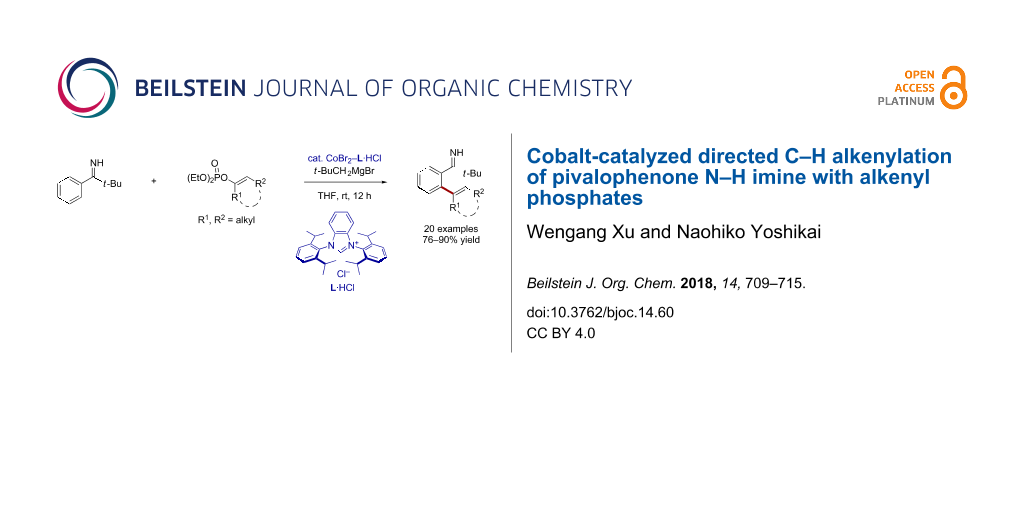
Abstract
A cobalt–N-heterocyclic carbene (NHC) catalyst efficiently promotes an ortho C–H alkenylation reaction of pivalophenone N–H imine with an alkenyl phosphate. The reaction tolerates various substituted pivalophenone N–H imines as well as cyclic and acyclic alkenyl phosphates.

Graphical Abstract
Introduction
Transition-metal-catalyzed, directing group-assisted arene C–H activation reactions have been extensively studied over the last few decades to offer a broad array of atom and step-economical methods for the synthesis of functionalized aromatic compounds [1-6]. Among various C–H transformations, the introduction of alkenyl groups into the ortho position of functionalized arenes has attracted significant attention because of the synthetic versatility of alkenyl groups. The C–H alkenylation has been achieved most extensively by way of the dehydrogenative Heck-type reaction of olefins [7-9]. Meanwhile, the hydroarylation of alkynes has also been explored as an alternative approach for C–H alkenylation [10]. Despite the significant progress made, each of these C–H alkenylation manifolds has some critical limitations. For example, the dehydrogenative Heck reaction is often limited to activated monosubstituted alkenes (e.g., acrylates), and is challenging with unactivated and multisubstituted alkenes [11]. The hydroarylation of alkynes does not allow for the introduction of cycloalkenyl groups because of the unavailability of the corresponding alkynes. In light of such limitations, a coupling between arene substrates and alkenyl electrophiles would offer a complementary approach for the C–H alkenylation [12]. In particular, C–H alkenylations by way of alkenyl C–O bond cleavage has attracted much attention because of the ready accessibility of the corresponding alkenyl electrophiles (e.g., acetate, phosphate) from ketones [13-17].
