimed.
![[1860-5397-14-262-i1]](/bjoc/content/inline/1860-5397-14-262-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pd-catalyzed C(sp2)–P couplings at C-2 or C-4 in the 13α-estrone series.
Scheme 1: Pd-catalyzed C(sp2)–P couplings at C-2 or C-4 in the 13α-estrone series.
Results and Discussion
The Hirao reaction is a powerful tool for the synthesis of arylphosphonates from aryl bromides or iodides using dialkyl phosphites (H-phosphonates) as the reagents, Pd(PPh3)4 as the catalyst and Et3N as the base [26]. Variations of the reaction have recently been published to prepare diverse functionalized phosphonates [27,28]. The C–P couplings have been performed not only under traditional thermal conditions, but the important benefits of microwave-irradiation have also been utilized in this field [29].
The optimization of reaction conditions was carried out using 2-bromo- or 2-iodo-13α-estrone 3-methyl ether (1 or 1I) as starting compounds and diethyl phosphite (7a) as the reagent (Table 1, Scheme 1). Two Pd sources, namely Pd(PPh3)4 or Pd(OAc)2 were used, the latter without the addition of any mono- or bidentate P-ligand. Although Et3N can usually be used successfully, the dealkylation of the phosphite reagent may occur. In order to avoid this unwanted side reaction, K2CO3 was also involved in optimization. The selection of acetonitrile as solvent was based on literature data [27-29]. Acetonitrile is one of the most extensively used solvents in the Hirao reaction. Couplings were carried out under microwave irradiation. The quantity of diethyl phosphite was selected according to literature data [29]; namely, 1 equiv in the case of Pd(PPh3)4 and 1.3 equiv when using Pd(OAc)2. It was earlier established that in the microwave-assisted Pd(II)-catalyzed C–P coupling of aryl bromides and dialkyl phosphites, an excess of the applied dialkyl phosphite may serve as phosphorus ligand and reducing agent [29].
Table 1: Effect of the reaction conditions on Hirao reaction of 2-bromo- or 2-iodo-13α-estrone 3-methyl ether (1 or 1I) with diethyl phosphite (7a).
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-262-i2.svg?max-width=637&scale=1.0)
|
||||||
| entry | (EtO)2P(O)H quantity [equiv] | Hlg | Pd source [mol %] | base | temperature [°C] | yielda [%] |
|---|---|---|---|---|---|---|
| 1 | 1.3 | Br | Pd(OAc)2 [10] | K2CO3 | 150 | 65 |
| 2 | 1.3 | Br | Pd(OAc)2 [10] | K2CO3 | 100 | 77 |
| 3 | 1.3 | I | Pd(OAc)2 [10] | K2CO3 | 150 | 61 |
| 4 | 1.3 | I | Pd(OAc)2 [10] | K2CO3 | 100 | 66 |
| 5 | 1.3 | Br | Pd(OAc)2 [10] | Et3N | 150 | 58 |
| 6 | 1.3 | Br | Pd(OAc)2 [10] | Et3N | 100 | 59 |
| 7 | 1.3 | I | Pd(OAc)2 [10] | Et3N | 150 | 60 |
| 8 | 1.3 | I | Pd(OAc)2 [10] | Et3N | 100 | 62 |
| 9 | 1.0 | Br | Pd(PPh3)4 [10] | K2CO3 | 150 | 73 |
| 10 | 1.0 | Br | Pd(PPh3)4 [10] | K2CO3 | 100 | 89 |
| 11 | 1.0 | I | Pd(PPh3)4 [10] | K2CO3 | 150 | 69 |
| 12 | 1.0 | I | Pd(PPh3)4 [10] | K2CO3 | 100 | 72 |
| 13 | 1.0 | Br | Pd(PPh3)4 [10] | Et3N | 150 | 70 |
| 14 | 1.0 | Br | Pd(PPh3)4 [10] | Et3N | 100 | 78 |
| 15 | 1.0 | I | Pd(PPh3)4 [10] | Et3N | 150 | 66 |
| 16 | 1.0 | I | Pd(PPh3)4 [10] | Et3N | 100 | 70 |
aObtained after flash chromatography.
