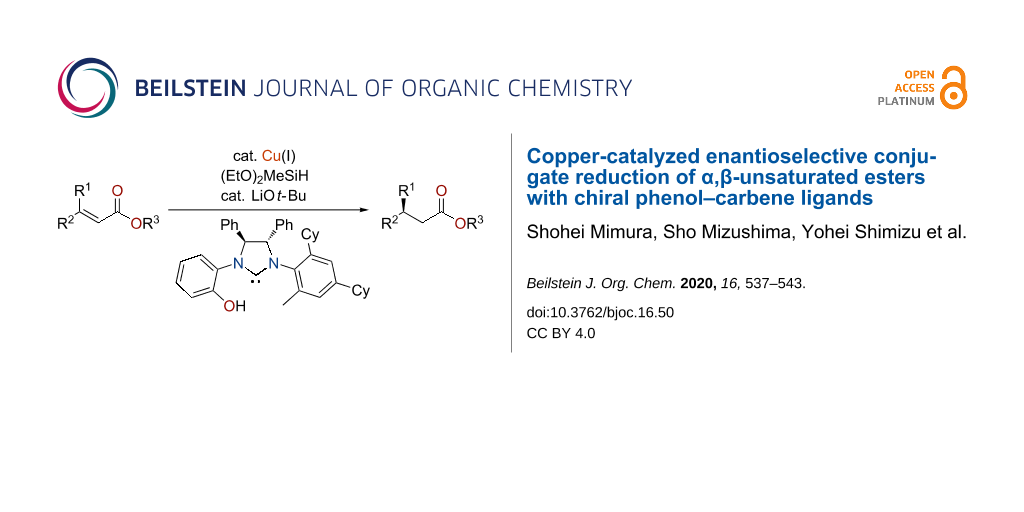
Abstract
A chiral phenol–NHC ligand enabled the copper-catalyzed enantioselective conjugate reduction of α,β-unsaturated esters. The phenol moiety of the chiral NHC ligand played a critical role in producing the enantiomerically enriched products. The catalyst worked well for various (Z)-isomer substrates. Opposite enantiomers were obtained from (Z)- and (E)-isomers, with a higher enantiomeric excess from the (Z)-isomer.

Graphical Abstract
Introduction
Since the leading work of Stryker and co-workers on triphenylphosphine-stabilized copper hydride complexes [1,2], copper hydrides have been widely used for conjugate reductions of α,β-unsaturated carbonyl compounds [3]. Especially a chiral copper catalyst combined with a stoichiometric amount of a silane reagent, which generated copper hydride in situ, has successfully been utilized for enantioselective reactions with β,β-disubstituted α,β-unsaturated carbonyl compounds [4-11]. The pioneering work of Buchwald and co-workers on the enantioselective conjugate reduction of α,β-unsaturated esters using a chiral p-tol-BINAP/copper catalyst established the excellent utility of chiral bisphosphine ligands for this type of reaction [4]. Surprisingly, however, chiral ligands based on N-heterocyclic carbenes (NHCs) [12] have not been applied to the conjugate reduction of α,β-unsaturated carbonyl compounds, while an achiral NHC/copper catalyst has successfully been utilized in this reaction [13].
Meanwhile, we devoted our effort to develop novel enantioselective C–C bond formation reactions utilizing chiral phenol–NHC/copper catalyst systems [14-18], in which the phenol group of the NHC ligand plays crucial roles in both the catalytic activity and stereoselectivity [19-21]. Notably, these catalyst systems were also applicable for three-component coupling reactions using hydrosilanes as hydride reagents [17]. Based on this knowledge, we decided to investigate the effects of the phenol–NHC ligand on the copper-catalyzed enantioselective conjugate reduction of α,β-unsaturated esters with hydrosilanes, placing a focus on (Z)-isomer substrates, which generally gave slightly lower enantiomeric excess with the chiral bisphosphines compared to the (E)-isomer substrates.
