Abstract
A practical three-step protocol for the assembly of triazolobenzodiazepine-fused diketopiperazines and hydantoins has been developed. The synthesis of these tetracyclic ring systems was initiated by an Ugi reaction, which brought together all necessary functionalities for further transformations. The Ugi adducts were then subjected to a base-induced ring closing and an intramolecular azide–alkyne cycloaddition reaction in succession to obtain highly fused benzodiazepine frameworks.

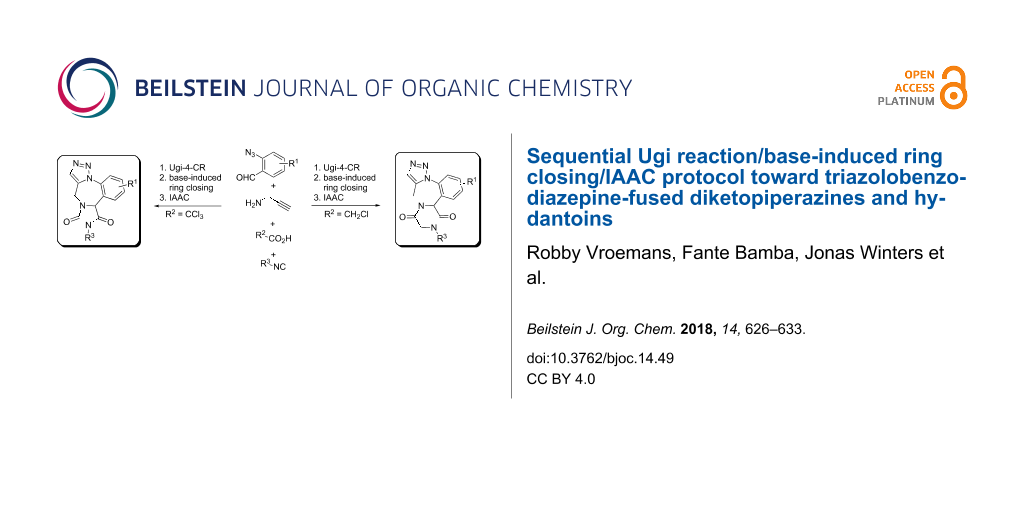
Graphical Abstract
Introduction
The versatile bioactivities of multiring-fused heterocyclic scaffolds continue to attract significant attention in developing new methods for their synthesis. A plethora of functionalized fused heterocycles can be easily accessed by designing sequential organic transformations incorporating multicomponent reactions [1,2] combined with secondary transformations [3-11]. Over the past decade, several groups of chemists interested in synthetic chemistry have efficiently utilized the strategy of linking multicomponent reactions with intramolecular azide–alkyne cycloaddition (IAAC) for the generation of triazole-fused heterocycles [12-23]. In this report we disclose our results on the development of a sequential synthetic approach involving Ugi 4-component reaction (4-CR) and two ring-closing steps toward triazolobenzodiazepine-fused diketopiperazines and hydantoins.
Benzodiazepine derivatives [24,25] form an important class of ‘psychoactive drugs’ which is being extensively used in the treatment of anxiety, insomnia, agitation, seizures, muscle spasms, alcohol withdrawal, etc. [26-34]. In addition, these azaheterocycles also exhibit anti-inflammatory, antitumor, antiparasitic and anxiolytic activities [35-42]. In particular, 1,4-benzodiazepine derivatives are proposed to serve as a structural mimic of peptide β-turns [43-45] and are also known to bind to a number of biological targets [46-48]. Interestingly, the fusion of a triazole ring to a 1,4-benzodiazepine core has resulted in an increase in the biological activity as evident from the different drugs with triazolobenzodiazepine structure such as alprazolam, etizolam, triazolam, etc., available for the treatment of CNS related ailments (Figure 1) [49-55].
![[1860-5397-14-49-1]](/bjoc/content/figures/1860-5397-14-49-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Another set of heterocycles are 2,5-diketopiperazines, which form the core structure of many natural products and have recently been thoroughly studied for their wide spectrum of biological activities [56-58]. Owing to our interest in the chemistry of 1,2,3-triazoles [59-72] and the interesting biological activities of benzodiazepines and 2,5-diketopiperazines, we were motivated to develop a facile route towards multiring-fused derivatives incorporating all these heterocyclic moieties. We hypothesized that 2,5-diketopiperazine-fused triazolobenzodiazepine could be obtained by proper functionalization of building blocks starting with an Ugi reaction; followed by two ring-closing steps, one being a base-induced heterocycle formation and the other an IAAC. Unlike some of the reported strategies using Cu-catalyzed IAAC [13,15], we postulated that the intramolecular dipolar cycloaddition would proceed under thermal conditions (metal-free) due to the close proximity of the azide and similar functionalities in the intermediate. The functionalities required for the secondary transformations like the leaving group (-Cl) for nucleophilic substitution, alkyne and azide for IAAC can be assembled via the Ugi reaction (Scheme 1).
![[1860-5397-14-49-i1]](/bjoc/content/inline/1860-5397-14-49-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis towards 2,5-diketopiperazine fused triazolobenzodiazepine.
Scheme 1: Retrosynthetic analysis towards 2,5-diketopiperazine fused triazolobenzodiazepine.
Results and Discussion
We initiated our studies with the Ugi 4-component reaction of o-azidobenzaldehyde (1a), propargylamine (2a), 2-chloroacetic acid (3a) and benzyl isocyanide (4a, Scheme 2). Initially, the condensation of the aldehyde and the amine was effected in MeOH in the presence of 4 Å MS. This was followed by the sequential addition of chloroacetic acid and the isocyanide which, after 24 hours at room temperature, afforded the expected Ugi adduct 5a in 96% yield.
![[1860-5397-14-49-i2]](/bjoc/content/inline/1860-5397-14-49-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The synthesis of fused heterocycles from the Ugi adduct 5a was attempted via two routes (Scheme 3). The first route involved the base-induced cyclisation of 5a at the amide end by treatment with KOH from which the diketopiperazine moiety 6 was obtained in 51% yield. Compound 6 bearing azide and alkyne functionalities was subjected to intramolecular azide–alkyne cycloaddition (IAAC) in EtOH at reflux conditions which afforded the target diketopiperazine-fused triazolobenzodiazepine 7a in 81% yield. The secondary path commenced with the intramolecular azide–alkyne cycloaddition of 5a which furnished the triazole-fused benzodiazepine 8 in 72% yield after 3 hours. The triazolobenzodiazepine 8 was then subjected to base-induced cyclisation with KOH which failed to afford the expected tetracyclic compound 7a. We believe that conformational restrictions prevent the side chains on the benzodiazepine bearing the amide and halogen to be in close proximity, which is needed for the reaction to proceed.