The aryl bromide or iodide 1 or 1I was dissolved in acetonitrile, the reagent, the catalyst and the base were added and then the mixture was irradiated in a microwave reactor for 5 min. The nature of the Pd source, the base, the reaction temperature and the halogen substantially influenced the yields of the C–P couplings. Reactions with catalyst precursor Pd(OAc)2 gave the desired phosphonate 8a in moderate yields (Table 1, entries 1–8). Starting from 2-iodo compound 1I, dehalogenation, an unwanted side reaction occurred, in particular, at elevated temperature (Table 1, entries 3, 4, 7, 8). Pd(PPh3)4, used as catalyst in the classical Hirao reaction [26], proved to be more efficient concerning the yields of the desired arylphosphonate (8a, Table 1, entries 9–16). K2CO3 as inorganic base seemed to be superior to organic Et3N (Table 1, entries 1–4, 9–12). The extent of dehalogenation could be suppressed by lowering the reaction temperature (Table 1, entries 2, 4, 6, 8, 10, 12, 14, 16). Conditions described in entry 10 proved to be optimal for coupling of compound 1 with diethyl phosphite (7a). These conditions were selected for extension of C–P couplings to other steroidal scaffolds 2–6, which differ in regioisomerism and/or nature of the C-3 substituent. In order to get certain functionalized 13α-estrone derivatives with a different substitution pattern for biological investigations, not only 2-bromo 1–3, but 4-bromo compounds 4–6 were also coupled with the two reagents 7a and 7b differing in size and polarity (Table 2, Scheme 1). In certain cases (Table 2, entries 2–6 and 8–12), however, it was necessary to change the optimal conditions (Table 1, entry 10) concerning reaction time and temperature. 2-Regioisomers 1–3 could successfully be transformed without significant changes in reaction conditions (Table 2, entries 1, 7), but the reaction time had to be prolonged in four cases (Table 2, entries 3, 5, 9, 11). However, in the reactions of 2-bromo 3-benzyl ether 3 and the 4-regioisomers 4–6 higher temperature (150 °C) was needed (Table 2, entries 2, 4, 5, 6, 8, 10, 11,12). The steric hindrance resulting from the presence of ring B near C-4 might be the main cause for the harsher reaction conditions. This is in agreement with our recently published Sonogashira coupling procedure, which required higher temperatures starting from 4-regioisomers in comparison with those applied for their 2-substituted counterparts [22]. Concerning the nature of the C-3 substituent, methyl ethers 1 and 4 were overally the most reactive. Compounds bearing a 3-OH group 2 and 5 reacted slower due to the more activated nature of the phenolic ring A. It can be stated that the reaction time and temperature greatly depends on the nature of the C-3 substituent and on the position of the bromine, not on the size and polarity of the P-coupling agent. In the case of 3-benzyl ethers 3 and 6, solvent change from acetonitrile to toluene was required. This might be attributed to the decreased polarity of the 3-benzyloxy compounds 3 and 6. We have recently disclosed our finding that toluene is an appropriate solvent for C(sp2)–N coupling reactions of 2- or 4-bromo-13α-estrone 3-benzyl ethers [30]. The structures of the newly-synthesized derivatives 8–13 were established through 1H, 13C, 31P, COSY, HSQC and/or HMBC measurements.
Table 2: Scope of the reactiona of 2- or 4-bromo-13α-estrones 1–6 with diethyl phosphite (7a) or diphenylphosphine oxide (7b).
