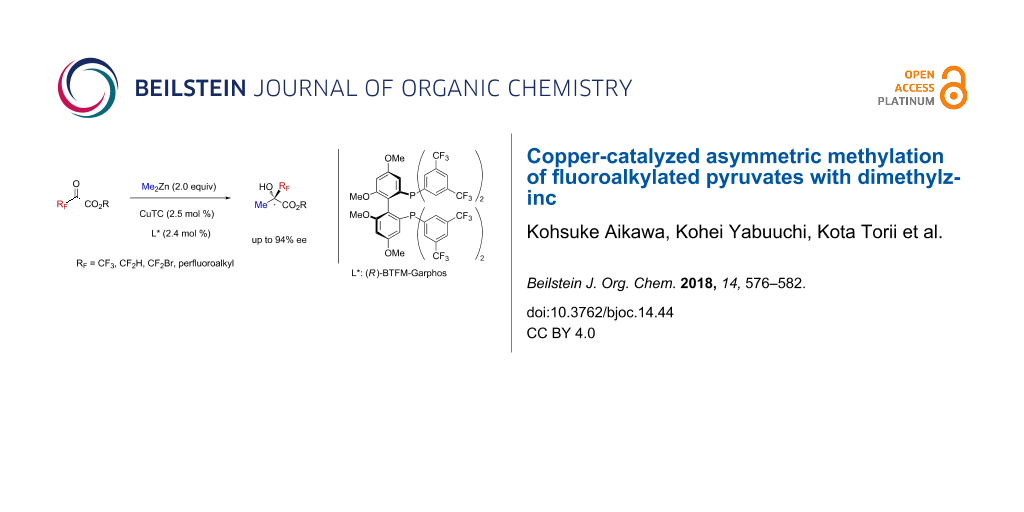
Abstract
The catalytic asymmetric methylation of fluoroalkylated pyruvates is shown with dimethylzinc as a methylating reagent in the presence of a copper catalyst bearing a chiral phosphine ligand. This is the first catalytic asymmetric methylation to synthesize various α-fluoroalkylated tertiary alcohols with CF3, CF2H, CF2Br, and n-CnF2n+1 (n = 2, 3, 8) groups in good-to-high yields and enantioselectivities. Axial backbones and substituents on phosphorus atoms of chiral phosphine ligands critically influence the enantioselectivity. Moreover, the methylation of simple perfluoroalkylated ketones is found to be facilitated by only chiral phosphines without copper.

Graphical Abstract
Introduction
The introduction of fluorine atoms into organic compounds plays an important role in the discovery of lead candidates with unique biological and physicochemical properties [1,2]. Therefore, the development of novel synthetic methods for the introduction of fluorinated fragments, such as trifluoromethyl (CF3), difluoromethyl (CF2H), and difluoromethylene (-CF2-), has attracted a great deal of attention from synthetic organic chemists [3-6]. Among these methods, many researchers including us have studied the catalytic asymmetric synthesis of optically active α-trifluoromethylated tertiary alcohols [7,8]. In these cases, one of commercially available and versatile trifluoromethyl sources, trifluoropyruvate, has been utilized for a variety of catalytic asymmetric carbon–carbon bond forming reactions, providing efficiently α-trifluoromethylated tertiary alcohols in high enantioselectivities [9-19]. Over the past decade we have also investigated several catalytic asymmetric reactions using trifluoropyruvate as an electrophile in the presence of a chiral Lewis acid complex [20-27]. However, the synthetic method for chiral α-trifluoromethylated tertiary alcohols via methylation of trifluoropyruvate is quite limited, although several drug candidates bearing this chiral trifluoromethylated moiety have so far been reported [7,28-30]. In 2007, Gosselin and Britton et al. reported that treatment of ethyl trifluoropyruvate (1a) with (R)-BINOL-mediated organozincate as a chiral methylating regent provided the corresponding methylated tertiary alcohol 2a in moderate enantioselectivity (Scheme 1, reaction 1) [31]. Kinetic resolution of racemic α-trifluoromethylated tertiary alcohols 2a by an enzyme is also reported to give the corresponding alcohols 2a in high enantioselectivity (Scheme 1, reaction 2) [32]. However, there has been no report for catalytic asymmetric methylation of trifluoropyruvate. Herein, we disclose the catalytic asymmetric methylation of trifluoropyruvate derivatives as electrophiles and dimethylzinc as a methylating nucleophile by a chiral copper catalyst. This method is also applicable to the asymmetric synthesis of various α-fluoroalkylated tertiary alcohols bearing CF2H, CF2Br, and n-CnF2n+1 (n = 2, 3, 8) groups.
![[1860-5397-14-44-i1]](/bjoc/content/inline/1860-5397-14-44-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of chiral α-fluoroalkylated tertiary alcohols.
Scheme 1: Synthesis of chiral α-fluoroalkylated tertiary alcohols.