![[1860-5397-14-49-i3]](/bjoc/content/inline/1860-5397-14-49-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of diketopiperazine-fused triazolobenzodiazepine 7a.
Scheme 3: Synthesis of diketopiperazine-fused triazolobenzodiazepine 7a.
The generality of the developed fused-heterocycle synthesis was studied with substituted azidoaldehyde, isocyanides and chloroacetic acid (Figure 2, see Supporting Information File 1 for full experimental data). In all the cases, the Ugi adduct precipitated from the reaction mixture within 24 hours, which was filtered, washed, dried and used as such without further purification. For all the examples, the ring-closing reactions were done in one-pot; which involved neutralization after base-induced diketopiperazine synthesis and dilution with EtOH before the IAAC step. Our first attempt was to study the effect of different substituents on the isocyanide (4a–d) moiety; from which we observed a slight decrease in the yield of the final product with increase in steric bulk of the substituent. Hence, an overall yield (over 3 steps) of 37% was obtained for benzyl-substituted polycyclic compound 7a, 34% for 7b (cyclohexyl) and 27% for 7c (tert-butyl). p-Methoxyphenyl-substituted compound 7d was obtained in a good overall yield of 67%. Next, we investigated the effect of the substitution pattern on the propargylamine 2. For this, we chose easily available propargylamines 2b and 2c which gave moderate to good yields (60 and 40%, respectively) of diketopiperazine-fused triazolobenzodiazepine 7e and 7f. Furthermore, halogen substituents were introduced on polyfused heterocycles 7 because these were shown to often have a positive effect on the bioactivity (see compounds in Figure 1). Starting with 2-azido-4-bromobenzaldehyde (1b), we were able to prepare two brominated tetracyclic derivatives 7g and 7h. In the same way, we were also successful in synthesizing chloro derivatives 7i and 7j from 2-azido-4-chlorobenzaldehyde (1c). The multiring-fused heterocycle synthesis was also tried with an azidobenzaldehyde bearing electron-donating substituents; the reaction with 2-azido-4,5-dimethoxybenzaldehyde (1d) yielded the product 7k in lesser yield (43%). Finally, an additional stereocenter was introduced in the diketopiperazine ring by carrying out the Ugi reaction with 2-chloropropanoic acid (3b). After the sequential ring-closing steps the expected diketopiperazine-fused triazolobenzodiazepine 7l was obtained in 41% yield as a 1:0.7 inseparable mixture of diastereomers. Various attempts were made with other azidobenzaldehydes (5-azido-3-methyl-1-phenyl-1H-pyrazole-4-carbaldehyde, 2-azidoquinoline-3-carbaldehyde and 2-azido-5-nitrobenzaldehyde) and isocyanides (2-morpholinoethyl isocyanide) towards the synthesis of diketopiperazine-fused triazolobenzodiazepine 7m–p. All of the mentioned substrates gave their respective Ugi adducts but the latter decomposed under basic treatment (in the case of 2-azidoquinoline-3-carbaldehyde, 2-azido-5-nitrobenzaldehyde, 2-morpholinoethyl isocyanide) or decomposed under refluxing conditions (in the case of 5-azido-3-methyl-1-phenyl-1H-pyrazole-4-carbaldehyde).
![[1860-5397-14-49-2]](/bjoc/content/figures/1860-5397-14-49-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Generality in the synthesis of diketopiperazine-fused triazolobenzodiazepine 7. Reaction conditions: 1. 2-azidobenzaldehyde 1 (1.85 mmol), propargylamine 2 (1.85 mmol), chloroacetic acid 3 (1.85 mmol), benzyl isocyanide 4 (1.85 mmol), 4 Å MS (100 mg), EtOH (7 mL), 20 h, rt; 2. Ugi adduct 5 (0.26 mmol), KOH (0.28 mmol), ethanol (1.5 mL), sonication for 30 minutes, rt; 3. neutralization with 1 M HCl, EtOH (20 mL) reflux, 3 h. Overall yields for 3 steps are given for all the examples.
Figure 2: Generality in the synthesis of diketopiperazine-fused triazolobenzodiazepine 7. Reaction conditions...
The success in the synthesis of diketopiperazine-fused triazolobenzodiazepine prompted us to examine the possibility of making the sequential synthetic route in ‘one-pot’ starting from the Ugi 4-CR (Scheme 4). Thus we initiated the reaction sequence from the Ugi reaction in EtOH, after completion of which, the ring-closing steps (base induced and IAAC) were performed in succession. Unfortunately, the final products were obtained in slightly lower yields; 30% for 7a and 26% for 7b, respectively.
![[1860-5397-14-49-i4]](/bjoc/content/inline/1860-5397-14-49-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: ‘One-pot’ synthesis of diketopiperazine-fused triazolobenzodiazepines 7a and 7b.
Scheme 4: ‘One-pot’ synthesis of diketopiperazine-fused triazolobenzodiazepines 7a and 7b.
Hydantoins belong to a unique class of heterocycles which can be found in a number of pharmaceuticals, cosmetics, insecticides, etc. [73-75]. These cyclic ureides are well known for their anticonvulsant activity [76-78]. With the assumption that these molecules when fused to a benzodiazepine core would enhance the anticonvulsant activity, we undertook the synthesis of hydantoin-fused triazolobenzodiazepines starting from the Ugi reaction of o-azidobenzaldehyde (1a), propargylamine (2a), trichloroacetic acid (9) and benzyl isocyanide (4a, Scheme 5). The Ugi adduct was subjected to base-induced ring closing with NaOEt in EtOH which furnished the hydantoin intermediates; which on subsequent neutralization–dilution–IAAC afforded the expected hydantoin-fused benzodiazepine derivative 10a in 45% overall yield. In a similar way the cyclohexyl and p-methoxyphenyl-substituted compounds 10b and 10c were obtained in 82% and 62% overall yield, respectively.