| entry | substrate | >P(O)H | solvent | temp [°C] | reaction time [min] | product | yieldb [%] | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-262-i3.svg?max-width=637&scale=1.0)
|
(EtO)2P(O)H | CH3CN | 100 | 5 | 8a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-262-i4.svg?max-width=637&scale=1.0)
|
89 |
| 2 | 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-262-i5.svg?max-width=637&scale=1.0)
|
(EtO)2P(O)H | CH3CN | 150 | 30 | 11a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-262-i6.svg?max-width=637&scale=1.0)
|
72 |
| 3 | 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-262-i7.svg?max-width=637&scale=1.0)
|
(EtO)2P(O)H | CH3CN | 100 | 15 | 9a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-262-i8.svg?max-width=637&scale=1.0)
|
87 |
| 4 | 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-262-i9.svg?max-width=637&scale=1.0)
|
(EtO)2P(O)H | CH3CN | 150 | 30 | 12a |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-262-i10.svg?max-width=637&scale=1.0)
|
71 |
| 5 | 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-262-i11.svg?max-width=637&scale=1.0)
|
(EtO)2P(O)H | toluene | 150 | 30 | 10a |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-262-i12.svg?max-width=637&scale=1.0)
|
90 |
| 6 | 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-262-i13.svg?max-width=637&scale=1.0)
|
(EtO)2P(O)H | toluene | 150 | 30 | 13a |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-262-i14.svg?max-width=637&scale=1.0)
|
79 |
| 7 | 1 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-262-i15.svg?max-width=637&scale=1.0)
|
Ph2P(O)H | CH3CN | 100 | 5 | 8b |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-262-i16.svg?max-width=637&scale=1.0)
|
93 |
| 8 | 4 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-262-i17.svg?max-width=637&scale=1.0)
|
Ph2P(O)H | CH3CN | 150 | 30 | 11b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-262-i18.svg?max-width=637&scale=1.0)
|
82 |
| 9 | 2 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-262-i19.svg?max-width=637&scale=1.0)
|
Ph2P(O)H | CH3CN | 100 | 15 | 9b |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-262-i20.svg?max-width=637&scale=1.0)
|
90 |
| 10 | 5 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-262-i21.svg?max-width=637&scale=1.0)
|
Ph2P(O)H | CH3CN | 150 | 30 | 12b |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-262-i22.svg?max-width=637&scale=1.0)
|
83 |
| 11 | 3 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-262-i23.svg?max-width=637&scale=1.0)
|
Ph2P(O)H | toluene | 150 | 30 | 10b |
![[Graphic 23]](/bjoc/content/inline/1860-5397-14-262-i24.svg?max-width=637&scale=1.0)
|
92 |
| 12 | 6 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-14-262-i25.svg?max-width=637&scale=1.0)
|
Ph2P(O)H | toluene | 150 | 30 | 13b |
![[Graphic 25]](/bjoc/content/inline/1860-5397-14-262-i26.svg?max-width=637&scale=1.0)
|
78 |
aReactions were performed on a 0.25 mmol scale with 10 mol % Pd(PPh3)4 and K2CO3 (1.5 equiv) under microwave irradiation. bFlash chromatography yields are reported.
The influence of the basic (13αE1OH, 13αE1OMe and 13αE1OBn) and new compounds 8–13 on the transport function of OATP2B1 was investigated by measuring Cascade Blue uptake (Table 3) [31]. 13α-Estrone (13αE1OH) itself displayed weak inhibitory action with an IC50 value above 50 μM. The presence of the methyl group at the phenolic OH function in compound 13αE1OMe increased inhibition dramatically. The 3-benzyl ether (13αE1OBn) exerted unexpected twofold inhibitory potency compared with that of its 3-methoxy counterpart 13αE1OMe. All newly-synthesized derivatives 8–13 displayed substantial inhibition on the transport function of OATP2B1, except for 4-substituted 3-OH compound 12b. Derivatives 9a, 9b and 12a bearing a 3-OH function and 2- or 4-substituted 3-MeO compounds 8 and 11 suppressed the transport in the low micromolar range. In the 3-OH series there appeared to be significant difference between the IC50 values of the potent derivatives 9a, 9b and 12a and that of 13αE1OH. Furthermore, the inhibitory action of the basic reference compound 13αE1OMe was improved in compounds 8 and 11. Surprisingly, but in accordance with the data obtained for compound 13αE1OBn, the most apolar 3-benzyl ethers 10 and 13 proved to be the best inhibitors with IC50 values in the submicromolar range. Accordingly, the inhibitory action of 13αE1OBn could be improved with one order of magnitude by the introduction of a diethyl phosphono group onto C-2 of 13αE1OBn. The most significant difference in IC50 values concerning the regioisomerism was observed among 3-OH test compounds 9b and 12b bearing the larger substituent. This might be attributed to certain intramolecular secondary interactions of the newly introduced substituent and the phenolic OH function, which may prevent the binding of the steroid to the transporter protein. Additionally, inhibitory potencies of the newly-synthesized compounds on human placental STS and 17β-HSD1 have been tested in vitro (Table 3). None of the newly-synthesized derivatives proved to be a potent STS inhibitor, since the compounds suppressed the estrone-sulfate to estrone conversion by less than 33% (applied in 10 μM test concentration). Henceforth we discuss the results of the 17β-HSD1 investigations. Compounds bearing phenolic OH function displayed the most potent inhibitory action, but exclusively the 2-regioisomers 9a and 9b. Introduction of a larger and less polar substituent onto C-2 seemed to be more advantageous, since compound 9b displayed one order of magnitude lower IC50 value than that of its 9a counterpart (IC50 values are 0.18 and 1.5 μM, respectively). A comparison of the IC50 values of compound 9b and 13αE1OH indicates that introduction of a large diphenylphosphine oxide moiety onto C-2 improves the binding of the steroid to the enzyme. C-4-substituted 3-OH derivatives 12a and 12b barely suppressed the estrone to 17β-estradiol conversion. The same tendency showed up in our previous works regarding the inhibitory potentials of certain 2- or 4-halogenated or phenylalkynylated 13α-estrone derivatives [22,23]. Among 3-ethers, only the methyl ether analog of compound 9b proved to be potent inhibitor with an IC50 value of 4.1 μM. The data obtained here for 3-methyl ethers do not correlate with those described earlier for 2- or 4-halogenated 3-MeO derivatives [21]. We established previously that 4-halo-3-methoxy derivatives are more potent 17β-HSD1 inhibitors than their 2-counterparts.
