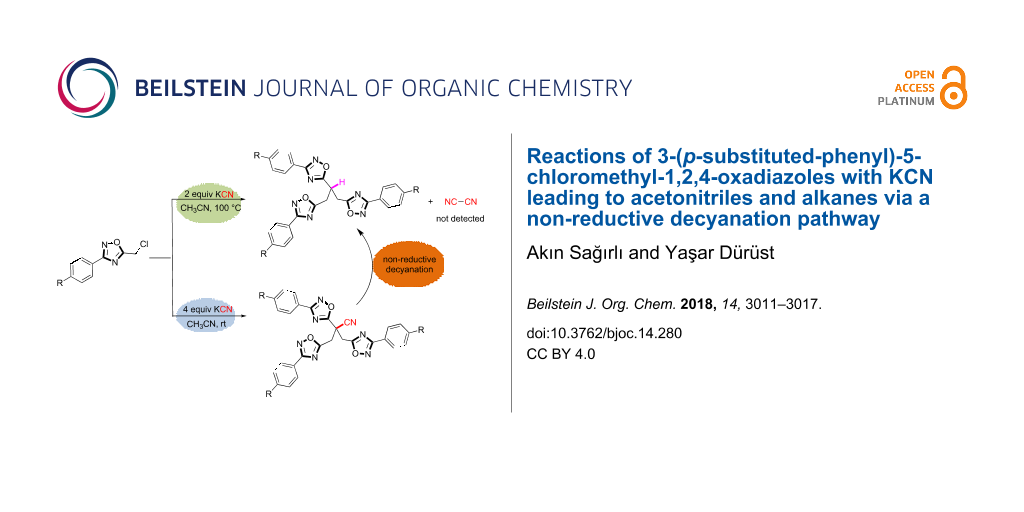
Abstract
The present work describes an unfamiliar reaction of 5-(chloromethyl)-3-substituted-phenyl-1,2,4-oxadiazoles with KCN affording trisubstituted 1,2,4-oxadiazol-5-ylacetonitriles and their parent alkanes, namely, 1,2,3-trisubstituted-1,2,4-oxadiazol-5-ylpropanes. To the best of our knowledge, the current synthetic route leading to decyanated products will be the first in terms of a decyanation process which allows the transformation of trisubstituted acetonitriles into alkanes by the incorporation of KCN with the association of in situ-formed HCN and most likely through the extrusion of cyanogen which could not be detected or isolated. In addition, the plausible mechanisms were proposed for both transformations. The structures of the title compounds were identified by means of IR, 1H NMR, 13C NMR, 2D NMR spectra, TOF–MS and X-ray measurements.

Graphical Abstract
Introduction
Heterocyclic scaffolds bearing 1,2,4-oxadiazole rings have been the subject of an increasing and remarkable attention due to their various bioactivities, such as anticancer [1], antimicrobial [2], antifungal [3], anti-inflammatory [4], tyrosine kinase inhibition [5] and histamine H3 antagonism properties [6]. In addition, these five-membered heterocycles were widely used as components of organic light emitting diodes (OLEDs), polymers, liquid crystals, and solar cells [7-10]. Taking into account the above considerations, new synthetic protocols to develop 1,2,4-oxadiazole-based heterocycles have gained an increasing attention over the recent decades [11-13].
On the other hand, arylacetonitriles are known as valuable intermediates that are generally obtained by the reaction of benzyl halides with appropriate cyanating agents such as KCN [14], TMSCN [15], K4(Fe(CN)6 [16]. Deprotonation of the α-carbon (adjacent to nitrile) by strong bases, especially lithiated ones, resulted in an anionic species that easily undergoes a substitution reaction with various alkyl halides to afford mono-, di- or trialkylated acetonitriles [17]. Most recently, Strzalko and co-workers disclose mono and dialkylation of the benzylic carbon of phenylacetonitrile with a poor selectivity by benzyl and methyl halides in the presence of LiHMDS, LDA or n-BuLi (Figure 1) [18]. However, such alkylation methods need harsh reaction conditions, most particularly lithiated bases and inert atmosphere.