![[1860-5397-14-49-i5]](/bjoc/content/inline/1860-5397-14-49-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of hydantoin-fused triazolobenzodiazepine 10. Reaction conditions: 1. 2-azidobenzaldehyde 1 (1.85 mmol), propargylamine (2a, 1.85 mmol), trichloroacetic acid (9, 1.85 mmol), benzyl isocyanide 4 (1.85 mmol), 4 Å MS (100 mg), EtOH (7 mL), 20 h, rt; 2. Ugi adduct 5 (0.26 mmol), NaOEt (0.28 mmol), ethanol (1.5 mL), 30 min, rt; 3. neutralization with 1 M HCl, EtOH (20 mL) reflux, 3 h. Overall yields for 3 steps are given for all the examples.
Scheme 5: Synthesis of hydantoin-fused triazolobenzodiazepine 10. Reaction conditions: 1. 2-azidobenzaldehyde ...
The structure of hydantoin-fused triazolobenzodiazepine was confirmed by a X-ray crystal structure determination of 10a (Figure 3).
![[1860-5397-14-49-3]](/bjoc/content/figures/1860-5397-14-49-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of hydantoin-fused triazolobenzodiazepine 10a. (Displacement ellipsoids are drawn at the 30% probability level.)
Figure 3: X-ray crystal structure of hydantoin-fused triazolobenzodiazepine 10a. (Displacement ellipsoids are...
The three-step synthesis of heterocycle-fused triazolobenzodiazepines involves the well-known Ugi 4-CR as the first step and the IAAC as the last, the mechanisms of which are well established (Scheme 6). The second stage is the base-induced ring closing of the Ugi adduct A and D to the respective heterocycles. In the case of Ugi adduct A, the base (KOH) abstracts the amide hydrogen which attacks the methylene carbon (expelling a chloride ion) and thereby furnishing the 2,5-diketopiperazine ring C. On the other hand, in the case of Ugi adduct D, a base induced intramolecular nucleophilic attack of the amide N-atom to the carbonyl carbon occurs (in intermediate E) and the hydantoin ring F forms by the elimination of the trichloromethyl anion.
![[1860-5397-14-49-i6]](/bjoc/content/inline/1860-5397-14-49-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Mechanism of formation of diketopiperazine and hydantoin-fused triazolobenzodiazepines.
Scheme 6: Mechanism of formation of diketopiperazine and hydantoin-fused triazolobenzodiazepines.
Conclusion
In conclusion, we have developed a sequential synthetic strategy to access diketopiperazine-fused triazolobenzodiazepines from simple starting materials. It is noteworthy to mention that halogenated (chloro and bromo) benzodiazepines could be synthesized following the developed protocol which might show analogues biological activity as that of halogenated triazolobenzodiazepine drugs available in market. We could also extend the strategy for the synthesis of hydantoin-fused triazolobenzodiazepines, other valuable multiring-fused heterocycles which might also exhibit potent bioactivities. The synthesized compounds are currently being evaluated for their biological properties, the results of which will be reported in due course.
Supporting Information
The Supporting Information features further experimental details, copies of 1H and 13C NMR spectra of compounds 5a, 6, 8, diketopiperazine-fused triazolobenzodiazepines 7 and hydantoin-fused triazolobenzodiazepines 10, and X-ray crystal structure details of 10a.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.8 MB | Download |
Acknowledgements
We thank the KU Leuven for financial support. Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225-7). RV thanks the Fonds Wetenschappelijk Onderzoek – Vlaanderen (FWO) for a Ph.D. fellowship (1S13516N). JJ thanks the Department of Science and Technology (DST)-SERB, India, for a Young Scientist Start-Up Research Grant (SB/FT/CS-107/2014). LVM thanks the Hercules Foundation for supporting the purchase of the diffractometer (grant AKUL/09/0035).
References
-
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527605118
Return to citation in text: [1] -
Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e
Return to citation in text: [1] -
Marcaccini, S.; Pepino, R.; Pozo, M. C. Tetrahedron Lett. 2001, 42, 2727–2728. doi:10.1016/S0040-4039(01)00232-5
Return to citation in text: [1] -
Marcaccini, S.; Miliciani, M.; Pepino, R. Tetrahedron Lett. 2005, 46, 711–713. doi:10.1016/j.tetlet.2004.11.083
Return to citation in text: [1] -