Table 3: OATP2B1, STS and 17β-HSD1 inhibition data of C–P coupled products 8–13 and their basic compounds 13αE1OMe, 13αE1OH, 13αE1OBn.
| compound | OATP2B1 IC50 ± SD [μM] | STS rel. conv. ± SDa [%] | 17β-HSD1 | |
|---|---|---|---|---|
| IC50 ± SD [μM] | rel. conv. ± SD [%] | |||
| 13αE1OMe | 3.4 ± 0.3 | 99 ± 3 | 5.5 ± 1.5 [20] | 99 ± 3 |
| 13αE1OH | >50 | 96 ± 1 [23] | 1.2 ± 0.2 [20] | 96 ± 1 [23] |
| 13αE1OBn | 1.7 ± 0.9 | 94 ± 3 | 94 ± 3 | |
| 8a | 1.8 ± 0.3 | 84 ± 4 | 60 ± 3 | |
| 11a | 2.8 ± 1.5 | 93 ± 7 | 100 ± 5 | |
| 9a | 2.1 ± 0.2 | 97 ± 5 | 1.5 ± 0.3 | |
| 12a | 1.4 ± 0.1 | 93 ± 6 | 84 ± 4 | |
| 10a | 0.2 ± 0.02 | 85 ± 7 | 64 ± 3 | |
| 13a | 0.3 ± 0.02 | 87 ± 5 | 92 ± 3 | |
| 8b | 1.2 ± 0.1 | 80 ± 8 | 4.1 ± 1.1 | |
| 11b | 2.5 ± 0.4 | 80 ± 5 | 106 ± 8 | |
| 9b | 2.6 ± 0.2 | 77 ± 10 | 0.18 ± 0.06 | |
| 12b | >50 | 93 ± 4 | 100 ± 5 | |
| 10b | 1 ± 0.1 | 85 ± 5 | 73 ± 3 | |
| 13b | 0.3 ± 0.02 | 105 ± 7 | 75 ± 2 | |
aMean ± standard deviation, n = 3.
Conclusion
In conclusion, we have developed an efficient microwave-assisted Pd-catalyzed C–P coupling procedure for the synthesis of 2- or 4-phosphonated 13α-estrone derivatives 8–13. The elaborated methodology proved to be suitable for the facile transformation of steroidal aryl bromides 1–6 containing OH, OMe or OBn functions in ortho positions. 2-Regioisomers of 3-OH derivatives 9a,b appeared to be dual OATP2B1 and 17β-HSD1 inhibitors. The most selective compounds are the 3-benzyl ethers 10, 13, which inhibit only OATP2B1 from the three investigated targets. To the best of our knowledge, there are no literature reports concerning potent estrone-based OATP2B1 inhibitors.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information
| Supporting Information File 1: General synthetic procedures, characterization data for the synthesized compounds and biological assay methods. | ||
| Format: PDF | Size: 449.8 KB | Download |
Acknowledgements
The work of Csilla Özvegy-Laczka was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. The work of Erzsébet Mernyák in this project was supported by the ÚNKP-17-4 „NEW NATIONAL EXCELLENCE PROGRAM OF THE MINISTRY OF HUMAN CAPACITIES”. The authors are grateful for financial support from the Hungarian Scientific Research Fund OTKA SNN124329 and FK128751.