Over the last several years, we and others have developed a series of directed arene C–H functionalization reactions with organic electrophiles under low-valent cobalt catalysis [18-21]. In particular, our group and the Ackermann group have independently demonstrated that the combination of a cobalt–N-heterocyclic carbene (NHC) catalyst and a Grignard reagent allows for the arene C–H functionalization with organic halides and pseudohalides under the assistance of nitrogen directing groups [17,22-27]. In this connection, Ackermann developed a mild and efficient C–H alkenylation of N-pyrimidylindoles and pyrroles with alkenyl acetates using a cobalt–NHC catalyst (Scheme 1a) [17]. The same catalytic system also promoted the alkenylation using alkenyl carbamates, carbonates, and phosphates. More recently, we have achieved an N-arylimine-directed arene C–H alkenylation reaction with alkenyl phosphates using a different cobalt–NHC catalyst (Scheme 1b) [28]. Meanwhile, we have also demonstrated that pivaloyl N–H imine serves as a powerful directing group for cobalt-catalyzed arene C–H functionalization reactions such as the hydroarylation of alkenes and alkylation/arylation using organic halides [29,30]. These previous studies have prompted us to expand the scope of cobalt catalysis for the C–H alkenylation and thus to develop an ortho C–H alkenylation reaction of pivalophenone N–H imine with alkenyl phosphates using a new cobalt–NHC catalyst, which is reported herein (Scheme 1c). The present alkenylation features a mild reaction temperature and displays applicability to a variety of substituted pivalophenone N–H imines and alkenyl phosphates. It should be emphasized that pivalophenone N–H imines and related bulky N–H imines can be readily prepared from the corresponding aryl nitriles and organolithium or Grignard reagents, while analogous N-substituted imines are nontrivial to synthesize because of sluggish ketone/amine condensation. As such, the present reaction would complement the N-arylimine-directed alkenylation.
![[1860-5397-14-60-i1]](/bjoc/content/inline/1860-5397-14-60-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cobalt–NHC-catalyzed C–H alkenylation reactions with alkenyl electrophiles.
Scheme 1: Cobalt–NHC-catalyzed C–H alkenylation reactions with alkenyl electrophiles.
Results and Discussion
The present study commenced with screening of the reaction conditions for the coupling between pivalophenone N–H imine 1a and cyclohexenyl phosphate 2a (Table 1). Thus, the reaction was performed in the presence of CoBr2 (10 mol %), ligand (10–20 mol %), and t-BuCH2MgBr (2 equiv) in THF at room temperature. While monodentate phosphines such as PPh3 and PCy3 were entirely ineffective (Table 1, entries 1 and 2), common bulky NHC precursors such as 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride (IMes·HCl) and 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (IPr·HCl) promoted the coupling reaction to afford the desired alkenylation product 3aa albeit in moderate yields (Table 1, entries 3 and 4). No significant improvement was observed using the saturated analogues of IMes·HCl and IPr·HCl (Table 1, entries 5 and 6) or the 2,6-diethylphenyl analogue (IEt·HCl, Table 1, entry 7). Furthermore, the NHC precursor featuring a cyclohexane backbone and 2,6-diethylphenyl groups (L1·HBr), which proved to be the optimal ligand for the C–H arylation of pivalophenone N–H imine as well as for the C–H alkenylation of N-arylimine (Scheme 1a, b) [28,29], was not particularly effective for the present reaction (Table 1, entry 8). To our delight, we observed a remarkable improvement in the reaction efficiency using the benzofused analogue of IPr·HCl (L2·HCl), affording 3aa in 88% yield without any trace of a dialkenylation product (Table 1, entry 9). It should be noted that, unlike the C–H arylation of pivalophenone N–H imine and the C–H alkenylation of N-arylimine (Scheme 1a, b), the addition of TMEDA was not necessary to achieve high reaction efficiency, while the reason for this remains unclear. Note also that the present reaction could employ the relatively inexpensive diethyl phosphate, whereas, in the N-arylimine-directed alkenylation, the use of diisopropyl phosphate was necessary to achieve higher and more reproducible yields (cf. Scheme 1b) [28].