Results and Discussion
Our initial investigation was focused on the methylation of ethyl trifluoropyruvate (1a) with Me2Zn in the presence of a copper salt bearing a chiral bidentate phosphine ligand (Table 1). We were delighted to find that the reaction proceeded smoothly in the presence of CuTC (TC: 2-thiophenecarboxylate, 2.5 mol %) and (R)-BINAP (2.7 mol %) in Et2O at −78 °C, furnishing the methylated product 2a in 99% yield with 38% ee (Table 1, entry 1). The effect of the Cu salt was also surveyed. The use of CuOAc resulted in slightly decreased enantioselectivities, and (CuOTf)·C6H6 and CuI led to a racemic product (Table 1, entries 2–4). Chiral phosphine ligands instead of BINAP were further assayed with the aim of enhancing the enantioselectivity. Indeed, the investigation of the effect of axial backbones and substituents on the phosphorus atoms led to an increase in the enantioselectivity. In the case of a biphenyl backbone, MeO-BIPHEP showed the same level of enantioselectivity as BINAP, while lower enantioselectivity was obtained by SEGPHOS (Table 1, entries 5 and 6). Exploring the effect of substituents on phosphorus, DM-BINAP slightly exceeded the level attained by BINAP, although Cy-BINAP and DTBM-BINAP decreased the enantioselectivities (Table 1, entries 7–9). In sharp contrast to BINAP derivatives, DTBM-SEGPHOS and DTBM-MeO-BIPHEP with extremely bulky aryl groups increased the enantioselectivities (Table 1, entries 10 and 11). Additionally, DTB-MeO-BIPHEP provided the desired alcohol in 84% yield with 60% ee (Table 1, entry 12). In toluene and CH2Cl2 as noncoordinating solvents (Table 1, entries 14 and 15), the reaction gave lower enantioselectivities, but TBME gave the best result in 90% yield and 67% ee (Table 1, entry 16). The use of methyl trifluoropyruvate (1b) instead of 1a resulted in a lower enantioselectivity (Table 1, entry 17). The absolute configuration of 2b was determined to be S by comparison with the optical rotation of reported data [32]. The absolute configurations of other alcohol products 2a and 2c–k were tentatively assigned by analogy to 2b.
Table 1: Copper-catalyzed asymmetric methylation.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-44-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry | ligand | Cu salt | solvent | yield (%)a | ee (%) |
| 1 | (R)-BINAP | CuTC | Et2O | 99 | 38 |
| 2 | (R)-BINAP | CuOAc | Et2O | 43 | 36 |
| 3 | (R)-BINAP | (CuOTf)·C6H6 | Et2O | 13 | 0 |
| 4 | (R)-BINAP | CuI | Et2O | 38 | 0 |
| 5 | (R)-SEGPHOS | CuTC | Et2O | 81 | 26 |
| 6 | (R)-MeO-BIPHEP | CuTC | Et2O | 85 | 38 |
| 7 | (R)-Cy-BINAP | CuTC | Et2O | 92 | 3 |
| 8 | (R)-DM-BINAP | CuTC | Et2O | 70 | 41 |
| 9 | (R)-DTBM-BINAP | CuTC | Et2O | 73 | 17 |
| 10 | (R)-DTBM-SEGPHOS | CuTC | Et2O | 67 | 55 |
| 11 | (R)-DTBM-MeO-BIPHEP | CuTC | Et2O | 99 | 50 |
| 12 | (R)-DTB-MeO-BIPHEP | CuTC | Et2O | 84 | 60 |
| 13 | (R)-DTB-MeO-BIPHEP | CuTC | THF | 85 | 57 |
| 14 | (R)-DTB-MeO-BIPHEP | CuTC | toluene | 98 | 38 |
| 15 | (R)-DTB-MeO-BIPHEP | CuTC | CH2Cl2 | 90 | 9 |
| 16 | (R)-DTB-MeO-BIPHEP | CuTC | TBME | 90 | 67 |
| 17b | (R)-DTB-MeO-BIPHEP | CuTC | TBME | 71 | 59 (S) |
aYields were determined by 19F NMR analysis using benzotrifluoride (BTF) as an internal standard. bMethyl trifluoropyruvate (1b) was used instead of ethyl trifluoropyruvate (1a).
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-44-i5.svg?max-width=637&scale=1.18182)
Additionally, the reaction conditions were fine-tuned as exemplified in Table 2. It was found that reactions without CuTC and phosphine ligand (Table 2, entry 1) or only without phosphine ligand (Table 2, entry 2) provided the alcohol as a racemic mixture even at −78 °C. In contrast, the chiral product was obtained in 64% yield in the absence of a copper salt but in low enantioselectivity (Table 2, entry 3). Therefore, decreasing the amount of phosphine ligand (2.4 mol %) to less than that of copper salt led to an enhancement of the enantioselectivity to 70% ee (Table 2, entries 4 vs 5). In addition, the selection of BTFM-Garphos instead of DTB-MeO-BIPHEP afforded a higher enantoselectivity (Table 2, entry 6), and consequently, a lower reaction temperature (−90 °C) gave the best result with 86% yield and 89% ee (Table 2, entry 7).
Table 2: Optimization of reaction conditions.