References
-
Miller, W. L.; Auchus, R. J. Endocr. Rev. 2011, 32, 81–151. doi:10.1210/er.2010-0013
Return to citation in text: [1] [2] [3] -
Geyer, J.; Bakhaus, K.; Bernhardt, R.; Blaschka, C.; Dezhkam, Y.; Fietz, D.; Grosser, G.; Hartmann, K.; Hartmann, M. F.; Neunzig, J.; Papadopoulos, D.; Sánchez-Guijo, A.; Scheiner-Bobis, G.; Schuler, G.; Shihan, M.; Wrenzycki, C.; Wudy, S. A.; Bergmann, M. J. Steroid Biochem. Mol. Biol. 2017, 172, 207–221. doi:10.1016/j.jsbmb.2016.07.002
Return to citation in text: [1] -
Izumi, S.; Nozaki, Y.; Komori, T.; Maeda, K.; Takenaka, O.; Kusano, K.; Yoshimura, Y.; Kusuhara, H.; Sugiyama, Y. Drug Metab. Dispos. 2013, 41, 1859–1866. doi:10.1124/dmd.113.052290
Return to citation in text: [1] -
Roth, M.; Obaidat, A.; Hagenbuch, B. Br. J. Pharmacol. 2012, 165, 1260–1287. doi:10.1111/j.1476-5381.2011.01724.x
Return to citation in text: [1] -
Hagenbuch, B.; Stieger, B. Mol. Aspects Med. 2013, 3, 396–412. doi:10.1016/j.mam.2012.10.009
Return to citation in text: [1] -
Grube, M.; Köck, K.; Karner, S.; Reuther, S.; Ritter, C. A.; Jedlitschky, G.; Kroemer, H. K. Mol. Pharmacol. 2006, 70, 1735–1741. doi:10.1124/mol.106.026450
Return to citation in text: [1] [2] -
Grube, M.; Reuther, S.; Meyer zu Schwabedissen, H.; Köck, K.; Draber, K.; Ritter, C. A.; Fusch, C.; Jedlitschky, G.; Kroemer, H. K. Drug Metab. Dispos. 2007, 35, 30–35. doi:10.1124/dmd.106.011411
Return to citation in text: [1] -
Knauer, M. J.; Girdwood, A. J.; Kim, R. B.; Tirona, R. G. Mol. Pharmacol. 2013, 83, 1218–1228. doi:10.1124/mol.112.083618
Return to citation in text: [1] -
Kovacsics, D.; Patik, I.; Özvegy-Laczka, C. Expert Opin. Drug Metab. Toxicol. 2017, 13, 409–424. doi:10.1080/17425255.2017.1253679
Return to citation in text: [1] -
Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] -
Schweigmann, H.; Sánchez-Guijo, A.; Ugele, B.; Hartmann, K.; Hartmann, M. F.; Bergmann, M.; Pfarrer, C.; Döring, B.; Wudy, S. A.; Petzinger, E.; Geyer, J.; Grosser, G. J. Steroid Biochem. Mol. Biol. 2014, 143, 259–265. doi:10.1016/j.jsbmb.2014.03.013
Return to citation in text: [1] -
Ugele, B.; Bahn, A.; Rex-Haffner, M. J. Steroid Biochem. Mol. Biol. 2008, 111, 1–6. doi:10.1016/j.jsbmb.2008.04.001
Return to citation in text: [1] -
Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; Frotscher, M.; Hartmann, R. W.; Adamski, J. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. doi:10.1016/j.jsbmb.2010.12.013
Return to citation in text: [1] -
Hong, Y.; Chen, S. Mol. Cell. Endocrinol. 2011, 340, 120–126. doi:10.1016/j.mce.2010.09.012
Return to citation in text: [1] [2] -
Buxhofer-Ausch, V.; Secky, L.; Wlcek, K.; Svoboda, M.; Kounnis, V.; Briasoulis, E.; Tzakos, A. G.; Jaeger, W.; Thalhammer, T. J. Drug Delivery 2013, No. 863539. doi:10.1155/2013/863539
Return to citation in text: [1] -
Arakawa, H.; Nakanishi, T.; Yanagihara, C.; Nishimoto, T.; Wakayama, T.; Mizokami, A.; Namiki, M.; Kawai, K.; Tamai, I. Biochem. Pharmacol. 2012, 84, 1070–1077. doi:10.1016/j.bcp.2012.07.026
Return to citation in text: [1] -
Yaremenko, F. G.; Khvat, A. V. Mendeleev Commun. 1994, 4, 187–188. doi:10.1070/mc1994v004n05abeh000409
Return to citation in text: [1] -
Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. J. Org. Chem. 2000, 65, 5487–5497. doi:10.1021/jo000108x