Results and Discussion
Optimization
The initial investigation of the reaction conditions was carried out with ethyl (Z)-3-phenylbut-2-enoate (1a) as a substrate (Table 1). When chiral NHC precursor L1·HBF4 (10 mol %) was used in combination with CuCl (10 mol %) and LiOt-Bu (20 mol %) for the conjugate reduction of 1a with diethoxymethylsilane (4 equiv) as a reductant and t-AmOH (1 equiv) as a protonation reagent in DMA as the solvent at 25 °C for 15 h, the product 2a was produced in 98% yield (1H NMR analysis) with a promising enantioselectivity of 69% ee (Table 1, entry 1). When the phenolic hydroxy group of L1 was changed to a methoxy group (in L2), the enantioselectivity drastically dropped to −10% ee, while the yield remained 99% (Table 1, entry 2). Similarly, N,N'-dimesityl-NHC L3, which lacked an oxygen functionality in the N-aryl group, showed poor enantioselectivity (9% ee) with high yield (99% yield, Table 1, entry 3). Thus, the hydroxy group of L1 was essential for the enantioselectivity by the catalyst. When the mesityl group of L1 was changed to a bulkier 2-Me-4,6-Cy2-C6H2 group in L4, the enantioselectivity was markedly improved to 90% ee, with a high yield (97%, Table 1, entry 4). A naphthol substituent on the nitrogen atom of the NHC (in L5 and L6) instead of the phenol substituent was not suitable, giving significantly lower enantioselectivities (Table 1, entry 5, 99% yield, 26% ee; entry 6, 97% yield, 58% ee).
Table 1: Optimization of the copper-catalyzed enantioselective conjugate reduction of 1a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-50-i2.svg?max-width=637&scale=1.0)
|
|||||
| entry | ligand | silane | alcohol | yield (%) | ee (%) |
| 1 | (S,S)-L1·HBF4 | (EtO)2MeSiH | t-AmOH | 98 | 69 |
| 2 | (S,S)-L2·HBF4 | (EtO)2MeSiH | t-AmOH | 99 | −10 |
| 3 | (S,S)-L3·HBF4 | (EtO)2MeSiH | t-AmOH | 99 | 9 |
| 4 | (S,S)-L4·HBF4 | (EtO)2MeSiH | t-AmOH | 97 | 90 |
| 5 | (S,S)-L5·HBF4 | (EtO)2MeSiH | t-AmOH | 99 | 26 |
| 6 | (S,S)-L6·HBF4 | (EtO)2MeSiH | t-AmOH | 97 | 58 |
| 7 | (S,S)-L4·HBF4 | (MeO)2MeSiH | t-AmOH | 98 | 84 |
| 8 | (S,S)-L4·HBF4 | (TMSO)2MeSiH | t-AmOH | 94 | 71 |
| 9 | (S,S)-L4·HBF4 | (EtO)3SiH | t-AmOH | 0 | – |
| 10 | (S,S)-L4·HBF4 | PMHS | t-AmOH | 5 | – |
| 11 | (S,S)-L4·HBF4 | (EtO)2MeSiH | t-BuOH | 99 | 82 |
| 12 | (S,S)-L4·HBF4 | (EtO)2MeSiH | iPrOH | 7 | – |
| 13 | (S,S)-L4·HBF4 | (EtO)2MeSiH | MeOH | 4 | – |
aThe yield was determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. The enantiomeric excess (ee) was determined by HPLC analysis with a chiral stationary phase column CHIRALCEL® OD-H.
Further optimization of the conditions was conducted with L4. Changing the silane group affected both the reactivity and selectivity (Table 1, entries 7–10), while the replacement of the ethoxy groups of (EtO)2MeSiH with methoxy or trimethylsilyloxy groups resulted in only moderate reductions in the enantioselectivity and high yields. At the same time, trialkoxysilane (EtO)3SiH and polymeric silane PMHS gave only trace amounts of the product.
The nature of the alcoholic protonation reagent also had a strong impact. The presence of a tertiary alcohol, t-AmOH or t-BuOH, was essential for the reaction to occur with a reasonable yield, while iPrOH and MeOH markedly suppressed the reaction (Table 1, entries 12 and 13). However, the bulkier t-AmOH was superior to t-BuOH in terms of enantioselectivity (Table 1, entries 4 and 11).