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-44-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | X (mol %/Cu) | Y (mol %/ligand) | ligand | yield (%)a | ee (%) |
| 1 | 0 | 0 | – | 17 | – |
| 2 | 2.5 | 0 | – | 15 | – |
| 3 | 0 | 2.5 | (R)-DTB-MeO-BIPHEP | 64 | 7 |
| 4 | 2.5 | 2.7 | (R)-DTB-MeO-BIPHEP | 90 | 67 |
| 5 | 2.5 | 2.4 | (R)-DTB-MeO-BIPHEP | 94 | 70 |
| 6 | 2.5 | 2.4 | (R)-BTFM-Garphos | 88 | 73 |
| 7b | 2.5 | 2.4 | (R)-BTFM-Garphos | 86 | 89 |
aYields were determined by 19F NMR analysis using benzotrifluoride (BTF) as an internal standard. bReaction temperature was −90 °C.
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-44-i7.svg?max-width=637&scale=1.18182)
Various fluoroalkylated pyruvates were applicable to this catalytic transformation under the optimized reaction conditions (Scheme 2). Alkyl substituents on the ester moiety of the trifluoropyruvate were found to influence the stereoselectivity drastically. The reaction of trifluoropyruvates (1c–e) bearing sterically demanding substituents such as isopropyl, cyclopentyl, and cyclohexyl led to a higher level of enantioselectivities (90–94% ee), compared to the corresponding ethyl ester 1a. In contrast, trifluoropyruvate 1f with an extremely bulky substituent caused a decrease of enantioselectivity. Ethyl difluoropyruvate (1g) and ethyl bromodifluoropyruvate (1h) also underwent the reactions in good enantioselectivities, although a slight decrease in yield was observed due to the steric hindrance of the CF2Br group. Significantly, ethyl perfluoropyruvates 1i–k with longer alkyl chains were also converted to the desired tertiary alcohols in good enantioselectivities.
![[1860-5397-14-44-i2]](/bjoc/content/inline/1860-5397-14-44-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of fluoroalkylated pyruvates. Yields were determined by 19F NMR analysis using benzotrifluoride (BTF) as an internal standard. aReaction temperature was −78 °C.
Scheme 2: Scope of fluoroalkylated pyruvates. Yields were determined by 19F NMR analysis using benzotrifluori...
The catalytic asymmetric methylation using the simple perfluoroalkylated ketone 3a instead of pyruvate derivatives was further examined (Scheme 3). In contrast to the pyruvate system, the combination of CuTC and BINAP did not facilitate the reaction even at −78 °C, but also afforded the racemic product (25% yield, 0% ee). Interestingly, the use of only BINAP without CuTC led to higher reactivity and enantioselectivity (54% yield, 8% ee), while BTFM-Garphos decreased the reactivity (7% yield, 8% ee). After screening of phosphines, DTB-MeO-BIPHEP was found to smoothly catalyze the asymmetric methylation to give the desired alcohol 4a in 87% yield and 24% ee.
![[1860-5397-14-44-i3]](/bjoc/content/inline/1860-5397-14-44-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Catalytic asymmetric methylation of the simple perfluoroalkylated ketone 3a. Yields were determined by 19F NMR analysis using benzotrifluoride (BTF) as an internal standard. aReaction was carried out without CuTC.
Scheme 3: Catalytic asymmetric methylation of the simple perfluoroalkylated ketone 3a. Yields were determined...
Conclusion
In summary, we have succeeded in the catalytic enantioselective methylation of fluoroalkylated pyruvates in the presence of chiral diphosphine–copper complexes, providing the corresponding tertiary alcohols with an RF group such as CF3, CF2H, CF2Br, and n-CnF2n+1 (n = 2, 3, 8) in good-to-high yields and enantioselectivities. This is the first report on catalytic asymmetric methylation with fluoroalkylated pyruvates. Moreover, a simple perfluoroalkyl ketone was also found to be methylated enantioselectively with dimethylzinc and a catalytic amount of a chiral diphosphine, but without copper.
Experimental
Typical procedure for copper-catalyzed asymmetric methylation of fluoroalkylated pyruvates: To a mixture of CuTC (1.0 mg, 0.005 mmol) and (R)-BTFM-Garphos (5.7 mg, 0.0048 mmol) was added CH2Cl2 (1.0 mL) at room temperature under an argon atmosphere, and the solution was stirred for 12 h. The solvent was removed under reduced pressure, and the prepared catalyst was dissolved in TBME (0.5 mL) under an argon atmosphere. After the solution was cooled to −90 °C, Me2Zn (1.0 M in heptane, 0.4 mL, 0.4 mmol) followed by fluoroalkylated pyruvate 1 (0.2 mmol) in TBME (0.5 mL) were added over 30 min. The reaction mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with saturated aq NH4Cl solution. The organic layer was separated and the aqueous layer was extracted twice with Et2O. The combined organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure (350 mmHg). The concentrated solution was used without purification for the next protection reaction. The yield of alcohol product 2 was determined by 19F NMR analysis using benzotrifluoride (BTF) as an internal standard.