Return to citation in text: [1] -
Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. doi:10.1016/j.jsbmb.2011.07.009
Return to citation in text: [1] -
Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610
Return to citation in text: [1] [2] [3] -
Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008
Return to citation in text: [1] [2] [3] -
Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B. E.; Szécsi, M.; Mernyák, E. Beilstein J. Org. Chem. 2017, 13, 1303–1309. doi:10.3762/bjoc.13.126
Return to citation in text: [1] [2] [3] [4] [5] -
Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. doi:10.1016/j.bmcl.2009.09.113
Return to citation in text: [1] -
Phan, C.-M.; Liu, Y.; Kim, B.-m.; Mostafa, Y.; Taylor, S. D. Bioorg. Med. Chem. 2011, 19, 5999–6005. doi:10.1016/j.bmc.2011.08.046
Return to citation in text: [1] -
Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Tetrahedron Lett. 1980, 21, 3595–3598. doi:10.1016/0040-4039(80)80245-0
Return to citation in text: [1] [2] -
Kegelvich, G.; Jablonkai, E.; Balázs, L. B. RSC Adv. 2014, 4, 22808–22816. doi:10.1039/c4ra03292f
Return to citation in text: [1] [2] -
Dziuganowska, Z. A.; Ślepokura, K.; Volle, J.-N.; Virieux, D.; Pirat, J.-L.; Kafarski, P. J. Org. Chem. 2016, 81, 4947–4954. doi:10.1021/acs.joc.6b00220
Return to citation in text: [1] [2] -
Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N. Z. Adv. Synth. Catal. 2017, 359, 4322–4331. doi:10.1002/adsc.201700895
Return to citation in text: [1] [2] [3] [4] -
Bacsa, I.; Szemerédi, D.; Wölfling, J.; Schneider, G.; Fekete, L.; Mernyák, E. Beilstein J. Org. Chem. 2018, 14, 998–1003. doi:10.3762/bjoc.14.85
Return to citation in text: [1] -
Patik, I.; Székely, V.; Német, O.; Szepesi, Á.; Kucsma, N.; Várady, G.; Szakács, G.; Bakos, É.; Özvegy-Laczka, C. Sci. Rep. 2018, 8, No. 2630. doi:10.1038/s41598-018-20815-1
Return to citation in text: [1]
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 21. | Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008 |
| 20. | Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610 |
| 31. | Patik, I.; Székely, V.; Német, O.; Szepesi, Á.; Kucsma, N.; Várady, G.; Szakács, G.; Bakos, É.; Özvegy-Laczka, C. Sci. Rep. 2018, 8, No. 2630. doi:10.1038/s41598-018-20815-1 |
| 22. | Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B. E.; Szécsi, M.; Mernyák, E. Beilstein J. Org. Chem. 2017, 13, 1303–1309. doi:10.3762/bjoc.13.126 |
| 23. | Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731 |
| 22. | Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B. E.; Szécsi, M.; Mernyák, E. Beilstein J. Org. Chem. 2017, 13, 1303–1309. doi:10.3762/bjoc.13.126 |
| 30. | Bacsa, I.; Szemerédi, D.; Wölfling, J.; Schneider, G.; Fekete, L.; Mernyák, E. Beilstein J. Org. Chem. 2018, 14, 998–1003. doi:10.3762/bjoc.14.85 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 26. | Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Tetrahedron Lett. 1980, 21, 3595–3598. doi:10.1016/0040-4039(80)80245-0 |
| 20. | Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610 |
| 23. | Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731 |
| 23. | Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731 |