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-60-i6.svg?max-width=637&scale=1.0)
|
||
| entry | ligand (mol %) | yield (%)b |
| 1 | PPh3 (20) | 0 |
| 2 | PCy3 (20) | 0 |
| 3 | IMes·HCl (10) | 29 |
| 4 | IPr·HCl (10) | 53 |
| 5 | SIMes·HCl (10) | 40 |
| 6 | SIPr·HCl (10) | 46 |
| 7 | IEt·HCl (10) | 38 |
| 8 | L1·HBr (10) | 37 |
| 9 | L2·HCl (10) | 88c |
aThe reaction was performed using 0.2 mmol of 1a and 0.3 mmol (1.5 equiv) of 2a. bDetermined by GC using n-tridecane as an internal standard. cIsolated yield.
With the optimized reaction conditions in hand, we explored the scope of the present alkenylation reaction. First, various substituted pivalophenone N–H imines were subjected to the reaction with 2a (Scheme 2). Pivalophenone N–H imines bearing a series of para-substituents all participated in the alkenylation reaction to afford the desired products 3ba–ga in good yields. The reaction of m-methyl-substituted imine took place preferentially at the less hindered position to afford 3ha as the major isomer with a moderate regioselectivity of 3:1. By contrast, imines bearing m-methoxy, m-fluoro, or a 3,4-methylenedioxy group underwent exclusive alkenylation at the proximity of the functional group to afford the products 3ia–ka in good yields. As was also observed in previously reported cobalt-catalyzed ortho C–H functionalization reactions [22,23,28,29], this regioselectivity may be ascribed to the role of the oxygen or fluorine atom as a secondary directing group to have an electrostatic interaction with the cobalt center during the C–H activation. For compound 3ja, an increased acidity of the ortho position of the fluorine atom could have also contributed to the observed regioselectivity [31]. Curiously, the reaction of 2-naphthylimine resulted in the preferential alkenylation of the more hindered 1-position rather than the 3-position, with a regioselectivity of 4:1 (see 3la).
![[1860-5397-14-60-i2]](/bjoc/content/inline/1860-5397-14-60-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of substituted pivalophenone N–H imines with 2a. aThe major regioisomer is shown (rr = regioisomer ratio).
Scheme 2: Reaction of substituted pivalophenone N–H imines with 2a. aThe major regioisomer is shown (rr = reg...
Next, the reaction of the parent pivalophenone N–H imine 1a with different alkenyl phosphates was explored (Scheme 3). The reaction of cyclopentenyl phosphate proceeded smoothly to afford the desired product 3ab in a high yield of 85%. This is in a sharp contrast to the poor reactivity of the analogous diisopropyl phosphate in the N–PMP imine-directed alkenylation (12% yield). Other cycloalkenyl phosphates with larger ring sizes also efficiently underwent the C–H alkenylation to afford the respective products 3ac–af in good yields. Notably, the cyclodecenylated product 3ae was obtained in an E-rich form from an E/Z mixture (1:1) of the starting alkenyl phosphate, demonstrating the E/Z isomerization during the C–C-bond formation. The E/Z isomerization was also observed for the conversion of cyclododecenyl phosphate (E/Z = 9:1) to the product 3af (E/Z = 3:1). Expectedly, 4-substituted cyclohexenyl phosphates reacted smoothly to afford the desired products 3ag and 3ah. Furthermore, a 6-methyl group on the cyclohexenyl phosphate did not interfere with the reaction (see 3ai). Finally an acyclic alkenyl phosphate derived from 4-heptanone (E isomer) was also amenable to the alkenylation reaction, affording the product 3aj with an E/Z ratio of 4:1.
![[1860-5397-14-60-i3]](/bjoc/content/inline/1860-5397-14-60-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of 1a with various alkenyl phosphates. aA mixture of E- and Z-alkenyl phosphate (ca. 1:1) was used. bZ-rich alkenyl phosphate (Z/E = ca. 9:1) was used. cE-alkenyl phosphate was used.
Scheme 3: Reaction of 1a with various alkenyl phosphates. aA mixture of E- and Z-alkenyl phosphate (ca. 1:1) ...