To a solution of DMAP (2.4 mg, 0.02 mmol) and the crude alcohol 2 in CH2Cl2 (2.0 mL) was added NEt3 (56 μL, 0.4 mmol) at room temperature under an argon atmosphere. After the reaction mixture was cooled to 0 °C, p-nitrobenzoyl chloride (56 mg, 0.3 mmol) was added. Then the mixture was warmed to room temperature and stirred for 1 h. After 1 N HCl (5.0 mL) was added to the reaction mixture, the organic layer was separated and the aqueous layer was extracted twice with Et2O. The combined organic layer was washed with saturated aq. NaHCO3, water, and brine, and then dried over anhydrous MgSO4 and evaporated under reduced pressure. The residue was purified by silica gel column chromatography to give p-nitrobenzoylated alcohol 2’. The enantiomeric excess was determined by chiral HPLC analysis.
(S)-3-Ethoxy-1,1,1-trifluoro-2-methyl-3-oxopropan-2-yl 4-nitrobenzoate (2a’)
The yield of alcohol 2a (86%) was determined by 19F NMR analysis. p-Nitrobenzoylated alcohol 2a’ was purified by silica-gel column chromatography (EtOAc/hexane 1:40) as a colorless liquid (53% yield for 2 steps, 89% ee). 1H NMR (300 MHz, CDCl3) δ 8.34–8.31 (m, 2H), 8.24–8.20 (m, 2H), 4.33 (q, 4H, J = 6.9 Hz), 1.97 (d, 3H, J = 0.9 Hz), 1.28 (t, 3H, J = 7.0 Hz); 13C NMR (75 MHz, CDCl3) δ 164.3, 162.3, 151.1, 134.0, 131.2, 123.7, 122.7 (q, J C-F = 282.9 Hz), 80.7 (q, J C-F = 30.4 Hz), 63.2, 16.6, 13.8; 19F NMR (282 MHz, CDCl3) δ −78.4 (s, 3F); HRMS (APCI-TOF): [M]−· calcd for C13H12F3NO6, 335.0617; found, 335.0623; FTIR (neat, cm−1) 784, 813, 849, 876, 927, 1011, 1109, 1149, 1273, 1342, 1387, 1452, 1525, 1602, 1740, 1763, 2857, 2920, 2952, 2996, 3087, 3116; [α]D22 −28.94 (c 0.20, CHCl3); HPLC (column, CHIRALCEL OJ-3, hexane/2-propanol 91:9, flow rate 0.6 mL/min, 20 °C detection UV 254 nm) tR of major isomer 13.1 min, tR of minor isomer 23.8 min.
(S)-1-Ethoxy-3,3-difluoro-2-methyl-1-oxopropan-2-yl benzoate (2g’)
Reaction temperature was −78 °C. The yield of alcohol 2g (89%) was determined by 19F NMR analysis. In the protection of alcohol, benzoyl chloride was used instead of p-nitrobenzoyl chloride. Benzoylated alcohol 2g’ was purified by silica gel column chromatography (EtOAc/hexane 1:40) as a colorless liquid (41% yield for 2 steps, 89% ee). 1H NMR (300 MHz, CDCl3) δ 8.05 (dd, J = 8.3, 1.3 Hz, 2H). 7.61 (tt, J = 6.7, 1.3 Hz, 1H), 7.49–7.44 (m, 2H), 6.30 (dd, JH-F = 56.8, 54.8 Hz, 1H), 4.29 (q, J = 7.1 Hz, 2H), 1.77 (t, JH-F = 1.6 Hz, 3H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 167.6, 164.8, 133.7, 130.0, 128.8, 128.5, 122.9 (dd, JC-F = 250.0, 245.0 Hz), 79.7 (dd, JC-F = 27.5, 21.9 Hz), 62.3, 14.6 (t, JC-F = 3.2 Hz) 13.9; 19F NMR (282 MHz, CDCl3) δ −128.40 (dd, J = 290.2 Hz, JF-H =54.7 Hz, 1F), −132.76 (dd, J = 289.90 Hz, JF-H = 56.4 Hz, 1F); HRMS (APCI-TOF): [M + Na]+ calcd for C13H14F2NaO4, 295.0758; found, 295.0761; FTIR (neat, cm−1) 1026, 1093, 1114, 1216, 1279, 1388, 1452, 1602, 1730, 1747, 2938, 2985, 3021; [α]D25 −7.47 (c 1.01, CHCl3); HPLC (column, CHIRALCEL OJ-3, hexane/2-propanol 99:1, flow rate 0.6 mL/min, 20 °C detection UV 220 nm) tR of major isomer 21.2 min, tR of minor isomer 22.6 min.