Ignacio, J. M.; Macho, S.; Marcaccini, S.; Pepino, R.; Torroba, T. Synlett 2005, 3051–3054. doi:10.1055/s-2005-922745
Return to citation in text: [1] -
Marcaccini, S.; Torroba, T. Nat. Protoc. 2007, 2, 632–639. doi:10.1038/nprot.2007.71
Return to citation in text: [1] -
Sunderhaus, J. D.; Dockendorff, C.; Martin, S. F. Org. Lett. 2007, 9, 4223–4226. doi:10.1021/ol7018357
Return to citation in text: [1] -
Hardy, S.; Martin, S. F. Org. Lett. 2011, 13, 3102–3105. doi:10.1021/ol201010s
Return to citation in text: [1] -
Sañudo, M.; Garcia-Valverde, M.; Marcaccini, S.; Torroba, T. Tetrahedron 2012, 68, 2621–2629. doi:10.1016/j.tet.2012.01.073
Return to citation in text: [1] -
Granger, B. A.; Wang, Z.; Kaneda, K.; Fang, Z.; Martin, S. F. ACS Comb. Sci. 2013, 15, 379–386. doi:10.1021/co400055b
Return to citation in text: [1] -
Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. J. Org. Chem. 2015, 80, 3411–3428. doi:10.1021/jo502829j
Return to citation in text: [1] -
Akritopoulou-Zanze, I.; Gracias, V.; Djuric, S. W. Tetrahedron Lett. 2004, 45, 8439–8441. doi:10.1016/j.tetlet.2004.09.117
Return to citation in text: [1] -
Gracias, V.; Darczak, D.; Gasiecki, A. F.; Djuric, S. W. Tetrahedron Lett. 2005, 46, 9053–9056. doi:10.1016/j.tetlet.2005.10.090
Return to citation in text: [1] [2] -
Pirali, T.; Tron, G. C.; Zhu, J. Org. Lett. 2006, 8, 4145–4148. doi:10.1021/ol061782p
Return to citation in text: [1] -
Bahulayan, D.; Arun, S. Tetrahedron Lett. 2012, 53, 2850–2855. doi:10.1016/j.tetlet.2012.03.116
Return to citation in text: [1] [2] -
Dhondge, A. P.; Afraj, S. N.; Nuzlia, C.; Chen, C.; Lee, G.-H. Eur. J. Org. Chem. 2013, 4119–4130. doi:10.1002/ejoc.201300226
Return to citation in text: [1] -
Nguyen, H. H.; Palazzo, T. A.; Kurth, M. J. Org. Lett. 2013, 15, 4492–4495. doi:10.1021/ol402045h
Return to citation in text: [1] -
Subba Reddy, B. V.; Majumder, N.; Prabhakar Rao, T. Synthesis 2014, 46, 3408–3414. doi:10.1055/s-0034-1379031
Return to citation in text: [1] -
De Moliner, F.; Bigatti, M.; De Rosa, C.; Banfi, L.; Riva, R.; Basso, A. Mol. Diversity 2014, 18, 473–482. doi:10.1007/s11030-014-9530-x
Return to citation in text: [1] -
De Moliner, F.; Bigatti, M.; Banfi, L.; Riva, R.; Basso, A. Org. Lett. 2014, 16, 2280–2283. doi:10.1021/ol500813p
Return to citation in text: [1] -
Barlow, T. M. A.; Jida, M.; Tourwé, D.; Ballet, S. Org. Biomol. Chem. 2014, 12, 6986–6989. doi:10.1039/C4OB01381F
Return to citation in text: [1] -
Salvador, C. E. M.; Pieber, B.; Neu, P. M.; Torvisco, A.; Andrade, C. K. Z.; Kappe, C. O. J. Org. Chem. 2015, 80, 4590–4602. doi:10.1021/acs.joc.5b00445
Return to citation in text: [1] -
Raj, P. J.; Bahulayan, D. Tetrahedron Lett. 2015, 56, 2451–2455. doi:10.1016/j.tetlet.2015.03.081
Return to citation in text: [1] -
Keating, T. A.; Armstrong, R. W. J. Am. Chem. Soc. 1996, 118, 2574–2583. doi:10.1021/ja953868b
Return to citation in text: [1] -
Keating, T. A.; Armstrong, R. W. J. Org. Chem. 1996, 61, 8935–8939. doi:10.1021/jo961517p
Return to citation in text: [1] -
Attwood, D.; Gibson, J. J. Pharm. Pharmacol. 1978, 30, 176–180. doi:10.1111/j.2042-7158.1978.tb13192.x
Return to citation in text: [1] -
Chakrabarti, J. K.; Hotten, T. M.; Pullar, I. A.; Steggles, D. J. J. Med. Chem. 1989, 32, 2375–2381. doi:10.1021/jm00130a025
Return to citation in text: [1] -
Hsu, M. C.; Schutt, A. D.; Holly, M.; Slice, L. W.; Sherman, M. I.; Richman, D. D.; Potash, M. J.; Volsky, D. J. Science 1991, 254, 1799–1802. doi:10.1126/science.1763331
Return to citation in text: [1] -
Liegeois, J. F. F.; Bruhwyler, J.; Damas, J.; Nguyen, T. P.; Chleide, E. M. G.; Mercier, M. G. A.; Rogister, F. A.; Delarge, J. E. J. Med. Chem. 1993, 36, 2107–2114. doi:10.1021/jm00067a009
Return to citation in text: [1] -
Liegeois, J. F. F.; Rogister, F. A.; Bruhwyler, J.; Damas, J.; Nguyen, T. P.; Inarejos, M. O.; Chleide, E. M. G.; Mercier, M. G. A.; Delarge, J. E. J. Med. Chem. 1994, 37, 519–525. doi:10.1021/jm00030a011
Return to citation in text: [1] -
Ballard, C. G.; O'Brien, J. T.; Swann, A. G.; Thompson, P.; Neill, D.; McKeith, I. G. J. Clin. Psychiatry 2001, 62, 46. doi:10.4088/JCP.v62n0110
Return to citation in text: [1] -
Nierenberg, A. A.; Gray, S. M.; Grandin, L. D. J. Clin. Psychiatry 2001, 62 (Suppl. 25), 27–30.
Return to citation in text: [1] -
Hanley, D. F., Jr.; Pozo, M. Int. J. Clin. Pract. 2000, 54, 30–35.