| 1. | Miller, W. L.; Auchus, R. J. Endocr. Rev. 2011, 32, 81–151. doi:10.1210/er.2010-0013 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 21. | Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008 |
| 22. | Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B. E.; Szécsi, M.; Mernyák, E. Beilstein J. Org. Chem. 2017, 13, 1303–1309. doi:10.3762/bjoc.13.126 |
| 23. | Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731 |
| 5. | Hagenbuch, B.; Stieger, B. Mol. Aspects Med. 2013, 3, 396–412. doi:10.1016/j.mam.2012.10.009 |
| 6. | Grube, M.; Köck, K.; Karner, S.; Reuther, S.; Ritter, C. A.; Jedlitschky, G.; Kroemer, H. K. Mol. Pharmacol. 2006, 70, 1735–1741. doi:10.1124/mol.106.026450 |
| 7. | Grube, M.; Reuther, S.; Meyer zu Schwabedissen, H.; Köck, K.; Draber, K.; Ritter, C. A.; Fusch, C.; Jedlitschky, G.; Kroemer, H. K. Drug Metab. Dispos. 2007, 35, 30–35. doi:10.1124/dmd.106.011411 |
| 8. | Knauer, M. J.; Girdwood, A. J.; Kim, R. B.; Tirona, R. G. Mol. Pharmacol. 2013, 83, 1218–1228. doi:10.1124/mol.112.083618 |
| 9. | Kovacsics, D.; Patik, I.; Özvegy-Laczka, C. Expert Opin. Drug Metab. Toxicol. 2017, 13, 409–424. doi:10.1080/17425255.2017.1253679 |
| 24. | Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. doi:10.1016/j.bmcl.2009.09.113 |
| 3. | Izumi, S.; Nozaki, Y.; Komori, T.; Maeda, K.; Takenaka, O.; Kusano, K.; Yoshimura, Y.; Kusuhara, H.; Sugiyama, Y. Drug Metab. Dispos. 2013, 41, 1859–1866. doi:10.1124/dmd.113.052290 |
| 4. | Roth, M.; Obaidat, A.; Hagenbuch, B. Br. J. Pharmacol. 2012, 165, 1260–1287. doi:10.1111/j.1476-5381.2011.01724.x |
| 19. | Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. doi:10.1016/j.jsbmb.2011.07.009 |
| 2. | Geyer, J.; Bakhaus, K.; Bernhardt, R.; Blaschka, C.; Dezhkam, Y.; Fietz, D.; Grosser, G.; Hartmann, K.; Hartmann, M. F.; Neunzig, J.; Papadopoulos, D.; Sánchez-Guijo, A.; Scheiner-Bobis, G.; Schuler, G.; Shihan, M.; Wrenzycki, C.; Wudy, S. A.; Bergmann, M. J. Steroid Biochem. Mol. Biol. 2017, 172, 207–221. doi:10.1016/j.jsbmb.2016.07.002 |
| 20. | Herman, B. E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. J. Enzyme Inhib. Med. Chem. 2016, 31, 61–69. doi:10.1080/14756366.2016.1204610 |
| 15. | Buxhofer-Ausch, V.; Secky, L.; Wlcek, K.; Svoboda, M.; Kounnis, V.; Briasoulis, E.; Tzakos, A. G.; Jaeger, W.; Thalhammer, T. J. Drug Delivery 2013, No. 863539. doi:10.1155/2013/863539 |
| 16. | Arakawa, H.; Nakanishi, T.; Yanagihara, C.; Nishimoto, T.; Wakayama, T.; Mizokami, A.; Namiki, M.; Kawai, K.; Tamai, I. Biochem. Pharmacol. 2012, 84, 1070–1077. doi:10.1016/j.bcp.2012.07.026 |
| 6. | Grube, M.; Köck, K.; Karner, S.; Reuther, S.; Ritter, C. A.; Jedlitschky, G.; Kroemer, H. K. Mol. Pharmacol. 2006, 70, 1735–1741. doi:10.1124/mol.106.026450 |
| 1. | Miller, W. L.; Auchus, R. J. Endocr. Rev. 2011, 32, 81–151. doi:10.1210/er.2010-0013 |
| 14. | Hong, Y.; Chen, S. Mol. Cell. Endocrinol. 2011, 340, 120–126. doi:10.1016/j.mce.2010.09.012 |
| 17. | Yaremenko, F. G.; Khvat, A. V. Mendeleev Commun. 1994, 4, 187–188. doi:10.1070/mc1994v004n05abeh000409 |
| 18. | Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. J. Org. Chem. 2000, 65, 5487–5497. doi:10.1021/jo000108x |