Substrate scope
Having established optimized conditions for the reaction of 1a (2a, 97% yield, 90% ee (R), Table 2, entry 1), the scope of α,β-unsaturated carbonyl compounds was examined. Because the separation of the product from silicon-based byproducts was troublesome, the isolated yields were lower than the yields determined by NMR spectroscopy (2a, 52%, Table 2, entry 1).
Table 2: Substrate scope of the copper-catalyzed enantioselective conjugate reduction.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-50-i3.svg?max-width=637&scale=1.0)
|
||||
| entry | substrate | product | yield (%) | ee (%) |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-50-i4.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-50-i5.svg?max-width=637&scale=1.0)
2a |
97 (52) | 90 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-50-i6.svg?max-width=637&scale=1.0)
(E)-1a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-50-i7.svg?max-width=637&scale=1.0)
(S)-2a |
>99 (75) | 82 |
| 3b |
![[Graphic 7]](/bjoc/content/inline/1860-5397-16-50-i8.svg?max-width=637&scale=1.0)
1b |
![[Graphic 8]](/bjoc/content/inline/1860-5397-16-50-i9.svg?max-width=637&scale=1.0)
2b |
>99 (60) | 75 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-16-50-i10.svg?max-width=637&scale=1.0)
1c |
![[Graphic 10]](/bjoc/content/inline/1860-5397-16-50-i11.svg?max-width=637&scale=1.0)
2c |
>99 (44) | 84 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-16-50-i12.svg?max-width=637&scale=1.0)
1d |
![[Graphic 12]](/bjoc/content/inline/1860-5397-16-50-i13.svg?max-width=637&scale=1.0)
2d |
96 (74) | 76 |
| 6b,c |
![[Graphic 13]](/bjoc/content/inline/1860-5397-16-50-i14.svg?max-width=637&scale=1.0)
1e |
![[Graphic 14]](/bjoc/content/inline/1860-5397-16-50-i15.svg?max-width=637&scale=1.0)
2e |
>99 (75) | 84 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-16-50-i16.svg?max-width=637&scale=1.0)
1f |
![[Graphic 16]](/bjoc/content/inline/1860-5397-16-50-i17.svg?max-width=637&scale=1.0)
2f |
>99 (45) | 85 |
| 8b,c |
![[Graphic 17]](/bjoc/content/inline/1860-5397-16-50-i18.svg?max-width=637&scale=1.0)
1g |
![[Graphic 18]](/bjoc/content/inline/1860-5397-16-50-i19.svg?max-width=637&scale=1.0)
2g |
>99 (55) | 70 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-16-50-i20.svg?max-width=637&scale=1.0)
1h |
![[Graphic 20]](/bjoc/content/inline/1860-5397-16-50-i21.svg?max-width=637&scale=1.0)
2h |
>99 (79) | 83 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-16-50-i22.svg?max-width=637&scale=1.0)
1i |
![[Graphic 22]](/bjoc/content/inline/1860-5397-16-50-i23.svg?max-width=637&scale=1.0)
2i |
81 (59) | 79 |
aThe yields were determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. Isolated yields are shown in parentheses. bt-BuOH was used instead of t-AmOH. c(EtO)2MeSiH (1 equiv) was used.
When (E)-1a was used as the substrate, the opposite enantiomer (S)-2a was obtained in >99% yield, with slightly lower enantioselectivity (Table 2, entry 2, 82% ee (S) vs 90% ee (R) in entry 1). The inversion of the absolute configuration of the product depended on the E/Z geometry of the substrates, and this was analogous to the reported results obtained with the chiral bisphosphine ligand systems, while the observation of a higher enantioselectivity for the (Z)-isomer substrate 1a was characteristic for the phenol–NHC chiral ligand [4,6,8]. The result suggested that the chiral catalyst may mainly discriminate the hydrogen atom and the ethoxycarbonyl group at the α-position rather than the two substituents at the β-position. In good agreement with this assumption is the reaction of substrate 1b, carrying a phenyl group and a p-tolyl group as β-substituents, which seemed to be difficult for the catalyst to differentiate, both sterically and electronically, affording the product 2b in good enantioselectivity (Table 2, entry 3, >99% yield, 75% ee).