Return to citation in text: [1] -
Zahradník, I.; Minarovič, I.; Zahradníková, A. J. Pharmacol. Exp. Ther. 2008, 324, 977–984. doi:10.1124/jpet.107.132456
Return to citation in text: [1] -
Coyne, W. E.; Cusic, J. W. J. Med. Chem. 1967, 10, 541–546. doi:10.1021/jm00316a009
Return to citation in text: [1] -
Wright, W. B., Jr.; Brabander, H. J.; Greenblatt, E. N.; Day, I. P.; Hardy, R. A., Jr. J. Med. Chem. 1978, 21, 1087–1089. doi:10.1021/jm00208a017
Return to citation in text: [1] -
Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s
Return to citation in text: [1] -
Kamal, A.; Shetti, R. V. C. R. N. C.; Ramaiah, M. J.; Swapna, P.; Reddy, K. S.; Mallareddy, A.; Rao, M. P. N.; Chourasia, M.; Sastry, G. N.; Juvekar, A.; Zingde, S.; Sarma, P.; Pushpavalli, S. N. C. V. L.; Pal-Bhadra, M. MedChemComm 2011, 2, 780–788. doi:10.1039/c1md00072a
Return to citation in text: [1] -
Gill, R. K.; Kaushik, S. O.; Chugh, J.; Bansal, S.; Shah, A.; Bariwal, J. Mini-Rev. Med. Chem. 2014, 14, 229–256. doi:10.2174/13895575113139990081
Return to citation in text: [1] -
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b
Return to citation in text: [1] -
Maheshwari, N.; Goyal, A.; Jain, S.; Jain, D.; Moorthy, N. S. H. N. Med. Chem. Res. 2014, 23, 765–774. doi:10.1007/s00044-013-0675-4
Return to citation in text: [1] -
Thurston, D. E.; Vassoler, H.; Jackson, P. J. M.; James, C. H.; Rahman, K. M. Org. Biomol. Chem. 2015, 13, 4031–4040. doi:10.1039/C4OB02405B
Return to citation in text: [1] -
Ripka, W. C.; De Lucca, G. V.; Bach, A. C., II; Pottorf, R. S.; Blaney, J. M. Tetrahedron 1993, 49, 3593–3608. doi:10.1016/S0040-4020(01)90217-0
Return to citation in text: [1] -
Ripka, A. S.; Rich, D. H. Curr. Opin. Chem. Biol. 1998, 2, 441–452. doi:10.1016/S1367-5931(98)80119-1
Return to citation in text: [1] -
Hata, M.; Marshall, G. R. J. Comput.-Aided Mol. Des. 2006, 20, 321–331. doi:10.1007/s10822-006-9059-x
Return to citation in text: [1] -
Mohler, H.; Okada, T. Science 1977, 198, 849–851. doi:10.1126/science.918669
Return to citation in text: [1] -
Braestrup, C.; Schmiechen, R.; Neef, G.; Nielsen, M.; Petersen, E. N. Science 1982, 216, 1241–1243. doi:10.1126/science.6281892
Return to citation in text: [1] -
Rudolph, U.; Knoflach, F. Nat. Rev. Drug Discovery 2011, 10, 685–697. doi:10.1038/nrd3502
Return to citation in text: [1] -
Scharf, M. B.; Roth, P. B.; Dominguez, R. A.; Ware, J. C. J. Clin. Pharmacol. 1990, 30, 461–467. doi:10.1002/j.1552-4604.1990.tb03486.x
Return to citation in text: [1] -
Post, G. L.; Patrick, R. O.; Crowder, J. E.; Houston, J.; Ferguson, J. M.; Bielski, R. J.; Bailey, L.; Pearlman, H. G.; Shu, V. S.; Pierce, M. W. J. Clin. Psychopharmacol. 1991, 11, 249–253. doi:10.1097/00004714-199108000-00005
Return to citation in text: [1] -
Gerecke, M.; Kyburz, E.; Borer, R.; Gassner, W. Heterocycles 1994, 39, 693–721. doi:10.3987/COM-94-S(B)61
Return to citation in text: [1] -
Levine, J.; Cole, D. P.; Chengappa, K. N. R.; Gershon, S. Depression Anxiety 2001, 14, 94–104. doi:10.1002/da.1051
Return to citation in text: [1] -
Snyder, P. J.; Werth, J.; Giordani, B.; Caveney, A. F.; Feltner, D.; Maruff, P. Hum. Psychopharmacol. Clin. Exp. 2005, 20, 263–273. doi:10.1002/hup.692
Return to citation in text: [1] -
Alajarín, M.; Cabrera, J.; Pastor, A.; Villalgordo, J. M. Tetrahedron Lett. 2007, 48, 3495–3499. doi:10.1016/j.tetlet.2007.03.121
Return to citation in text: [1] -
Mohapatra, D. K.; Maity, P. K.; Shabab, M.; Khan, M. I. Bioorg. Med. Chem. Lett. 2009, 19, 5241–5245. doi:10.1016/j.bmcl.2009.06.107
Return to citation in text: [1] -
Martins, M. B.; Carvalho, I. Tetrahedron 2007, 63, 9923–9932. doi:10.1016/j.tet.2007.04.105
Return to citation in text: [1] -
Hulme, C.; Morrissette, M. M.; Volz, F. A.; Burns, C. J. Tetrahedron Lett. 1998, 39, 1113–1116. doi:10.1016/S0040-4039(97)10795-X
Return to citation in text: [1] -
Nixey, T.; Kelly, M.; Hulme, C. Tetrahedron Lett. 2000, 41, 8729–8733. doi:10.1016/S0040-4039(00)01563-X
Return to citation in text: [1] -
Merckx, T.; Verwilst, P.; Dehaen, W. Tetrahedron Lett. 2013, 54, 4237–4240. doi:10.1016/j.tetlet.2013.05.133
Return to citation in text: [1] -
Thomas, J.; John, J.; Parekh, N.; Dehaen, W. Angew. Chem., Int. Ed. 2014, 53, 10155–10159. doi:10.1002/anie.201403453
Return to citation in text: [1] -
Efimov, I.; Bakulev, V.; Beliaev, N.; Beryozkina, T.; Knippschild, U.; Leban, J.; Zhi-Jin, F.; Eltsov, O.; Slepukhin, P.; Ezhikova, M.; Dehaen, W. Eur. J. Org. Chem. 2014, 3684–3689. doi:10.1002/ejoc.201402130