| 13. | Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; Frotscher, M.; Hartmann, R. W.; Adamski, J. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. doi:10.1016/j.jsbmb.2010.12.013 |
| 11. | Schweigmann, H.; Sánchez-Guijo, A.; Ugele, B.; Hartmann, K.; Hartmann, M. F.; Bergmann, M.; Pfarrer, C.; Döring, B.; Wudy, S. A.; Petzinger, E.; Geyer, J.; Grosser, G. J. Steroid Biochem. Mol. Biol. 2014, 143, 259–265. doi:10.1016/j.jsbmb.2014.03.013 |
| 12. | Ugele, B.; Bahn, A.; Rex-Haffner, M. J. Steroid Biochem. Mol. Biol. 2008, 111, 1–6. doi:10.1016/j.jsbmb.2008.04.001 |
| 1. | Miller, W. L.; Auchus, R. J. Endocr. Rev. 2011, 32, 81–151. doi:10.1210/er.2010-0013 |
| 14. | Hong, Y.; Chen, S. Mol. Cell. Endocrinol. 2011, 340, 120–126. doi:10.1016/j.mce.2010.09.012 |
| 25. | Phan, C.-M.; Liu, Y.; Kim, B.-m.; Mostafa, Y.; Taylor, S. D. Bioorg. Med. Chem. 2011, 19, 5999–6005. doi:10.1016/j.bmc.2011.08.046 |
| 21. | Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B. E.; Szécsi, M.; Mernyák, E. Steroids 2015, 104, 230–236. doi:10.1016/j.steroids.2015.10.008 |
| 22. | Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B. E.; Szécsi, M.; Mernyák, E. Beilstein J. Org. Chem. 2017, 13, 1303–1309. doi:10.3762/bjoc.13.126 |
| 23. | Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731 |
| 22. | Bacsa, I.; Jójárt, R.; Wölfling, J.; Schneider, G.; Herman, B. E.; Szécsi, M.; Mernyák, E. Beilstein J. Org. Chem. 2017, 13, 1303–1309. doi:10.3762/bjoc.13.126 |
| 29. | Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N. Z. Adv. Synth. Catal. 2017, 359, 4322–4331. doi:10.1002/adsc.201700895 |
| 10. | Matsumoto, J.; Ariyoshi, N.; Sakakibara, M.; Nakanishi, T.; Okubo, Y.; Shiina, N.; Fujisaki, K.; Nagashima, T.; Nakatani, Y.; Tamai, I.; Yamada, H.; Takeda, H.; Ishiia, I. Drug Metab. Pharmacokinet. 2015, 30, 133–141. doi:10.1016/j.dmpk.2014.10.005 |
| 27. | Kegelvich, G.; Jablonkai, E.; Balázs, L. B. RSC Adv. 2014, 4, 22808–22816. doi:10.1039/c4ra03292f |
| 28. | Dziuganowska, Z. A.; Ślepokura, K.; Volle, J.-N.; Virieux, D.; Pirat, J.-L.; Kafarski, P. J. Org. Chem. 2016, 81, 4947–4954. doi:10.1021/acs.joc.6b00220 |
| 29. | Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N. Z. Adv. Synth. Catal. 2017, 359, 4322–4331. doi:10.1002/adsc.201700895 |
| 29. | Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N. Z. Adv. Synth. Catal. 2017, 359, 4322–4331. doi:10.1002/adsc.201700895 |
| 27. | Kegelvich, G.; Jablonkai, E.; Balázs, L. B. RSC Adv. 2014, 4, 22808–22816. doi:10.1039/c4ra03292f |
| 28. | Dziuganowska, Z. A.; Ślepokura, K.; Volle, J.-N.; Virieux, D.; Pirat, J.-L.; Kafarski, P. J. Org. Chem. 2016, 81, 4947–4954. doi:10.1021/acs.joc.6b00220 |
| 29. | Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N. Z. Adv. Synth. Catal. 2017, 359, 4322–4331. doi:10.1002/adsc.201700895 |
| 23. | Bacsa, I.; Herman, B. E.; Jójárt, R.; Herman, K. S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanisnik-Rizner, T.; Szécsi, M.; Mernyák, E. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. doi:10.1080/14756366.2018.1490731 |
| 26. | Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. Tetrahedron Lett. 1980, 21, 3595–3598. doi:10.1016/0040-4039(80)80245-0 |
© 2018 Jójárt et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)