Next, the effects of the β-substituent (R1) were examined. Both electron-donating and -withdrawing substituents on the para-position of the β-aryl substituent gave excellent reactivities and good enantioselectivities (Table 2, entry 4, 2c: >99% yield, 84% ee; entry 5, 2d: 96% yield, 76% ee). Further, a heteroaryl substituent, 2-ethoxycarbonylthiophene, was tolerated (Table 2, entry 6, 2e: >99% yield, 84% ee), and a β-alkyl-substituted substrate 1f was also competent (entry 7, 2f: >99% yield, 85% ee).
The structure of the ester moiety affected the stereoselectivities: Lower enantioselectivities were observed with benzyl ester 2g (Table 2, entry 8, >99% yield, 70% ee) and isopropyl ester 2h (Table 2, entry 9, >99% yield, 83% ee). In addition to the α,β-unsaturated esters, an α,β-unsaturated Weinreb amide 1i afforded the corresponding product 2i with comparable results (Table 2, entry 10, 81% yield, 79% ee).
Proposed catalytic cycle
Based on the experimental observations and previous knowledge of the catalysis of phenol–NHC chiral ligands [14-18], we propose a catalytic cycle as shown in Scheme 1. LiOt-Bu abstracts the two protons of the acidic imidazolium C–H and phenol O–H groups of the L4·HBF4 adduct in the presence of CuCl to generate a phenoxy copper(I) species A. As a result, all LiOt-Bu (20 mol %) is consumed in this step. Thus, the system is neutral. Transmetalation between A and (EtO)2MeSiH produces the copper hydride species B. This transmetalation adds the silyl group to the phenoxy oxygen atom of the NHC ligand. The coordination of an α,β-unsaturated carbonyl compound 1 to the copper atom occurs in such a way that the bulky O-silyl group of the copper catalyst can avoid steric repulsions with the alkoxycarbonyl group of the substrate 1, forming π-complex C, and thus explaining the marked influence of the hydrosilane structure on the enantioselectivity. During this stereodetermining step, coordination of the phenoxy oxygen atom to the copper atom may render the chiral environment better defined. Then, the π-complex C undergoes 1,4-hydrocupration to afford copper enolate D. During our initial investigations, we observed configurational isomerization from 1a to (E)-1a when the reaction was conducted in the absence of alcoholic protonation reagents. This observation implied that the 1,4-hydrocupration step (C to D) is reversible. Finally, protonation of D by t-AmOH gives the product 2 and silyl ether (EtO)2MeSiOt-Am, regenerating the phenoxy copper(I) complex A. Due to the reversibility of the 1,4-hydrocupration, the choice of the alcoholic protonation reagent affects both the reactivity and enantioselectivity.
![[1860-5397-16-50-i1]](/bjoc/content/inline/1860-5397-16-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
A chiral phenol–NHC ligand efficiently promoted the enantioselective conjugate reduction of α,β-unsaturated esters with a hydrosilane. To the best of our knowledge, this is the first demonstration of the applicability of chiral NHC ligands in Cu-catalyzed enantioselective conjugate reductions. The phenolic N-substituent of the chiral NHC ligand was essential for the high enantioselectivity. Good enantioselectivities were observed for the various (Z)-configured substrates with different β-substituents. Further investigations on the catalyst development based on this knowledge are ongoing in our laboratory.