(S)-1-Bromo-3-ethoxy-1,1-difluoro-2-methyl-3-oxopropan-2-yl 4-nitrobenzoate (2h’)
Reaction temperature was −78 °C. The yield of alcohol 2h (53%) was determined by 19F NMR analysis. p-Nitrobenzoylated alcohol 2h’ was purified by silica gel column chromatography (EtOAc/hexane 1:50) as a white solid (32% yield for 2 steps, 82% ee). 1H NMR (300 MHz, CDCl3) δ 8.34–8.31 (m, 2H), 8.25–8.21 (m, 2H), 4.32 (q, 2H, J = 7.2 Hz), 2.02 (s, 3H), 1.29 (t, 3H, J = 7.0 Hz); 13C NMR (75 MHz, CDCl3) δ 164.3, 162.4, 151.2, 134.3, 131.3, 123.9, 121.0 (t, JC-F = 311.6 Hz), 84.8 (dd, JC-F = 25.6, 23.4 Hz), 63.4, 18.4, 14.0; 19F NMR (282 MHz, CDCl3) δ −56.9 (d, 1F, J = 168.6 Hz), −58.9 (d, 1F, J = 165.3 Hz); HRMS (APCI-TOF): [M]−· calcd for C13H12BrF2NO6, 394.9816; found, 394.9835; FTIR (KBr pellet, cm−1) 716, 843, 876, 961, 1020, 1106, 1146, 1280, 1347, 1446, 1528, 1610, 1751, 2866, 2936, 2988; [α]D22 −11.99 (c 1.55, CHCl3); HPLC (column, CHIRALCEL OD-3, hexane/2-propanol 91:9, flow rate 0.6 mL/min, 20 °C detection UV 254 nm) tR of major isomer 18.2 min, tR of minor isomer 12.5 min.
(S)-1-Ethoxy-3,3,4,4,4-pentafluoro-2-methyl-1-oxobutan-2-yl p-nitrobenzoate (2i’)
The yield of alcohol 2i (87%) was determined by 19F NMR analysis. p-Nitrobenzoylated alcohol 2i’ was purified by silica gel column chromatography (EtOAc/hexane 1:40) as a white solid (48% yield for 2 steps, 86% ee). 1H NMR (300 MHz, CDCl3) δ 8.35–8.30 (m, 2H) 8.21–8.16 (m, 2H), 4.37–4.27 (m, 2H), 2.04 (q, JH-F = 0.6 Hz, 3H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.2, 162.2 (d, JC-F = 2.0 Hz), 151.0, 134.1, 131.0, 123.8, 118.6 (qt, JC-F = 286.1, 35.6 Hz), 112.0 (tq, JC-F = 263.0, 36.8 Hz), 81.3 (t, JC-F = 25.4 Hz), 63.3, 16.6, 13.7; 19F NMR (282 MHz, CDCl3) δ −79.19 (s, 3F), −121.42 (d, J = 280.9 Hz, 1F), −122.98 (d, J = 279.7 Hz, 1F); HRMS (APCI-TOF): [M]−· calcd for C14H12F5NO6, 385.0585; found, 385.0582; FTIR (KBr pellet, cm−1) 1014, 1142, 1208, 1222, 1281, 1350, 1385, 1533, 1747, 2942, 2987, 3059; [α]D25 −27.75 (c 1.02, CHCl3); HPLC (column, CHIRALCEL OJ-3, hexane/2-propanol 99:1, flow rate 0.6 mL/min, 20 °C detection UV 220 nm) tR of major isomer 16.2 min, tR of minor isomer 31.4 min.
(S)-1-Ethoxy-3,3,4,4,5,5,5-heptafluoro-2-methyl-1-oxopentan-2-yl p-nitrobenzoate (2j’)
The yield of alcohol 2j (98%) was determined by 19F NMR analysis. p-Nitrobenzoylated alcohol 2j’ was purified by silica-gel column chromatography (EtOAc/hexane 1:50) as a colorless oil (48% yield for 2 steps, 78% ee). 1H NMR (300 MHz, CDCl3) δ 8.35–8.31 (m, 2H) 8.21–8.16 (m, 2H), 4.36–4.29 (m, 2H), 2.07 (q, JH-F = 1.3 Hz, 3H), 1.28 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.2 162.2, 151.0, 134.1, 131.0, 123.8, 117.6 (qt, JC-F = 286.9, 33.9 Hz), 113.6 (tt, JC-F = 263.9, 30.9 Hz), 122.9 (tq, JC-F = 266.9, 37.4 Hz), 82.2 (t, JC-F = 25.7 Hz), 63.4, 16.8, 13.7; 19F NMR (282 MHz, CDCl3) δ −80.65 (m, 3F), −117.75 (d, J = 288.5 Hz, 1F), −119.60 (d, J = 288.2 Hz, 1F), −123.882 (s, 2F); HRMS (APCI-TOF): [M]−· calcd for C15H12F7NO6, 435.0553; found, 435.0547; FTIR (neat, cm−1) 1090, 1140, 1200, 1233, 1349, 1387, 1534, 1609, 1744, 1761, 2942, 2988, 3059; [α]D25 −22.60 (c 0.94, CHCl3); HPLC (column, CHIRALCEL OJ-3, hexane/2-propanol 99:1, flow rate 0.6 mL/min, 20 °C detection UV 220 nm) tR of major isomer 12.2 min, tR of minor isomer 15.5 min.