![[1860-5397-14-280-1]](/bjoc/content/figures/1860-5397-14-280-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthesis of mono- or dialkylated acetonitriles.
Figure 1: Synthesis of mono- or dialkylated acetonitriles.
In order to get a deeper understanding of the limitation step of alkylation processes and to extend their scope in the synthesis of mono-, di- or trialkylated structures, cyanation attempts of 5-(chloromethyl)-3-phenyl-1,2,4-oxadiazoles 1 with excess KCN at room temperature in CH3CN have been investigated leading to trisubstituted acetonitrile 3 instead of anticipated product 2. This result is in accord with a previous report where only one example (3a) has been exploited by providing very limited structural data [19]. Interestingly, increasing the reaction temperature to 100 °C yielded 1,2,4-oxadiazole-substituted propanes 4 as the major products which can only be interpreted via a decyanation pathway of cyanated oxadiazoles 3 (Figure 2).
![[1860-5397-14-280-2]](/bjoc/content/figures/1860-5397-14-280-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyanation through 5-chloromethyl-3-(p-substituted-phenyl)-1,2,4-oxadiazole.
Figure 2: Cyanation through 5-chloromethyl-3-(p-substituted-phenyl)-1,2,4-oxadiazole.
Up to date, various methods have been reported for the conversion of organic nitriles into the parent alkanes; such as oxidative decyanation [20], dehydrocyanation [21] and more commonly, reductive decyanation [22]. Among them, the reductive decyanation is a widely used method employing metal hydrides [23], electrolysis [24], transition metal complexes [25], and alkali metals in a variety of solvents [26]. Generally, they require mostly an inert atmosphere as well as harsh reaction conditions. These facts constitute challenges to the development of mild reaction conditions in the reduction of nitrilated compounds.
In the light of the above considerations, we report here an unfamiliar example of non-reductive decyanation through the reaction of 5-(chloromethyl)-3-(substituted-phenyl)-1,2,4-oxadiazoles 1 with 2 equiv of KCN at 100 °C in a single step transformation.
Results and Discussion
In the first part of this work, 5-(chloromethyl)-3-(substituted-phenyl)-1,2,4-oxadiazoles 1 were obtained via a previously published literature procedure by reacting p-substituted benzamidoximes with chloroacetyl chloride in refluxing benzene [27]. After having been obtained the starting oxadiazoles, our goal was to replace the chlorine atom with a CN− anion by a simple SN2 reaction. For this purpose, 5-chloromethyl-1,2,4-oxadiazole 1a was reacted with 10 equiv of KCN in CH3CN at reflux, after completion of the reaction on the basis of TLC, surprisingly we obtained a mixture of products 4 (major) and 3 (minor, Table 1, entry 1). This unexpected product distribution of the reaction prompted us to investigate the reaction of 5-(chloromethyl)-3-(p-substituted-phenyl)-1,2,4-oxadiazoles 1 with KCN.
Our initial effort was to investigate the limitation of the reaction. For this, the effect of temperature on this reaction was examined in CH3CN with various equivalents of KCN and varying reaction times (Table 1, entries 2–6). Treatment of 1a with 10 equiv of KCN at 100 °C gave mainly product 4a with a yield of 75%, as well as product 3a in trace amounts even when heating at 110 °C. Due to the toxic nature of KCN, the minimum amount of KCN was determined for optimal formation of 4a. When 2 equiv of KCN was used instead of 10 equiv at 100 °C in CH3CN, the reaction proceeded to give compound 4a with a yield of 75% again, but 24 h for completion of the reaction were needed.
Table 1: Optimisation of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-280-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | KCN (equiv)a | Conditions | Yield (%)b | |
| 4a | 3a | |||
| 1 | 10 | CH3CN, 80 °C, 3 h | 55 | 32 |
| 2 | 10 | CH3CN, 80 °C, 12 h | 70 | 15 |
| 3 | 10 | CH3CN, 100 °C, 12 h | 75 | trace |
| 4 | 10 | CH3CN, 110 °C, 12 h | 75 | trace |
| 5c | 2 | CH3CN, 100 °C, 24 h | 75 | trace |
| 6 | 10 | CH3CN, rt, 12 h | trace | 85 |
| 7 | 6 | CH3CN, rt, 20 h | trace | 85 |
| 8d | 4 | CH3CN, rt, 24 h | trace | 85 |
| 9 | 2 | CH3CN, rt, 24 h | – | 52 |
aEquivalent of KCN respect to 1a. bAll yields were calculated after purification by flash chromatography. cThe most effective method for the formation of 4a. dThe most effective method for the formation of 3a.