In our previous study on the C–H alkylation and arylation of pivalophenone N–H imines, we demonstrated that the pivaloyl imine readily undergoes fragmentation into a cyano group via an iminyl radical under peroxide photolysis or copper-catalyzed aerobic conditions [29]. Under the same peroxide photolysis conditions (t-BuOOt-Bu with UV (254 nm) irradiation), the ortho-alkenylated imine 3aa underwent a C–N bond-forming cyclization to afford the spirocyclic imine 4 in 81% yield (Scheme 4). The reaction likely involves the initial formation of an iminyl radical from 3aa and a tert-butoxyl radical and its intramolecular addition to the cyclohexenyl group.
![[1860-5397-14-60-i4]](/bjoc/content/inline/1860-5397-14-60-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The cyclization of o-alkenylpivalophenone N–H imine.
Scheme 4: The cyclization of o-alkenylpivalophenone N–H imine.
On the basis of our previous studies on the N-arylimine-directed C–H alkenylation and the N–H imine-directed C–H alkylation/arylation [28,29], we are tempted to propose the catalytic cycle illustrated in Scheme 5. An alkylcobalt species A, generated from the cobalt precatalyst and the Grignard reagent, would undergo cyclometalation of magnesium alkylidene amide 1·MgX, generated from imine 1 and the Grignard reagent, to give a cobaltacycle species B while liberating an alkane R–H. The species B would then undergo a single-electron transfer (SET) to the alkenyl phosphate 2 to generate a pair of an oxidized cobaltacycle B+ and a radical anion 2•−. This would be followed by the elimination of a phosphate anion and immediate recombination of the cobalt center and the alkenyl radical to give a diorganocobalt intermediate C. The C–C-bond rotation of the radical anion 2•− or the transiently formed alkenyl radical might be responsible for the stereochemical mutation of the C=C bond observed in some cases. The reductive elimination of C and subsequent transmetalation with the Grignard reagent would furnish the alkenylation product 3·MgX and regenerate the species A. While the relationship between the ligand and the catalytic activity remains unclear, we speculate that a strong σ-donating ability of NHC ligands would facilitate the SET step among others.
![[1860-5397-14-60-i5]](/bjoc/content/inline/1860-5397-14-60-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed catalytic cycle (R = t-BuCH2, R' = P(O)(OEt)2).
Scheme 5: Proposed catalytic cycle (R = t-BuCH2, R' = P(O)(OEt)2).
Conclusion
In summary, we have developed an ortho C–H alkenylation reaction of pivalophenone N–H imines with alkenyl phosphates using a cobalt–NHC catalyst. The reaction takes place smoothly at room temperature and is applicable to a variety of substituted pivalophenone N–H imines and alkenyl phosphates. The NHC ligand architecture proved to have a significant impact on the efficiency of the present C–H/electrophile coupling. We anticipate that the elaboration of NHC ligands would also be instrumental to the improvement of other C–H activation and related transformations promoted by low-valent cobalt complexes [32-40].
Experimental
Typical procedure: Cobalt-catalyzed alkenylation of pivalophenone N–H imine 1a with alkenyl phosphate 2a. A 10 mL Schlenk tube equipped with a magnetic stirring bar was charged with L2·HCl (9.5 mg, 0.020 mmol), CoBr2 (4.4 mg, 0.020 mmol), and THF (0.30 mL). The resulting solution was cooled in an ice bath, followed by the addition of t-BuCH2MgBr (2.0 M in THF, 0.20 mL, 0.40 mmol). After stirring for 30 min, 2,2-dimethyl-1-phenylpropan-1-imine (1a, 33 mg, 0.20 mmol) and cyclohex-1-en-1-yl diethyl phosphate (2a, 70 mg, 0.30 mmol) were added. The resulting mixture was warmed to room temperature, stirred for 12 h, and then filtered through a short silica-gel column, which was washed with ethyl acetate (5 mL). The filtrate was concentrated under reduced pressure. Silica gel chromatography (eluent: hexane/EtOAc/NEt3 50:1:1) of the crude product afforded the desired alkenylation product as a colorless oil (43 mg, 88%).