Experimental
A typical procedure for the copper-catalyzed enantioselective conjugate reduction (Table 1, entry 4; Table 2, entry 1): In a glove box, CuCl (1.5 mg, 0.015 mmol), L4⋅HBF4 (9.8 mg, 0.015 mmol), and LiOt-Bu (2.4 mg, 0.03 mmol) were placed in a vial containing a magnetic stirring bar. The vial was sealed with a teflon-coated silicon rubber septum. N,N-Dimethylacetamide (0.90 mL) was added to the vial, and then the mixture was stirred at room temperature for 5 min. Next, t-AmOH (16.3 μL, 0.15 mmol) was added, and the vial was taken out of the glove box. To the reaction mixture was added 1a (28.5 mg, 0.15 mmol). After stirring for 5 min at room temperature, diethoxymethylsilane (101 μL, 0.60 mmol) was added to the mixture. After stirring for 15 h at 25 °C, the reaction mixture was diluted with diethyl ether (1.0 mL) and quenched with H2O (0.6 mL). The organic layer was separated, and the aqueous layer was extracted three times with diethyl ether. The combined organic layer was filtered through a short plug of silica gel by using diethyl ether as an eluent. After the solvent was removed under reduced pressure, the yield of the product was determined to be 97% by 1H NMR analysis with 1,1,2,2-tetrachloroethane as an internal standard. After a rough purification of the crude product by silica gel chromatography (eluent: 0 to 1% EtOAc/hexane), the collected residue was further purified by GPC (eluent: CHCl3) to afford the pure product 2a as colorless oil (15.1 mg, 52% yield, 90% ee). 1H NMR (400 MHz, CDCl3) δ 1.17 (t, J = 7.2 Hz, 3H), 1.29 (d, J = 7.2 Hz, 3H), 2.50–2.63 (m, 2H), 3.27 (sext, J = 7.2 Hz, 1H), 4.06 (q, J = 7.2 Hz, 2H), 7.16–7.22 (m, 3H), 7.28 (t, J = 7.2 Hz, 2H); 13C NMR (100.5 Hz, CDCl3) δ 14.1, 21.7, 36.4, 42.9, 60.2, 126.3, 126.7, 128.4, 145.6, 172.3; [α]D27.3 −23.2 (c 1.25, CHCl3). The spectral data matched those reported in the literature [22].
The ee value was determined by HPLC analysis with a chiral stationary column CHIRALCEL® OD-H (Daicel Chemical Industries, 4.6 mm, 250 mm, hexane/2-propanol, 98:2, v/v, 0.5 mL/min, 40 °C, 220 nm UV detector, retention time = 9.1 min for the (R)-isomer and 13.3 min for the (S)-isomer). The absolute configuration of 2a was assigned by the comparison of the optical rotation with the same compound prepared by a reported method [4].
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, HPLC charts, and NMR spectra (1H, 13C) for the new compounds. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291–293. doi:10.1021/ja00209a048
Return to citation in text: [1] -
Mahoney, W. S.; Stryker, J. M. J. Am. Chem. Soc. 1989, 111, 8818–8823. doi:10.1021/ja00206a008
Return to citation in text: [1] -
Deutsch, C.; Krause, N.; Lipshutz, B. H. Chem. Rev. 2008, 108, 2916–2927. doi:10.1021/cr0684321
Return to citation in text: [1] -
Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9473–9474. doi:10.1021/ja992366l
Return to citation in text: [1] [2] [3] [4] -
Moritani, Y.; Appella, D. H.; Jurkauskas, V.; Buchwald, S. L. J. Am. Chem. Soc. 2000, 122, 6797–6798. doi:10.1021/ja0009525
Return to citation in text: [1] -
Lipshutz, B. H.; Servesko, J. M. Angew. Chem., Int. Ed. 2003, 42, 4789–4792. doi:10.1002/anie.200352313
Return to citation in text: [1] [2] -