In further trials, we aimed to decrease the reaction temperature from 100 °C to room temperature in CH3CN using 10 equiv of KCN. In this case, the reaction ended with the formation of compound 3a as a major product (85%) and decyanated product 4a was obtained in trace amounts. To optimize the reaction conditions for compound 3a, different equivalents of KCN have been tried. We were pleased to find that the use of 4 equiv KCN in CH3CN gave the desired product 3a in high yield with a trace amount of decyanated product 4a.
After having the optimized reaction conditions, 5-(chloromethyl)-3-(p-substituted-phenyl)-1,2,4-oxadiazoles 1 were reacted sequentially with 2 equiv of KCN in CH3CN at 100 °C and 4 equiv of KCN in CH3CN at room temperature giving the title compounds 3 and 4 as major products in high yields (75–85%, Table 2). Interestingly, incorporation of electron-poor substituents in compounds 1f and 1g with 2 equiv of KCN at 100 °C (method A) afforded only the products 4f and 4g, respectively, in a shorter reaction time (Table 2, entries 6 and 7).
Table 2: Formation of 3 and their parent alkanes 4.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-280-i4.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Substrate | 4 | 3 | Yield (%) | |||
| Method Aa | Method Bb | ||||||
| 3 | 4 | 3 | 4 | ||||
| 1 | 1a: R = H | 4a: R = H; X = H | 3a: R = H; X = CN | trace | 75 | 85 | 5 |
| 2 | 1b: R = Cl | 4b: R = Cl; X = H | 3b: R = Cl; X = CN | 7 | 70 | 78 | trace |
| 3 | 1c: R = I | 4c: R = I; X = H | 3c: R = I; X = CN | 5 | 68 | 72 | trace |
| 4 | 1d: R = F | 4d: R = F; X = H | 3d: R = F; X = CN | 7 | 65 | 70 | trace |
| 5 | 1e: R = CH3 | 4e: R = CH3; X = H | 3e: R = CH3; X = CN | trace | 72 | 82 | 5 |
| 6 | 1f: R = NO2 | 4f: R = NO2; X = H | 3f: R = NO2; X = CN | – | 82c | 78 | 15 |
| 7 | 1g: R = CF3 | 4g: R = CF3; X = H | 3g: R = CF3; X = CN | – | 78d | 75 | 12 |
| 8 | 1h: R = OCH3 | 4h: R = OCH3; X = H | 3h: R = OCH3; X = CN | 5 | 70 | 78 | trace |
| 9 | 1j: R = SCH3 | 4j: R = SCH3; X = H | 3j: R = SCH3; X = CN | 7 | 72 | 76 | trace |
aCH3CN, 2 equiv KCN, 100 °C, 12 h bCH3CN, 4 equiv KCN, rt, 24 h cCH3CN, 2 equiv KCN, 100 °C, 6 h dCH3CN, 2 equiv KCN, 100 °C, 8 h.
However, due to the replacement of the nitrile group with hydrogen after decyanation in product 4, the methylenic protons and the methinic proton resonates at around 4–5 ppm as doublets and 5 ppm as pentet, respectively (Figure 3). The aliphatic protons of title compounds 3 and 4 were also assigned on the basis of HSQC and HMBC NMR experiments. The expanded HSQC spectrum of 3a showed the C2 carbon at 35 ppm which corresponds to the methylenic hydrogens H2. In addition, the HSQC spectrum of 4a showed that the H1 proton located between the methylene coupled with the C1 carbon and that the methylene protons H2 were incorporated in the C2 carbon signal.
![[1860-5397-14-280-3]](/bjoc/content/figures/1860-5397-14-280-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Expanded HSQC spectrum of 4a and 3a.