Return to citation in text: [1] -
Merckx, T.; Haynes, C. J. E.; Karagiannidis, L. E.; Clarke, H. J.; Holder, K.; Kelly, A.; Tizzard, G. J.; Coles, S. J.; Verwilst, P.; Gale, P. A.; Dehaen, W. Org. Biomol. Chem. 2015, 13, 1654–1661. doi:10.1039/C4OB02236J
Return to citation in text: [1] -
John, J.; Thomas, J.; Parekh, N.; Dehaen, W. Eur. J. Org. Chem. 2015, 4922–4930. doi:10.1002/ejoc.201500459
Return to citation in text: [1] -
Beryozkina, T. V.; Efimov, I. V.; Fabian, W. M. F.; Beliaev, N. A.; Slepukhin, P. A.; Isenov, M. L.; Dehaen, W.; Lubec, G.; Eltsov, O. S.; Fan, Z.; Thomas, J.; Bakulev, V. A. Tetrahedron 2015, 71, 6189–6195. doi:10.1016/j.tet.2015.06.088
Return to citation in text: [1] -
John, J.; Thomas, J.; Dehaen, W. Chem. Commun. 2015, 51, 10797–10806. doi:10.1039/C5CC02319J
Return to citation in text: [1] -
Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885–2888. doi:10.1039/C5CC08347H
Return to citation in text: [1] -
Verbelen, B.; Dehaen, W. Org. Lett. 2016, 18, 6412–6415. doi:10.1021/acs.orglett.6b03309
Return to citation in text: [1] -
Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966–9970. doi:10.1002/chem.201601928
Return to citation in text: [1] -
Jana, S.; Thomas, J.; Dehaen, W. J. Org. Chem. 2016, 81, 12426–12432. doi:10.1021/acs.joc.6b02607
Return to citation in text: [1] -
Jalani, H. B.; Karagöz, A. C.; Tsogoeva, S. V. Synthesis 2016, 49, 29–41. doi:10.1055/s-0036-1588904
Return to citation in text: [1] -
Raiguel, S.; Depuydt, D.; Vander Hoogerstraete, T.; Thomas, J.; Dehaen, W.; Binnemans, K. Dalton Trans. 2017, 46, 5269–5278. doi:10.1039/C7DT00624A
Return to citation in text: [1] -
Jana, S.; Iram, S.; Thomas, J.; Hayat, M. Q.; Pannecouque, C.; Dehaen, W. Molecules 2017, 22, 303. doi:10.3390/molecules22020303
Return to citation in text: [1] -
Ware, E. Chem. Rev. 1950, 46, 403–470. doi:10.1021/cr60145a001
Return to citation in text: [1] -
Avendaño López, C.; González Trigo, G. Adv. Heterocycl. Chem. 1985, 38, 177–228. doi:10.1016/S0065-2725(08)60920-4
Return to citation in text: [1] -
Meusel, M.; Gütschow, M. Org. Prep. Proced. Int. 2004, 36, 391–443. doi:10.1080/00304940409356627
Return to citation in text: [1] -
Mehta, N. B.; Diuguid, C. A. R.; Soroko, F. E. J. Med. Chem. 1981, 24, 465–468. doi:10.1021/jm00136a018
Return to citation in text: [1] -
Kutt, H.; Harden, C. L. Phenytoin and Congeners. In Antiepileptic Drugs; Eadie, M. J.; Vajda, F. J. E., Eds.; Handb. Exp. Pharmacol., Vol. 138; Springer: Berlin, Heidelberg, 1999; pp 229–265. doi:10.1007/978-3-642-60072-2_9
Return to citation in text: [1] -
Rogawski, M. A.; Löscher, W. Nat. Rev. Neurosci. 2004, 5, 553–564. doi:10.1038/nrn1430
Return to citation in text: [1]
| 1. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527605118 |
| 2. | Brauch, S.; van Berkel, S. S.; Westermann, B. Chem. Soc. Rev. 2013, 42, 4948–4962. doi:10.1039/c3cs35505e |
| 26. | Attwood, D.; Gibson, J. J. Pharm. Pharmacol. 1978, 30, 176–180. doi:10.1111/j.2042-7158.1978.tb13192.x |
| 27. | Chakrabarti, J. K.; Hotten, T. M.; Pullar, I. A.; Steggles, D. J. J. Med. Chem. 1989, 32, 2375–2381. doi:10.1021/jm00130a025 |
| 28. | Hsu, M. C.; Schutt, A. D.; Holly, M.; Slice, L. W.; Sherman, M. I.; Richman, D. D.; Potash, M. J.; Volsky, D. J. Science 1991, 254, 1799–1802. doi:10.1126/science.1763331 |
| 29. | Liegeois, J. F. F.; Bruhwyler, J.; Damas, J.; Nguyen, T. P.; Chleide, E. M. G.; Mercier, M. G. A.; Rogister, F. A.; Delarge, J. E. J. Med. Chem. 1993, 36, 2107–2114. doi:10.1021/jm00067a009 |
| 30. | Liegeois, J. F. F.; Rogister, F. A.; Bruhwyler, J.; Damas, J.; Nguyen, T. P.; Inarejos, M. O.; Chleide, E. M. G.; Mercier, M. G. A.; Delarge, J. E. J. Med. Chem. 1994, 37, 519–525. doi:10.1021/jm00030a011 |
| 31. | Ballard, C. G.; O'Brien, J. T.; Swann, A. G.; Thompson, P.; Neill, D.; McKeith, I. G. J. Clin. Psychiatry 2001, 62, 46. doi:10.4088/JCP.v62n0110 |
| 32. | Nierenberg, A. A.; Gray, S. M.; Grandin, L. D. J. Clin. Psychiatry 2001, 62 (Suppl. 25), 27–30. |
| 33. | Hanley, D. F., Jr.; Pozo, M. Int. J. Clin. Pract. 2000, 54, 30–35. |
| 34. | Zahradník, I.; Minarovič, I.; Zahradníková, A. J. Pharmacol. Exp. Ther. 2008, 324, 977–984. doi:10.1124/jpet.107.132456 |
| 24. | Keating, T. A.; Armstrong, R. W. J. Am. Chem. Soc. 1996, 118, 2574–2583. doi:10.1021/ja953868b |
| 25. | Keating, T. A.; Armstrong, R. W. J. Org. Chem. 1996, 61, 8935–8939. doi:10.1021/jo961517p |