(S)-1-Ethoxy-3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluoro-2-methyl-1-oxodecan-2-yl p-nitrobenzoate (3k’)
The yield of alcohol 2k (92%) was determined by 19F NMR analysis. p-Nitrobenzoylated alcohol 2k’ was purified by silica-gel column chromatography (EtOAc/hexane 1:50) as a white solid (85% yield for 2 steps, 73% ee). 1H NMR (300 MHz, CDCl3) δ 8.36–8.31 (m, 2H), 8.20–8.16 (m, 2H), 4.36–4.29 (m, 2H), 2.08 (s, 3H), 1.28 (t, 3H, J = 7.3 Hz); 13C NMR (75 MHz, CDCl3) δ 164.3, 162.4, 151.2, 134.3, 131.2, 123.9, 118.4–104.7 (m), 117.3 (qt, JC-F = 288.4, 33.2 Hz), 82.8 (t, JC-F = 25.4 Hz), 63.5, 17.1, 13.8; 19F NMR (282 MHz, CDCl3) δ –80.7 (m, 3F), –116.4 to −126.0 (m, 14F); HRMS (APCI-TOF): [M]−· calcd for C20H12F17NO6, 685.0393; found, 685.0362; FTIR (KBr pellet cm−1) 847, 969, 1009, 1142, 1214, 1246, 1297, 1472, 1530, 1613, 1732, 1757, 2339, 2360, 2860, 2922, 2997, 3112, 3454, 3493; [α]D22 −11.93 (c 0.48, CHCl3); HPLC (column, CHIRALPAK AD-3 and AD-H, hexane/2-propanol 99.5:0.5, flow rate 0.6 mL/min, 20 °C detection UV 254 nm) tR of major isomer 16.8 min, tR of minor isomer 24.4 min.
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of new compounds with copies of 1H, 13C and 19F NMR spectra. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351
Return to citation in text: [1] -
Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Sugiishi, T.; Amii, H.; Aikawa, K.; Mikami, K. Beilstein J. Org. Chem. 2015, 11, 2661–2670. doi:10.3762/bjoc.11.286
Return to citation in text: [1] -
Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a
Return to citation in text: [1] [2] -
Mikami, K.; Itoh, Y.; Yamanaka, M. Chem. Rev. 2004, 104, 1–16. doi:10.1021/cr030685w
Return to citation in text: [1] -
Gathergood, N.; Zhuang, W.; Jørgensen, K. A. J. Am. Chem. Soc. 2000, 122, 12517–12522. doi:10.1021/ja002593j
Return to citation in text: [1] -
Zhuang, W.; Gathergood, N.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2001, 66, 1009–1013. doi:10.1021/jo001176m
Return to citation in text: [1] -
Török, B.; Abid, M.; London, G.; Esquibel, J.; Török, M.; Mhadgut, S. C.; Yan, P.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2005, 44, 3086–3089. doi:10.1002/anie.200462877
Return to citation in text: [1] -
Suri, J. T.; Mitsumori, S.; Albertshofer, K.; Tanaka, F.; Barbas, C. F., III. J. Org. Chem. 2006, 71, 3822–3828. doi:10.1021/jo0602017
Return to citation in text: [1] -
Doherty, S.; Knight, J. G.; Smyth, C. H.; Harrington, R. W.; Clegg, W. J. Org. Chem. 2006, 71, 9751–9764. doi:10.1021/jo062023n
Return to citation in text: [1] -
Rueping, M.; Theissmann, T.; Kuenkel, A.; Koenigs, R. M. Angew. Chem., Int. Ed. 2008, 47, 6798–6801. doi:10.1002/anie.200802139
Return to citation in text: [1] -
Zhao, J.-F.; Tjan, T.-B. W.; Tan, B.-H.; Loh, T.-P. Org. Lett. 2009, 11, 5714–5716. doi:10.1021/ol902507x
Return to citation in text: [1] -
Ogawa, S.; Iida, N.; Tokunaga, E.; Shiro, M.; Shibata, N. Chem. – Eur. J. 2010, 16, 7090–7095. doi:10.1002/chem.201000911
Return to citation in text: [1] -
Ohshima, T.; Kawabata, T.; Takeuchi, Y.; Kakinuma, T.; Iwasaki, T.; Yonezawa, T.; Murakami, H.; Nishiyama, H.; Mashima, K. Angew. Chem., Int. Ed. 2011, 50, 6296–6300. doi:10.1002/anie.201100252
Return to citation in text: [1] -
Dong, X.; Sun, J. Org. Lett. 2014, 16, 2450–2453. doi:10.1021/ol500830a
Return to citation in text: [1] -
Morisaki, K.; Morimoto, H.; Mashima, K.; Ohshima, T. Heterocycles 2017, 95, 637–661. doi:10.3987/REV-16-SR(S)4
Return to citation in text: [1] -
Aikawa, K.; Kainuma, S.; Hatano, M.; Mikami, K. Tetrahedron Lett. 2004, 45, 183–185. doi:10.1016/j.tetlet.2003.10.137