Figure 3: Expanded HSQC spectrum of 4a and 3a.
Fortunately, the exact structures of compounds 3a and 4e were also confirmed by X-ray structures (Figure 4). The CIF data of 3a and 4e have been deposited at Cambridge Crystallographic Data Centre with the deposition numbers 1844118 and 1832133.
![[1860-5397-14-280-4]](/bjoc/content/figures/1860-5397-14-280-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray ORTEP plots of 3a and 4e.
Figure 4: X-ray ORTEP plots of 3a and 4e.
We propose a plausible mechanism for the transformation of chloromethyloxadiazoles 1 into the title products 3 and 4. Accordingly, the reaction of 1 with a CN− anion gives cyanomethyloxadiazole by simple SN2 reaction. Then, the acidic hydrogen adjacent to the nitrile group in the intermediate product is sequentially abstracted by a CN− anion with the extrusion of an HCN molecule and a carbanion alpha to the nitrile group bearing a 1,2,4-oxadiazole ring is formed. The resulted carbanion undergoes a substitution reaction with another molecule of 1, affording di-1,2,4-oxadiazole-substituted acetonitrile, which then undergoes a second substitution reaction with another molecule of 1, in the same manner, finally affording title compound 3 (Scheme 1).
![[1860-5397-14-280-i1]](/bjoc/content/inline/1860-5397-14-280-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Plausible mechanism for the formation of 3.
Scheme 1: Plausible mechanism for the formation of 3.
Considering the formation of product 4, as we believe, the temperature plays a significant role upon formation of it and in situ-formed HCN facilitates the decyanation (nitrile–alkane conversion) possibly since it is the only proton source in the reaction medium. This unfamiliar decyanation reaction presumably proceeds through product 3. First, in the presence of CN− anions, cyanoimine salt intermediate A forms by addition to a nitrile. Then, a possible cyanogen release would occur to form intermediate B. The imine–enamine tautomerization of B by the effect of in situ-generated HCN results in the desired decyanated product 4 (Scheme 2).
![[1860-5397-14-280-i2]](/bjoc/content/inline/1860-5397-14-280-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible mechanism for the formation of 4 via decyanation of 3.
Scheme 2: Plausible mechanism for the formation of 4 via decyanation of 3.
Conclusion
In summary, we reported a convenient one-pot protocol for the synthesis of 3 and its transformation into 4 via a decyanation pathway by the reaction of 5-(chloromethyl)-3-(p-substituted-phenyl)-1,2,4-oxadiazoles 1 with KCN at different temperatures. This decyanation method is the first example for the conversion of a nitrile group into an alkane by using KCN–HCN associates through a possible release of cyanogen which is not detected or isolated. This synthetic protocol seems to be applicable for further decyanation processes containing a 1,2,4-oxadiazole moiety.
Supporting Information