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of new compounds. | ||
| Format: PDF | Size: 5.3 MB | Download |
References
-
Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624–655. doi:10.1021/cr900005n
Return to citation in text: [1] -
Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j
Return to citation in text: [1] -
Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788–802. doi:10.1021/ar200185g
Return to citation in text: [1] -
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e
Return to citation in text: [1] -
Kakiuchi, F.; Kochi, T.; Murai, S. Synlett 2014, 25, 2390–2414. doi:10.1055/s-0034-1379210
Return to citation in text: [1] -
De Sarkar, S.; Liu, W.; Kozhushkov, S. I.; Ackermann, L. Adv. Synth. Catal. 2014, 356, 1461–1479. doi:10.1002/adsc.201400110
Return to citation in text: [1] -
Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170–1214. doi:10.1021/cr100209d
Return to citation in text: [1] -
Kozhushkov, S. I.; Ackermann, L. Chem. Sci. 2013, 4, 886–896. doi:10.1039/C2SC21524A
Return to citation in text: [1] -
Zhou, L.; Lu, W. Chem. – Eur. J. 2014, 20, 634–642. doi:10.1002/chem.201303670
Return to citation in text: [1] -
Boyarskiy, V. P.; Ryabukhin, D. S.; Bokach, N. A.; Vasilyev, A. V. Chem. Rev. 2016, 116, 5894–5986. doi:10.1021/acs.chemrev.5b00514
Return to citation in text: [1] -
Deb, A.; Maiti, D. Eur. J. Org. Chem. 2017, 1239–1252. doi:10.1002/ejoc.201601253
Return to citation in text: [1] -
Rossi, R.; Bellina, F.; Lessi, M. Synthesis 2010, 4131–4153. doi:10.1055/s-0030-1258262
Return to citation in text: [1] -
Matsuura, Y.; Tamura, M.; Kochi, T.; Sato, M.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2007, 129, 9858–9859. doi:10.1021/ja071965m
Return to citation in text: [1] -
Ogiwara, Y.; Tamura, M.; Kochi, T.; Matsuura, Y.; Chatani, N.; Kakiuchi, F. Organometallics 2014, 33, 402–420. doi:10.1021/om401204h
Return to citation in text: [1] -
Meng, L.; Kamada, Y.; Muto, K.; Yamaguchi, J.; Itami, K. Angew. Chem., Int. Ed. 2013, 52, 10048–10051. doi:10.1002/anie.201304492
Return to citation in text: [1] -
Ackermann, L.; Barfüsser, S.; Pospech, J. Org. Lett. 2010, 12, 724–726. doi:10.1021/ol9028034
Return to citation in text: [1] -
Moselage, M.; Sauermann, N.; Richter, S. C.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 6352–6355. doi:10.1002/anie.201412319
Return to citation in text: [1] [2] [3] -
Gao, K.; Yoshikai, N. Acc. Chem. Res. 2014, 47, 1208–1219. doi:10.1021/ar400270x
Return to citation in text: [1] -
Yoshikai, N. Bull. Chem. Soc. Jpn. 2014, 87, 843–857. doi:10.1246/bcsj.20140149
Return to citation in text: [1] -
Ackermann, L. J. Org. Chem. 2014, 79, 8948–8954. doi:10.1021/jo501361k