Return to citation in text: [1] -
Mikami, K.; Kawakami, Y.; Akiyama, K.; Aikawa, K. J. Am. Chem. Soc. 2007, 129, 12950–12951. doi:10.1021/ja076539f
Return to citation in text: [1] -
Aikawa, K.; Hioki, Y.; Mikami, K. Org. Lett. 2010, 12, 5716–5719. doi:10.1021/ol102541s
Return to citation in text: [1] -
Mikami, K.; Aikawa, K.; Aida, J. Synlett 2011, 2719–2724. doi:10.1055/s-0031-1289540
Return to citation in text: [1] -
Aikawa, K.; Hioki, Y.; Shimizu, N.; Mikami, K. J. Am. Chem. Soc. 2011, 133, 20092–20095. doi:10.1021/ja2085299
Return to citation in text: [1] -
Aikawa, K.; Asai, Y.; Hioki, Y.; Mikami, K. Tetrahedron: Asymmetry 2014, 25, 1104–1115. doi:10.1016/j.tetasy.2014.06.013
Return to citation in text: [1] -
Aikawa, K.; Kondo, D.; Honda, K.; Mikami, K. Chem. – Eur. J. 2015, 21, 17565–17569. doi:10.1002/chem.201503631
Return to citation in text: [1] -
Aikawa, K.; Yoshida, S.; Kondo, D.; Asai, Y.; Mikami, K. Org. Lett. 2015, 17, 5108–5111. doi:10.1021/acs.orglett.5b02617
Return to citation in text: [1] -
Aicher, T. D.; Anderson, R. C.; Bebernitz, G. R.; Coppola, G. M.; Jewell, C. F.; Knorr, D. C.; Liu, C.; Sperbeck, D. M.; Brand, L. J.; Strohschein, R. J.; Gao, J.; Vinluan, C. C.; Shetty, S. S.; Dragland, C.; Kaplan, E. L.; DelGrande, D.; Islam, A.; Liu, X.; Lozito, R. J.; Maniara, W. M.; Walter, R. E.; Mann, W. R. J. Med. Chem. 1999, 42, 2741–2746. doi:10.1021/jm9902584
Return to citation in text: [1] -
Aicher, T. D.; Anderson, R. C.; Gao, J.; Shetty, S. S.; Coppola, G. M.; Stanton, J. L.; Knorr, D. C.; Sperbeck, D. M.; Brand, L. J.; Vinluan, C. C.; Kaplan, E. L.; Dragland, C. J.; Tomaselli, H. C.; Islam, A.; Lozito, R. J.; Liu, X.; Maniara, W. M.; Fillers, W. S.; DelGrande, D.; Walter, R. E.; Mann, W. R. J. Med. Chem. 2000, 43, 236–249. doi:10.1021/jm990358+
Return to citation in text: [1] -
Bebernitz, G. R.; Aicher, T. D.; Stanton, J. L.; Gao, J.; Shetty, S. S.; Knorr, D. C.; Strohschein, R. J.; Tan, J.; Brand, L. J.; Liu, C.; Wang, W. H.; Vinluan, C. C.; Kaplan, E. L.; Dragland, C. J.; DelGrande, D.; Islam, A.; Lozito, R. J.; Liu, X.; Maniara, W. M.; Mann, W. R. J. Med. Chem. 2000, 43, 2248–2257. doi:10.1021/jm0000923
Return to citation in text: [1] -
Gosselin, F.; Britton, R. A.; Mowat, J.; O’Shea, P. D.; Davies, I. W. Synlett 2007, 2193–2196. doi:10.1055/s-2007-984911
Return to citation in text: [1] -
Konigsberger, K.; Prasad, K.; Repič, O. Tetrahedron: Asymmetry 1999, 10, 679–687. doi:10.1016/S0957-4166(99)00017-8
Return to citation in text: [1] [2]
| 1. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 2. | Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f |
| 20. | Aikawa, K.; Kainuma, S.; Hatano, M.; Mikami, K. Tetrahedron Lett. 2004, 45, 183–185. doi:10.1016/j.tetlet.2003.10.137 |
| 21. | Mikami, K.; Kawakami, Y.; Akiyama, K.; Aikawa, K. J. Am. Chem. Soc. 2007, 129, 12950–12951. doi:10.1021/ja076539f |
| 22. | Aikawa, K.; Hioki, Y.; Mikami, K. Org. Lett. 2010, 12, 5716–5719. doi:10.1021/ol102541s |
| 23. | Mikami, K.; Aikawa, K.; Aida, J. Synlett 2011, 2719–2724. doi:10.1055/s-0031-1289540 |
| 24. | Aikawa, K.; Hioki, Y.; Shimizu, N.; Mikami, K. J. Am. Chem. Soc. 2011, 133, 20092–20095. doi:10.1021/ja2085299 |
| 25. | Aikawa, K.; Asai, Y.; Hioki, Y.; Mikami, K. Tetrahedron: Asymmetry 2014, 25, 1104–1115. doi:10.1016/j.tetasy.2014.06.013 |
| 26. | Aikawa, K.; Kondo, D.; Honda, K.; Mikami, K. Chem. – Eur. J. 2015, 21, 17565–17569. doi:10.1002/chem.201503631 |
| 27. | Aikawa, K.; Yoshida, S.; Kondo, D.; Asai, Y.; Mikami, K. Org. Lett. 2015, 17, 5108–5111. doi:10.1021/acs.orglett.5b02617 |