| Supporting Information File 1: Experimental details, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 3.5 MB | Download |
References
-
Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G. S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; Pellicciari, R.; Mai, A.; Steegborn, C.; Rotili, D. J. Med. Chem. 2017, 60, 2344–2360. doi:10.1021/acs.jmedchem.6b01609
Return to citation in text: [1] -
O’Daniel, P. I.; Peng, Z.; Pi, H.; Testero, S. A.; Ding, D.; Spink, E.; Leemans, E.; Boudreau, M. A.; Yamaguchi, T.; Schroeder, V. A.; Wolter, W. R.; Llarrull, L. I.; Song, W.; Lastochkin, E.; Kumarasiri, M.; Antunes, N. T.; Espahbodi, M.; Lichtenwalter, K.; Suckow, M. A.; Vakulenko, S.; Mobashery, S.; Chang, M. J. Am. Chem. Soc. 2014, 136, 3664–3672. doi:10.1021/ja500053x
Return to citation in text: [1] -
Karad, S. C.; Purohit, V. B.; Thummar, R. P.; Vaghasiya, B. K.; Kamani, R. D.; Thakor, P.; Thakkar, V. R.; Thakkar, S. S.; Ray, A.; Raval, D. K. Eur. J. Med. Chem. 2017, 126, 894–909. doi:10.1016/j.ejmech.2016.12.016
Return to citation in text: [1] -
Heimann, D.; Börgel, F.; de Vries, H.; Bachmann, K.; Rose, V. E.; Frehland, B.; Schepmann, D.; Heitman, L. H.; Wünsch, B. Eur. J. Med. Chem. 2018, 143, 1436–1447. doi:10.1016/j.ejmech.2017.10.049
Return to citation in text: [1] -
Leite, A. C. L.; Vieira, R. F.; de Faria, A. R.; Wanderley, A. G.; Afiatpour, P.; Ximenes, E. C. P. A.; Srivastava, R. M.; de Oliveira, C. F.; Medeiros, M. V.; Antunes, E.; Brondani, D. J. Farmaco 2000, 55, 719–724. doi:10.1016/s0014-827x(00)00099-9
Return to citation in text: [1] -
Yang, X.; Liu, G.; Li, H.; Zhang, Y.; Song, D.; Li, C.; Wang, R.; Liu, B.; Liang, W.; Jing, Y.; Zhao, G. J. Med. Chem. 2010, 53, 1015–1022. doi:10.1021/jm9011565
Return to citation in text: [1] -
Wei, H.; He, C.; Zhang, J.; Shreeve, J. N. M. Angew. Chem., Int. Ed. 2015, 54, 9367–9371. doi:10.1002/anie.201503532
Return to citation in text: [1] -
Li, Q.; Cui, L.-S.; Zhong, C.; Jiang, Z.-Q.; Liao, L.-S. Org. Lett. 2014, 16, 1622–1625. doi:10.1021/ol5002494
Return to citation in text: [1] -
Guo, J.; Hua, R.; Sui, Y.; Cao, J. Tetrahedron Lett. 2014, 55, 1557–1560. doi:10.1016/j.tetlet.2014.01.066
Return to citation in text: [1] -
Li, Q.; Cui, L.-S.; Zhong, C.; Yuan, X.-D.; Dong, S.-C.; Jiang, Z.-Q.; Liao, L.-S. Dyes Pigm. 2014, 101, 142–149. doi:10.1016/j.dyepig.2013.09.029
Return to citation in text: [1] -
Wang, W.; Xu, H.; Xu, Y.; Ding, T.; Zhang, W.; Ren, Y.; Chang, H. Org. Biomol. Chem. 2016, 14, 9814–9822. doi:10.1039/c6ob01794k
Return to citation in text: [1] -
Kuram, M. R.; Kim, W. G.; Myung, K.; Hong, S. Y. Eur. J. Org. Chem. 2016, 438–442. doi:10.1002/ejoc.201501502
Return to citation in text: [1] -
Grant, D.; Dahl, R.; Cosford, N. D. P. J. Org. Chem. 2008, 73, 7219–7223. doi:10.1021/jo801152c
Return to citation in text: [1] -
Chidambaram, M.; Sonavane, S. U.; de la Zerda, J.; Sasson, Y. Tetrahedron 2007, 63, 7696–7701. doi:10.1016/j.tet.2007.05.017
Return to citation in text: [1] -