Return to citation in text: [1] -
Moselage, M.; Li, J.; Ackermann, L. ACS Catal. 2016, 6, 498–525. doi:10.1021/acscatal.5b02344
Return to citation in text: [1] -
Gao, K.; Lee, P.-S.; Long, C.; Yoshikai, N. Org. Lett. 2012, 14, 4234–4237. doi:10.1021/ol301934y
Return to citation in text: [1] [2] -
Gao, K.; Yoshikai, N. J. Am. Chem. Soc. 2013, 135, 9279–9282. doi:10.1021/ja403759x
Return to citation in text: [1] [2] -
Gao, K.; Yamakawa, T.; Yoshikai, N. Synthesis 2014, 46, 2024–2039. doi:10.1055/s-0033-1338658
Return to citation in text: [1] -
Song, W.; Ackermann, L. Angew. Chem., Int. Ed. 2012, 51, 8251–8254. doi:10.1002/anie.201202466
Return to citation in text: [1] -
Punji, B.; Song, W.; Shevchenko, G. A.; Ackermann, L. Chem. – Eur. J. 2013, 19, 10605–10610. doi:10.1002/chem.201301409
Return to citation in text: [1] -
Mei, R.; Ackermann, L. Adv. Synth. Catal. 2016, 358, 2443–2448. doi:10.1002/adsc.201600384
Return to citation in text: [1] -
Lee, P.-S.; Xu, W.; Yoshikai, N. Adv. Synth. Catal. 2017, 359, 4340–4347. doi:10.1002/adsc.201701105
Return to citation in text: [1] [2] [3] [4] [5] -
Xu, W.; Yoshikai, N. Chem. Sci. 2017, 8, 5299–5304. doi:10.1039/C7SC01732D
Return to citation in text: [1] [2] [3] [4] [5] -
Xu, W.; Yoshikai, N. Angew. Chem., Int. Ed. 2016, 55, 12731–12735. doi:10.1002/anie.201605877
Return to citation in text: [1] -
Eisenstein, O.; Milani, J.; Perutz, R. N. Chem. Rev. 2017, 117, 8710–8753. doi:10.1021/acs.chemrev.7b00163
Return to citation in text: [1] -
Someya, H.; Ohmiya, H.; Yorimitsu, H.; Oshima, K. Org. Lett. 2007, 9, 1565–1567. doi:10.1021/ol070392f
Return to citation in text: [1] -
Hatakeyama, T.; Hashimoto, S.; Ishizuka, K.; Nakamura, M. J. Am. Chem. Soc. 2009, 131, 11949–11963. doi:10.1021/ja9039289
Return to citation in text: [1] -
Kobayashi, T.; Yorimitsu, H.; Oshima, K. Chem. – Asian J. 2009, 4, 1078–1083. doi:10.1002/asia.200900111
Return to citation in text: [1] -
Mo, Z.; Liu, Y.; Deng, L. Angew. Chem., Int. Ed. 2013, 52, 10845–10849. doi:10.1002/anie.201304596
Return to citation in text: [1] -
Mo, Z.; Mao, J.; Gao, Y.; Deng, L. J. Am. Chem. Soc. 2014, 136, 17414–17417. doi:10.1021/ja510924v
Return to citation in text: [1] -
Röse, P.; Hilt, G. Synthesis 2016, 48, 463–492. doi:10.1055/s-0035-1560378
Return to citation in text: [1] -
Cahiez, G.; Moyeux, A. Chem. Rev. 2010, 110, 1435–1462. doi:10.1021/cr9000786
Return to citation in text: [1] -
Hess, W.; Treutwein, J.; Hilt, G. Synthesis 2008, 3537–3562. doi:10.1055/s-0028-1083210
Return to citation in text: [1] -
Gosmini, C.; Bégouin, J.-M.; Moncomble, A. Chem. Commun. 2008, 3221–3233. doi:10.1039/b805142a
Return to citation in text: [1]
| 28. | Lee, P.-S.; Xu, W.; Yoshikai, N. Adv. Synth. Catal. 2017, 359, 4340–4347. doi:10.1002/adsc.201701105 |