Hughes, G.; Kimura, M.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11253–11258. doi:10.1021/ja0351692
Return to citation in text: [1] -
Lipshutz, B. H.; Servesko, J. M.; Taft, B. R. J. Am. Chem. Soc. 2004, 126, 8352–8353. doi:10.1021/ja049135l
Return to citation in text: [1] [2] -
Huang, S.; Voigtritter, K. R.; Unger, J. B.; Lipshutz, B. H. Synlett 2010, 2041–2042. doi:10.1055/s-0030-1258540
Return to citation in text: [1] -
Wu, Y.; Qi, S.-B.; Wu, F.-F.; Zhang, X.-C.; Li, M.; Wu, J.; Chan, A. S. C. Org. Lett. 2011, 13, 1754–1757. doi:10.1021/ol200287z
Return to citation in text: [1] -
Hou, C.-J.; Guo, W.-L.; Hu, X.-P.; Deng, J.; Zheng, Z. Tetrahedron: Asymmetry 2011, 22, 195–199. doi:10.1016/j.tetasy.2011.01.022
Return to citation in text: [1] -
Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Chem. Soc. Rev. 2017, 46, 4845–4854. doi:10.1039/c7cs00200a
Return to citation in text: [1] -
Jurkauskas, V.; Sadighi, J. P.; Buchwald, S. L. Org. Lett. 2003, 5, 2417–2420. doi:10.1021/ol034560p
Return to citation in text: [1] -
Harada, A.; Makida, Y.; Sato, T.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2014, 136, 13932–13939. doi:10.1021/ja5084333
Return to citation in text: [1] [2] -
Ohmiya, H.; Zhang, H.; Shibata, S.; Harada, A.; Sawamura, M. Angew. Chem., Int. Ed. 2016, 55, 4777–4780. doi:10.1002/anie.201600619
Return to citation in text: [1] [2] -
Yasuda, Y.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2016, 55, 10816–10820. doi:10.1002/anie.201605125
Return to citation in text: [1] [2] -
Hojoh, K.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2017, 139, 2184–2187. doi:10.1021/jacs.6b12881
Return to citation in text: [1] [2] [3] -
Yasuda, Y.; Ohmiya, H.; Sawamura, M. Synthesis 2018, 50, 2235–2246. doi:10.1055/s-0036-1591548
Return to citation in text: [1] [2] -
Clavier, H.; Coutable, L.; Guillemin, J.-C.; Mauduit, M. Tetrahedron: Asymmetry 2005, 16, 921–924. doi:10.1016/j.tetasy.2005.01.015
Return to citation in text: [1] -
Martin, D.; Kehrli, S.; d'Augustin, M.; Clavier, H.; Mauduit, M.; Alexakis, A. J. Am. Chem. Soc. 2006, 128, 8416–8417. doi:10.1021/ja0629920
Return to citation in text: [1] -
Shintani, R.; Takatsu, K.; Takeda, M.; Hayashi, T. Angew. Chem., Int. Ed. 2011, 50, 8656–8659. doi:10.1002/anie.201103581
Return to citation in text: [1] -
Guo, S.; Zhou, J. Org. Lett. 2016, 18, 5344–5347. doi:10.1021/acs.orglett.6b02662
Return to citation in text: [1]
| 1. | Mahoney, W. S.; Brestensky, D. M.; Stryker, J. M. J. Am. Chem. Soc. 1988, 110, 291–293. doi:10.1021/ja00209a048 |
| 2. | Mahoney, W. S.; Stryker, J. M. J. Am. Chem. Soc. 1989, 111, 8818–8823. doi:10.1021/ja00206a008 |
| 12. | Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Chem. Soc. Rev. 2017, 46, 4845–4854. doi:10.1039/c7cs00200a |
| 4. | Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9473–9474. doi:10.1021/ja992366l |
| 4. | Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9473–9474. doi:10.1021/ja992366l |
| 5. | Moritani, Y.; Appella, D. H.; Jurkauskas, V.; Buchwald, S. L. J. Am. Chem. Soc. 2000, 122, 6797–6798. doi:10.1021/ja0009525 |
| 6. | Lipshutz, B. H.; Servesko, J. M. Angew. Chem., Int. Ed. 2003, 42, 4789–4792. doi:10.1002/anie.200352313 |