| 12. | Akritopoulou-Zanze, I.; Gracias, V.; Djuric, S. W. Tetrahedron Lett. 2004, 45, 8439–8441. doi:10.1016/j.tetlet.2004.09.117 |
| 13. | Gracias, V.; Darczak, D.; Gasiecki, A. F.; Djuric, S. W. Tetrahedron Lett. 2005, 46, 9053–9056. doi:10.1016/j.tetlet.2005.10.090 |
| 14. | Pirali, T.; Tron, G. C.; Zhu, J. Org. Lett. 2006, 8, 4145–4148. doi:10.1021/ol061782p |
| 15. | Bahulayan, D.; Arun, S. Tetrahedron Lett. 2012, 53, 2850–2855. doi:10.1016/j.tetlet.2012.03.116 |
| 16. | Dhondge, A. P.; Afraj, S. N.; Nuzlia, C.; Chen, C.; Lee, G.-H. Eur. J. Org. Chem. 2013, 4119–4130. doi:10.1002/ejoc.201300226 |
| 17. | Nguyen, H. H.; Palazzo, T. A.; Kurth, M. J. Org. Lett. 2013, 15, 4492–4495. doi:10.1021/ol402045h |
| 18. | Subba Reddy, B. V.; Majumder, N.; Prabhakar Rao, T. Synthesis 2014, 46, 3408–3414. doi:10.1055/s-0034-1379031 |
| 19. | De Moliner, F.; Bigatti, M.; De Rosa, C.; Banfi, L.; Riva, R.; Basso, A. Mol. Diversity 2014, 18, 473–482. doi:10.1007/s11030-014-9530-x |
| 20. | De Moliner, F.; Bigatti, M.; Banfi, L.; Riva, R.; Basso, A. Org. Lett. 2014, 16, 2280–2283. doi:10.1021/ol500813p |
| 21. | Barlow, T. M. A.; Jida, M.; Tourwé, D.; Ballet, S. Org. Biomol. Chem. 2014, 12, 6986–6989. doi:10.1039/C4OB01381F |
| 22. | Salvador, C. E. M.; Pieber, B.; Neu, P. M.; Torvisco, A.; Andrade, C. K. Z.; Kappe, C. O. J. Org. Chem. 2015, 80, 4590–4602. doi:10.1021/acs.joc.5b00445 |
| 23. | Raj, P. J.; Bahulayan, D. Tetrahedron Lett. 2015, 56, 2451–2455. doi:10.1016/j.tetlet.2015.03.081 |
| 73. | Ware, E. Chem. Rev. 1950, 46, 403–470. doi:10.1021/cr60145a001 |
| 74. | Avendaño López, C.; González Trigo, G. Adv. Heterocycl. Chem. 1985, 38, 177–228. doi:10.1016/S0065-2725(08)60920-4 |
| 75. | Meusel, M.; Gütschow, M. Org. Prep. Proced. Int. 2004, 36, 391–443. doi:10.1080/00304940409356627 |
| 3. | Marcaccini, S.; Pepino, R.; Pozo, M. C. Tetrahedron Lett. 2001, 42, 2727–2728. doi:10.1016/S0040-4039(01)00232-5 |
| 4. | Marcaccini, S.; Miliciani, M.; Pepino, R. Tetrahedron Lett. 2005, 46, 711–713. doi:10.1016/j.tetlet.2004.11.083 |
| 5. | Ignacio, J. M.; Macho, S.; Marcaccini, S.; Pepino, R.; Torroba, T. Synlett 2005, 3051–3054. doi:10.1055/s-2005-922745 |
| 6. | Marcaccini, S.; Torroba, T. Nat. Protoc. 2007, 2, 632–639. doi:10.1038/nprot.2007.71 |
| 7. | Sunderhaus, J. D.; Dockendorff, C.; Martin, S. F. Org. Lett. 2007, 9, 4223–4226. doi:10.1021/ol7018357 |
| 8. | Hardy, S.; Martin, S. F. Org. Lett. 2011, 13, 3102–3105. doi:10.1021/ol201010s |
| 9. | Sañudo, M.; Garcia-Valverde, M.; Marcaccini, S.; Torroba, T. Tetrahedron 2012, 68, 2621–2629. doi:10.1016/j.tet.2012.01.073 |
| 10. | Granger, B. A.; Wang, Z.; Kaneda, K.; Fang, Z.; Martin, S. F. ACS Comb. Sci. 2013, 15, 379–386. doi:10.1021/co400055b |
| 11. | Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. J. Org. Chem. 2015, 80, 3411–3428. doi:10.1021/jo502829j |
| 76. | Mehta, N. B.; Diuguid, C. A. R.; Soroko, F. E. J. Med. Chem. 1981, 24, 465–468. doi:10.1021/jm00136a018 |
| 77. | Kutt, H.; Harden, C. L. Phenytoin and Congeners. In Antiepileptic Drugs; Eadie, M. J.; Vajda, F. J. E., Eds.; Handb. Exp. Pharmacol., Vol. 138; Springer: Berlin, Heidelberg, 1999; pp 229–265. doi:10.1007/978-3-642-60072-2_9 |
| 78. | Rogawski, M. A.; Löscher, W. Nat. Rev. Neurosci. 2004, 5, 553–564. doi:10.1038/nrn1430 |
| 49. | Scharf, M. B.; Roth, P. B.; Dominguez, R. A.; Ware, J. C. J. Clin. Pharmacol. 1990, 30, 461–467. doi:10.1002/j.1552-4604.1990.tb03486.x |
| 50. | Post, G. L.; Patrick, R. O.; Crowder, J. E.; Houston, J.; Ferguson, J. M.; Bielski, R. J.; Bailey, L.; Pearlman, H. G.; Shu, V. S.; Pierce, M. W. J. Clin. Psychopharmacol. 1991, 11, 249–253. doi:10.1097/00004714-199108000-00005 |
| 51. | Gerecke, M.; Kyburz, E.; Borer, R.; Gassner, W. Heterocycles 1994, 39, 693–721. doi:10.3987/COM-94-S(B)61 |
| 52. | Levine, J.; Cole, D. P.; Chengappa, K. N. R.; Gershon, S. Depression Anxiety 2001, 14, 94–104. doi:10.1002/da.1051 |
| 53. | Snyder, P. J.; Werth, J.; Giordani, B.; Caveney, A. F.; Feltner, D.; Maruff, P. Hum. Psychopharmacol. Clin. Exp. 2005, 20, 263–273. doi:10.1002/hup.692 |
| 54. | Alajarín, M.; Cabrera, J.; Pastor, A.; Villalgordo, J. M. Tetrahedron Lett. 2007, 48, 3495–3499. doi:10.1016/j.tetlet.2007.03.121 |
| 55. | Mohapatra, D. K.; Maity, P. K.; Shabab, M.; Khan, M. I. Bioorg. Med. Chem. Lett. 2009, 19, 5241–5245. doi:10.1016/j.bmcl.2009.06.107 |
| 59. | Merckx, T.; Verwilst, P.; Dehaen, W. Tetrahedron Lett. 2013, 54, 4237–4240. doi:10.1016/j.tetlet.2013.05.133 |