| 9. | Gathergood, N.; Zhuang, W.; Jørgensen, K. A. J. Am. Chem. Soc. 2000, 122, 12517–12522. doi:10.1021/ja002593j |
| 10. | Zhuang, W.; Gathergood, N.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2001, 66, 1009–1013. doi:10.1021/jo001176m |
| 11. | Török, B.; Abid, M.; London, G.; Esquibel, J.; Török, M.; Mhadgut, S. C.; Yan, P.; Prakash, G. K. S. Angew. Chem., Int. Ed. 2005, 44, 3086–3089. doi:10.1002/anie.200462877 |
| 12. | Suri, J. T.; Mitsumori, S.; Albertshofer, K.; Tanaka, F.; Barbas, C. F., III. J. Org. Chem. 2006, 71, 3822–3828. doi:10.1021/jo0602017 |
| 13. | Doherty, S.; Knight, J. G.; Smyth, C. H.; Harrington, R. W.; Clegg, W. J. Org. Chem. 2006, 71, 9751–9764. doi:10.1021/jo062023n |
| 14. | Rueping, M.; Theissmann, T.; Kuenkel, A.; Koenigs, R. M. Angew. Chem., Int. Ed. 2008, 47, 6798–6801. doi:10.1002/anie.200802139 |
| 15. | Zhao, J.-F.; Tjan, T.-B. W.; Tan, B.-H.; Loh, T.-P. Org. Lett. 2009, 11, 5714–5716. doi:10.1021/ol902507x |
| 16. | Ogawa, S.; Iida, N.; Tokunaga, E.; Shiro, M.; Shibata, N. Chem. – Eur. J. 2010, 16, 7090–7095. doi:10.1002/chem.201000911 |
| 17. | Ohshima, T.; Kawabata, T.; Takeuchi, Y.; Kakinuma, T.; Iwasaki, T.; Yonezawa, T.; Murakami, H.; Nishiyama, H.; Mashima, K. Angew. Chem., Int. Ed. 2011, 50, 6296–6300. doi:10.1002/anie.201100252 |
| 18. | Dong, X.; Sun, J. Org. Lett. 2014, 16, 2450–2453. doi:10.1021/ol500830a |
| 19. | Morisaki, K.; Morimoto, H.; Mashima, K.; Ohshima, T. Heterocycles 2017, 95, 637–661. doi:10.3987/REV-16-SR(S)4 |
| 7. | Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a |
| 8. | Mikami, K.; Itoh, Y.; Yamanaka, M. Chem. Rev. 2004, 104, 1–16. doi:10.1021/cr030685w |
| 3. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351 |
| 4. | Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 5. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 6. | Sugiishi, T.; Amii, H.; Aikawa, K.; Mikami, K. Beilstein J. Org. Chem. 2015, 11, 2661–2670. doi:10.3762/bjoc.11.286 |
| 32. | Konigsberger, K.; Prasad, K.; Repič, O. Tetrahedron: Asymmetry 1999, 10, 679–687. doi:10.1016/S0957-4166(99)00017-8 |
| 32. | Konigsberger, K.; Prasad, K.; Repič, O. Tetrahedron: Asymmetry 1999, 10, 679–687. doi:10.1016/S0957-4166(99)00017-8 |
| 31. | Gosselin, F.; Britton, R. A.; Mowat, J.; O’Shea, P. D.; Davies, I. W. Synlett 2007, 2193–2196. doi:10.1055/s-2007-984911 |
| 7. | Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a |
| 28. | Aicher, T. D.; Anderson, R. C.; Bebernitz, G. R.; Coppola, G. M.; Jewell, C. F.; Knorr, D. C.; Liu, C.; Sperbeck, D. M.; Brand, L. J.; Strohschein, R. J.; Gao, J.; Vinluan, C. C.; Shetty, S. S.; Dragland, C.; Kaplan, E. L.; DelGrande, D.; Islam, A.; Liu, X.; Lozito, R. J.; Maniara, W. M.; Walter, R. E.; Mann, W. R. J. Med. Chem. 1999, 42, 2741–2746. doi:10.1021/jm9902584 |
| 29. | Aicher, T. D.; Anderson, R. C.; Gao, J.; Shetty, S. S.; Coppola, G. M.; Stanton, J. L.; Knorr, D. C.; Sperbeck, D. M.; Brand, L. J.; Vinluan, C. C.; Kaplan, E. L.; Dragland, C. J.; Tomaselli, H. C.; Islam, A.; Lozito, R. J.; Liu, X.; Maniara, W. M.; Fillers, W. S.; DelGrande, D.; Walter, R. E.; Mann, W. R. J. Med. Chem. 2000, 43, 236–249. doi:10.1021/jm990358+ |
| 30. | Bebernitz, G. R.; Aicher, T. D.; Stanton, J. L.; Gao, J.; Shetty, S. S.; Knorr, D. C.; Strohschein, R. J.; Tan, J.; Brand, L. J.; Liu, C.; Wang, W. H.; Vinluan, C. C.; Kaplan, E. L.; Dragland, C. J.; DelGrande, D.; Islam, A.; Lozito, R. J.; Liu, X.; Maniara, W. M.; Mann, W. R. J. Med. Chem. 2000, 43, 2248–2257. doi:10.1021/jm0000923 |
© 2018 Aikawa et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)