| 7. | Hughes, G.; Kimura, M.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 11253–11258. doi:10.1021/ja0351692 |
| 8. | Lipshutz, B. H.; Servesko, J. M.; Taft, B. R. J. Am. Chem. Soc. 2004, 126, 8352–8353. doi:10.1021/ja049135l |
| 9. | Huang, S.; Voigtritter, K. R.; Unger, J. B.; Lipshutz, B. H. Synlett 2010, 2041–2042. doi:10.1055/s-0030-1258540 |
| 10. | Wu, Y.; Qi, S.-B.; Wu, F.-F.; Zhang, X.-C.; Li, M.; Wu, J.; Chan, A. S. C. Org. Lett. 2011, 13, 1754–1757. doi:10.1021/ol200287z |
| 11. | Hou, C.-J.; Guo, W.-L.; Hu, X.-P.; Deng, J.; Zheng, Z. Tetrahedron: Asymmetry 2011, 22, 195–199. doi:10.1016/j.tetasy.2011.01.022 |
| 4. | Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9473–9474. doi:10.1021/ja992366l |
| 3. | Deutsch, C.; Krause, N.; Lipshutz, B. H. Chem. Rev. 2008, 108, 2916–2927. doi:10.1021/cr0684321 |
| 17. | Hojoh, K.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2017, 139, 2184–2187. doi:10.1021/jacs.6b12881 |
| 14. | Harada, A.; Makida, Y.; Sato, T.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2014, 136, 13932–13939. doi:10.1021/ja5084333 |
| 15. | Ohmiya, H.; Zhang, H.; Shibata, S.; Harada, A.; Sawamura, M. Angew. Chem., Int. Ed. 2016, 55, 4777–4780. doi:10.1002/anie.201600619 |
| 16. | Yasuda, Y.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2016, 55, 10816–10820. doi:10.1002/anie.201605125 |
| 17. | Hojoh, K.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2017, 139, 2184–2187. doi:10.1021/jacs.6b12881 |
| 18. | Yasuda, Y.; Ohmiya, H.; Sawamura, M. Synthesis 2018, 50, 2235–2246. doi:10.1055/s-0036-1591548 |
| 19. | Clavier, H.; Coutable, L.; Guillemin, J.-C.; Mauduit, M. Tetrahedron: Asymmetry 2005, 16, 921–924. doi:10.1016/j.tetasy.2005.01.015 |
| 20. | Martin, D.; Kehrli, S.; d'Augustin, M.; Clavier, H.; Mauduit, M.; Alexakis, A. J. Am. Chem. Soc. 2006, 128, 8416–8417. doi:10.1021/ja0629920 |
| 21. | Shintani, R.; Takatsu, K.; Takeda, M.; Hayashi, T. Angew. Chem., Int. Ed. 2011, 50, 8656–8659. doi:10.1002/anie.201103581 |
| 22. | Guo, S.; Zhou, J. Org. Lett. 2016, 18, 5344–5347. doi:10.1021/acs.orglett.6b02662 |
| 14. | Harada, A.; Makida, Y.; Sato, T.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2014, 136, 13932–13939. doi:10.1021/ja5084333 |
| 15. | Ohmiya, H.; Zhang, H.; Shibata, S.; Harada, A.; Sawamura, M. Angew. Chem., Int. Ed. 2016, 55, 4777–4780. doi:10.1002/anie.201600619 |
| 16. | Yasuda, Y.; Ohmiya, H.; Sawamura, M. Angew. Chem., Int. Ed. 2016, 55, 10816–10820. doi:10.1002/anie.201605125 |
| 17. | Hojoh, K.; Ohmiya, H.; Sawamura, M. J. Am. Chem. Soc. 2017, 139, 2184–2187. doi:10.1021/jacs.6b12881 |
| 18. | Yasuda, Y.; Ohmiya, H.; Sawamura, M. Synthesis 2018, 50, 2235–2246. doi:10.1055/s-0036-1591548 |
| 13. | Jurkauskas, V.; Sadighi, J. P.; Buchwald, S. L. Org. Lett. 2003, 5, 2417–2420. doi:10.1021/ol034560p |
| 4. | Appella, D. H.; Moritani, Y.; Shintani, R.; Ferreira, E. M.; Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 9473–9474. doi:10.1021/ja992366l |
| 6. | Lipshutz, B. H.; Servesko, J. M. Angew. Chem., Int. Ed. 2003, 42, 4789–4792. doi:10.1002/anie.200352313 |
| 8. | Lipshutz, B. H.; Servesko, J. M.; Taft, B. R. J. Am. Chem. Soc. 2004, 126, 8352–8353. doi:10.1021/ja049135l |
© 2020 Mimura et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)