| 60. | Thomas, J.; John, J.; Parekh, N.; Dehaen, W. Angew. Chem., Int. Ed. 2014, 53, 10155–10159. doi:10.1002/anie.201403453 |
| 61. | Efimov, I.; Bakulev, V.; Beliaev, N.; Beryozkina, T.; Knippschild, U.; Leban, J.; Zhi-Jin, F.; Eltsov, O.; Slepukhin, P.; Ezhikova, M.; Dehaen, W. Eur. J. Org. Chem. 2014, 3684–3689. doi:10.1002/ejoc.201402130 |
| 62. | Merckx, T.; Haynes, C. J. E.; Karagiannidis, L. E.; Clarke, H. J.; Holder, K.; Kelly, A.; Tizzard, G. J.; Coles, S. J.; Verwilst, P.; Gale, P. A.; Dehaen, W. Org. Biomol. Chem. 2015, 13, 1654–1661. doi:10.1039/C4OB02236J |
| 63. | John, J.; Thomas, J.; Parekh, N.; Dehaen, W. Eur. J. Org. Chem. 2015, 4922–4930. doi:10.1002/ejoc.201500459 |
| 64. | Beryozkina, T. V.; Efimov, I. V.; Fabian, W. M. F.; Beliaev, N. A.; Slepukhin, P. A.; Isenov, M. L.; Dehaen, W.; Lubec, G.; Eltsov, O. S.; Fan, Z.; Thomas, J.; Bakulev, V. A. Tetrahedron 2015, 71, 6189–6195. doi:10.1016/j.tet.2015.06.088 |
| 65. | John, J.; Thomas, J.; Dehaen, W. Chem. Commun. 2015, 51, 10797–10806. doi:10.1039/C5CC02319J |
| 66. | Thomas, J.; Jana, S.; John, J.; Liekens, S.; Dehaen, W. Chem. Commun. 2016, 52, 2885–2888. doi:10.1039/C5CC08347H |
| 67. | Verbelen, B.; Dehaen, W. Org. Lett. 2016, 18, 6412–6415. doi:10.1021/acs.orglett.6b03309 |
| 68. | Thomas, J.; Goyvaerts, V.; Liekens, S.; Dehaen, W. Chem. – Eur. J. 2016, 22, 9966–9970. doi:10.1002/chem.201601928 |
| 69. | Jana, S.; Thomas, J.; Dehaen, W. J. Org. Chem. 2016, 81, 12426–12432. doi:10.1021/acs.joc.6b02607 |
| 70. | Jalani, H. B.; Karagöz, A. C.; Tsogoeva, S. V. Synthesis 2016, 49, 29–41. doi:10.1055/s-0036-1588904 |
| 71. | Raiguel, S.; Depuydt, D.; Vander Hoogerstraete, T.; Thomas, J.; Dehaen, W.; Binnemans, K. Dalton Trans. 2017, 46, 5269–5278. doi:10.1039/C7DT00624A |
| 72. | Jana, S.; Iram, S.; Thomas, J.; Hayat, M. Q.; Pannecouque, C.; Dehaen, W. Molecules 2017, 22, 303. doi:10.3390/molecules22020303 |
| 46. | Mohler, H.; Okada, T. Science 1977, 198, 849–851. doi:10.1126/science.918669 |
| 47. | Braestrup, C.; Schmiechen, R.; Neef, G.; Nielsen, M.; Petersen, E. N. Science 1982, 216, 1241–1243. doi:10.1126/science.6281892 |
| 48. | Rudolph, U.; Knoflach, F. Nat. Rev. Drug Discovery 2011, 10, 685–697. doi:10.1038/nrd3502 |
| 13. | Gracias, V.; Darczak, D.; Gasiecki, A. F.; Djuric, S. W. Tetrahedron Lett. 2005, 46, 9053–9056. doi:10.1016/j.tetlet.2005.10.090 |
| 15. | Bahulayan, D.; Arun, S. Tetrahedron Lett. 2012, 53, 2850–2855. doi:10.1016/j.tetlet.2012.03.116 |
| 43. | Ripka, W. C.; De Lucca, G. V.; Bach, A. C., II; Pottorf, R. S.; Blaney, J. M. Tetrahedron 1993, 49, 3593–3608. doi:10.1016/S0040-4020(01)90217-0 |
| 44. | Ripka, A. S.; Rich, D. H. Curr. Opin. Chem. Biol. 1998, 2, 441–452. doi:10.1016/S1367-5931(98)80119-1 |
| 45. | Hata, M.; Marshall, G. R. J. Comput.-Aided Mol. Des. 2006, 20, 321–331. doi:10.1007/s10822-006-9059-x |
| 35. | Coyne, W. E.; Cusic, J. W. J. Med. Chem. 1967, 10, 541–546. doi:10.1021/jm00316a009 |
| 36. | Wright, W. B., Jr.; Brabander, H. J.; Greenblatt, E. N.; Day, I. P.; Hardy, R. A., Jr. J. Med. Chem. 1978, 21, 1087–1089. doi:10.1021/jm00208a017 |
| 37. | Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s |
| 38. | Kamal, A.; Shetti, R. V. C. R. N. C.; Ramaiah, M. J.; Swapna, P.; Reddy, K. S.; Mallareddy, A.; Rao, M. P. N.; Chourasia, M.; Sastry, G. N.; Juvekar, A.; Zingde, S.; Sarma, P.; Pushpavalli, S. N. C. V. L.; Pal-Bhadra, M. MedChemComm 2011, 2, 780–788. doi:10.1039/c1md00072a |
| 39. | Gill, R. K.; Kaushik, S. O.; Chugh, J.; Bansal, S.; Shah, A.; Bariwal, J. Mini-Rev. Med. Chem. 2014, 14, 229–256. doi:10.2174/13895575113139990081 |
| 40. | Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b |
| 41. | Maheshwari, N.; Goyal, A.; Jain, S.; Jain, D.; Moorthy, N. S. H. N. Med. Chem. Res. 2014, 23, 765–774. doi:10.1007/s00044-013-0675-4 |
| 42. | Thurston, D. E.; Vassoler, H.; Jackson, P. J. M.; James, C. H.; Rahman, K. M. Org. Biomol. Chem. 2015, 13, 4031–4040. doi:10.1039/C4OB02405B |
| 56. | Martins, M. B.; Carvalho, I. Tetrahedron 2007, 63, 9923–9932. doi:10.1016/j.tet.2007.04.105 |
| 57. | Hulme, C.; Morrissette, M. M.; Volz, F. A.; Burns, C. J. Tetrahedron Lett. 1998, 39, 1113–1116. doi:10.1016/S0040-4039(97)10795-X |
| 58. | Nixey, T.; Kelly, M.; Hulme, C. Tetrahedron Lett. 2000, 41, 8729–8733. doi:10.1016/S0040-4039(00)01563-X |
© 2018 Vroemans et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)