| 29. | Xu, W.; Yoshikai, N. Chem. Sci. 2017, 8, 5299–5304. doi:10.1039/C7SC01732D |
| 32. | Someya, H.; Ohmiya, H.; Yorimitsu, H.; Oshima, K. Org. Lett. 2007, 9, 1565–1567. doi:10.1021/ol070392f |
| 33. | Hatakeyama, T.; Hashimoto, S.; Ishizuka, K.; Nakamura, M. J. Am. Chem. Soc. 2009, 131, 11949–11963. doi:10.1021/ja9039289 |
| 34. | Kobayashi, T.; Yorimitsu, H.; Oshima, K. Chem. – Asian J. 2009, 4, 1078–1083. doi:10.1002/asia.200900111 |
| 35. | Mo, Z.; Liu, Y.; Deng, L. Angew. Chem., Int. Ed. 2013, 52, 10845–10849. doi:10.1002/anie.201304596 |
| 36. | Mo, Z.; Mao, J.; Gao, Y.; Deng, L. J. Am. Chem. Soc. 2014, 136, 17414–17417. doi:10.1021/ja510924v |
| 37. | Röse, P.; Hilt, G. Synthesis 2016, 48, 463–492. doi:10.1055/s-0035-1560378 |
| 38. | Cahiez, G.; Moyeux, A. Chem. Rev. 2010, 110, 1435–1462. doi:10.1021/cr9000786 |
| 39. | Hess, W.; Treutwein, J.; Hilt, G. Synthesis 2008, 3537–3562. doi:10.1055/s-0028-1083210 |
| 40. | Gosmini, C.; Bégouin, J.-M.; Moncomble, A. Chem. Commun. 2008, 3221–3233. doi:10.1039/b805142a |
| 1. | Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624–655. doi:10.1021/cr900005n |
| 2. | Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j |
| 3. | Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788–802. doi:10.1021/ar200185g |
| 4. | Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e |
| 5. | Kakiuchi, F.; Kochi, T.; Murai, S. Synlett 2014, 25, 2390–2414. doi:10.1055/s-0034-1379210 |
| 6. | De Sarkar, S.; Liu, W.; Kozhushkov, S. I.; Ackermann, L. Adv. Synth. Catal. 2014, 356, 1461–1479. doi:10.1002/adsc.201400110 |
| 12. | Rossi, R.; Bellina, F.; Lessi, M. Synthesis 2010, 4131–4153. doi:10.1055/s-0030-1258262 |
| 31. | Eisenstein, O.; Milani, J.; Perutz, R. N. Chem. Rev. 2017, 117, 8710–8753. doi:10.1021/acs.chemrev.7b00163 |
| 11. | Deb, A.; Maiti, D. Eur. J. Org. Chem. 2017, 1239–1252. doi:10.1002/ejoc.201601253 |
| 10. | Boyarskiy, V. P.; Ryabukhin, D. S.; Bokach, N. A.; Vasilyev, A. V. Chem. Rev. 2016, 116, 5894–5986. doi:10.1021/acs.chemrev.5b00514 |
| 28. | Lee, P.-S.; Xu, W.; Yoshikai, N. Adv. Synth. Catal. 2017, 359, 4340–4347. doi:10.1002/adsc.201701105 |
| 7. | Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170–1214. doi:10.1021/cr100209d |
| 8. | Kozhushkov, S. I.; Ackermann, L. Chem. Sci. 2013, 4, 886–896. doi:10.1039/C2SC21524A |
| 9. | Zhou, L.; Lu, W. Chem. – Eur. J. 2014, 20, 634–642. doi:10.1002/chem.201303670 |
| 22. | Gao, K.; Lee, P.-S.; Long, C.; Yoshikai, N. Org. Lett. 2012, 14, 4234–4237. doi:10.1021/ol301934y |
| 23. | Gao, K.; Yoshikai, N. J. Am. Chem. Soc. 2013, 135, 9279–9282. doi:10.1021/ja403759x |