Ren, Y.; Dong, C.; Zhao, S.; Sun, Y.; Wang, J.; Ma, J.; Hou, C. Tetrahedron Lett. 2012, 53, 2825–2827. doi:10.1016/j.tetlet.2012.03.109
Return to citation in text: [1] -
Brunel, J.-M.; Holmes, I. P. Angew. Chem., Int. Ed. 2004, 43, 2752–2778. doi:10.1002/anie.200300604
Return to citation in text: [1] -
Tsao, J.-P.; Tsai, T.-Y.; Chen, I.-C.; Liu, H.-J.; Zhu, J.-L.; Tsao, S.-W. Synthesis 2010, 4242–4250. doi:10.1055/s-0030-1258301
Return to citation in text: [1] -
Strzalko, T.; Wartski, L.; Corset, J.; Castellà-Ventura, M.; Froment, F. J. Org. Chem. 2012, 77, 6431–6442. doi:10.1021/jo300758g
Return to citation in text: [1] -
Jaunin, R. Helv. Chim. Acta 1966, 49, 412–419. doi:10.1002/hlca.660490146
Return to citation in text: [1] -
Parker, K. A.; Kallmerten, J. L. Tetrahedron Lett. 1979, 20, 1197–1200. doi:10.1016/s0040-4039(01)86101-3
Return to citation in text: [1] -
Ahlbrecht, H.; Raab, W.; Vonderheid, C. Synthesis 1979, 127–129. doi:10.1055/s-1979-28587
Return to citation in text: [1] -
Too, P. C.; Chan, G. H.; Tnay, Y. L.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2016, 55, 3719–3723. doi:10.1002/anie.201600305
Return to citation in text: [1] -
Marshall, J. A.; Bierenbaum, R. J. Org. Chem. 1977, 42, 3309–3311. doi:10.1021/jo00440a029
Return to citation in text: [1] -
Franck-Neumann, M.; Miesch, M.; Lacroix, E.; Mertz, B.; Ken, J.-M. Tetrahedron 1992, 48, 1911–1926. doi:10.1016/s0040-4020(01)88514-8
Return to citation in text: [1] -
Chang, K.-J.; Rayabarapu, D. K.; Cheng, C.-H. J. Org. Chem. 2004, 69, 4781–4787. doi:10.1021/jo049506g
Return to citation in text: [1] -
Yamada, S.-i.; Tomioka, K.; Koga, K. Tetrahedron Lett. 1976, 17, 61–64. doi:10.1016/s0040-4039(00)71323-2
Return to citation in text: [1] -
Ağırbaş, H.; Sümengen, D.; Dürüst, Y.; Dürüst, N. Synth. Commun. 1992, 22, 209–217. doi:10.1080/00397919208021295
Return to citation in text: [1]
| 24. | Franck-Neumann, M.; Miesch, M.; Lacroix, E.; Mertz, B.; Ken, J.-M. Tetrahedron 1992, 48, 1911–1926. doi:10.1016/s0040-4020(01)88514-8 |
| 22. | Too, P. C.; Chan, G. H.; Tnay, Y. L.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2016, 55, 3719–3723. doi:10.1002/anie.201600305 |
| 23. | Marshall, J. A.; Bierenbaum, R. J. Org. Chem. 1977, 42, 3309–3311. doi:10.1021/jo00440a029 |
| 1. | Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G. S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; Pellicciari, R.; Mai, A.; Steegborn, C.; Rotili, D. J. Med. Chem. 2017, 60, 2344–2360. doi:10.1021/acs.jmedchem.6b01609 |
| 5. | Leite, A. C. L.; Vieira, R. F.; de Faria, A. R.; Wanderley, A. G.; Afiatpour, P.; Ximenes, E. C. P. A.; Srivastava, R. M.; de Oliveira, C. F.; Medeiros, M. V.; Antunes, E.; Brondani, D. J. Farmaco 2000, 55, 719–724. doi:10.1016/s0014-827x(00)00099-9 |
| 20. | Parker, K. A.; Kallmerten, J. L. Tetrahedron Lett. 1979, 20, 1197–1200. doi:10.1016/s0040-4039(01)86101-3 |