| 28. | Lee, P.-S.; Xu, W.; Yoshikai, N. Adv. Synth. Catal. 2017, 359, 4340–4347. doi:10.1002/adsc.201701105 |
| 29. | Xu, W.; Yoshikai, N. Chem. Sci. 2017, 8, 5299–5304. doi:10.1039/C7SC01732D |
| 17. | Moselage, M.; Sauermann, N.; Richter, S. C.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 6352–6355. doi:10.1002/anie.201412319 |
| 29. | Xu, W.; Yoshikai, N. Chem. Sci. 2017, 8, 5299–5304. doi:10.1039/C7SC01732D |
| 30. | Xu, W.; Yoshikai, N. Angew. Chem., Int. Ed. 2016, 55, 12731–12735. doi:10.1002/anie.201605877 |
| 17. | Moselage, M.; Sauermann, N.; Richter, S. C.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 6352–6355. doi:10.1002/anie.201412319 |
| 22. | Gao, K.; Lee, P.-S.; Long, C.; Yoshikai, N. Org. Lett. 2012, 14, 4234–4237. doi:10.1021/ol301934y |
| 23. | Gao, K.; Yoshikai, N. J. Am. Chem. Soc. 2013, 135, 9279–9282. doi:10.1021/ja403759x |
| 24. | Gao, K.; Yamakawa, T.; Yoshikai, N. Synthesis 2014, 46, 2024–2039. doi:10.1055/s-0033-1338658 |
| 25. | Song, W.; Ackermann, L. Angew. Chem., Int. Ed. 2012, 51, 8251–8254. doi:10.1002/anie.201202466 |
| 26. | Punji, B.; Song, W.; Shevchenko, G. A.; Ackermann, L. Chem. – Eur. J. 2013, 19, 10605–10610. doi:10.1002/chem.201301409 |
| 27. | Mei, R.; Ackermann, L. Adv. Synth. Catal. 2016, 358, 2443–2448. doi:10.1002/adsc.201600384 |
| 28. | Lee, P.-S.; Xu, W.; Yoshikai, N. Adv. Synth. Catal. 2017, 359, 4340–4347. doi:10.1002/adsc.201701105 |
| 29. | Xu, W.; Yoshikai, N. Chem. Sci. 2017, 8, 5299–5304. doi:10.1039/C7SC01732D |
| 18. | Gao, K.; Yoshikai, N. Acc. Chem. Res. 2014, 47, 1208–1219. doi:10.1021/ar400270x |
| 19. | Yoshikai, N. Bull. Chem. Soc. Jpn. 2014, 87, 843–857. doi:10.1246/bcsj.20140149 |
| 20. | Ackermann, L. J. Org. Chem. 2014, 79, 8948–8954. doi:10.1021/jo501361k |
| 21. | Moselage, M.; Li, J.; Ackermann, L. ACS Catal. 2016, 6, 498–525. doi:10.1021/acscatal.5b02344 |
| 13. | Matsuura, Y.; Tamura, M.; Kochi, T.; Sato, M.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2007, 129, 9858–9859. doi:10.1021/ja071965m |
| 14. | Ogiwara, Y.; Tamura, M.; Kochi, T.; Matsuura, Y.; Chatani, N.; Kakiuchi, F. Organometallics 2014, 33, 402–420. doi:10.1021/om401204h |
| 15. | Meng, L.; Kamada, Y.; Muto, K.; Yamaguchi, J.; Itami, K. Angew. Chem., Int. Ed. 2013, 52, 10048–10051. doi:10.1002/anie.201304492 |
| 16. | Ackermann, L.; Barfüsser, S.; Pospech, J. Org. Lett. 2010, 12, 724–726. doi:10.1021/ol9028034 |
| 17. | Moselage, M.; Sauermann, N.; Richter, S. C.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 6352–6355. doi:10.1002/anie.201412319 |
| 28. | Lee, P.-S.; Xu, W.; Yoshikai, N. Adv. Synth. Catal. 2017, 359, 4340–4347. doi:10.1002/adsc.201701105 |
© 2018 Xu and Yoshikai; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)