| 4. | Heimann, D.; Börgel, F.; de Vries, H.; Bachmann, K.; Rose, V. E.; Frehland, B.; Schepmann, D.; Heitman, L. H.; Wünsch, B. Eur. J. Med. Chem. 2018, 143, 1436–1447. doi:10.1016/j.ejmech.2017.10.049 |
| 21. | Ahlbrecht, H.; Raab, W.; Vonderheid, C. Synthesis 1979, 127–129. doi:10.1055/s-1979-28587 |
| 3. | Karad, S. C.; Purohit, V. B.; Thummar, R. P.; Vaghasiya, B. K.; Kamani, R. D.; Thakor, P.; Thakkar, V. R.; Thakkar, S. S.; Ray, A.; Raval, D. K. Eur. J. Med. Chem. 2017, 126, 894–909. doi:10.1016/j.ejmech.2016.12.016 |
| 18. | Strzalko, T.; Wartski, L.; Corset, J.; Castellà-Ventura, M.; Froment, F. J. Org. Chem. 2012, 77, 6431–6442. doi:10.1021/jo300758g |
| 2. | O’Daniel, P. I.; Peng, Z.; Pi, H.; Testero, S. A.; Ding, D.; Spink, E.; Leemans, E.; Boudreau, M. A.; Yamaguchi, T.; Schroeder, V. A.; Wolter, W. R.; Llarrull, L. I.; Song, W.; Lastochkin, E.; Kumarasiri, M.; Antunes, N. T.; Espahbodi, M.; Lichtenwalter, K.; Suckow, M. A.; Vakulenko, S.; Mobashery, S.; Chang, M. J. Am. Chem. Soc. 2014, 136, 3664–3672. doi:10.1021/ja500053x |
| 14. | Chidambaram, M.; Sonavane, S. U.; de la Zerda, J.; Sasson, Y. Tetrahedron 2007, 63, 7696–7701. doi:10.1016/j.tet.2007.05.017 |
| 16. | Brunel, J.-M.; Holmes, I. P. Angew. Chem., Int. Ed. 2004, 43, 2752–2778. doi:10.1002/anie.200300604 |
| 27. | Ağırbaş, H.; Sümengen, D.; Dürüst, Y.; Dürüst, N. Synth. Commun. 1992, 22, 209–217. doi:10.1080/00397919208021295 |
| 11. | Wang, W.; Xu, H.; Xu, Y.; Ding, T.; Zhang, W.; Ren, Y.; Chang, H. Org. Biomol. Chem. 2016, 14, 9814–9822. doi:10.1039/c6ob01794k |
| 12. | Kuram, M. R.; Kim, W. G.; Myung, K.; Hong, S. Y. Eur. J. Org. Chem. 2016, 438–442. doi:10.1002/ejoc.201501502 |
| 13. | Grant, D.; Dahl, R.; Cosford, N. D. P. J. Org. Chem. 2008, 73, 7219–7223. doi:10.1021/jo801152c |
| 17. | Tsao, J.-P.; Tsai, T.-Y.; Chen, I.-C.; Liu, H.-J.; Zhu, J.-L.; Tsao, S.-W. Synthesis 2010, 4242–4250. doi:10.1055/s-0030-1258301 |
| 7. | Wei, H.; He, C.; Zhang, J.; Shreeve, J. N. M. Angew. Chem., Int. Ed. 2015, 54, 9367–9371. doi:10.1002/anie.201503532 |
| 8. | Li, Q.; Cui, L.-S.; Zhong, C.; Jiang, Z.-Q.; Liao, L.-S. Org. Lett. 2014, 16, 1622–1625. doi:10.1021/ol5002494 |
| 9. | Guo, J.; Hua, R.; Sui, Y.; Cao, J. Tetrahedron Lett. 2014, 55, 1557–1560. doi:10.1016/j.tetlet.2014.01.066 |
| 10. | Li, Q.; Cui, L.-S.; Zhong, C.; Yuan, X.-D.; Dong, S.-C.; Jiang, Z.-Q.; Liao, L.-S. Dyes Pigm. 2014, 101, 142–149. doi:10.1016/j.dyepig.2013.09.029 |
| 25. | Chang, K.-J.; Rayabarapu, D. K.; Cheng, C.-H. J. Org. Chem. 2004, 69, 4781–4787. doi:10.1021/jo049506g |
| 6. | Yang, X.; Liu, G.; Li, H.; Zhang, Y.; Song, D.; Li, C.; Wang, R.; Liu, B.; Liang, W.; Jing, Y.; Zhao, G. J. Med. Chem. 2010, 53, 1015–1022. doi:10.1021/jm9011565 |
| 15. | Ren, Y.; Dong, C.; Zhao, S.; Sun, Y.; Wang, J.; Ma, J.; Hou, C. Tetrahedron Lett. 2012, 53, 2825–2827. doi:10.1016/j.tetlet.2012.03.109 |
| 26. | Yamada, S.-i.; Tomioka, K.; Koga, K. Tetrahedron Lett. 1976, 17, 61–64. doi:10.1016/s0040-4039(00)71323-2 |
© 2018 Sağırlı and Dürüst; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)