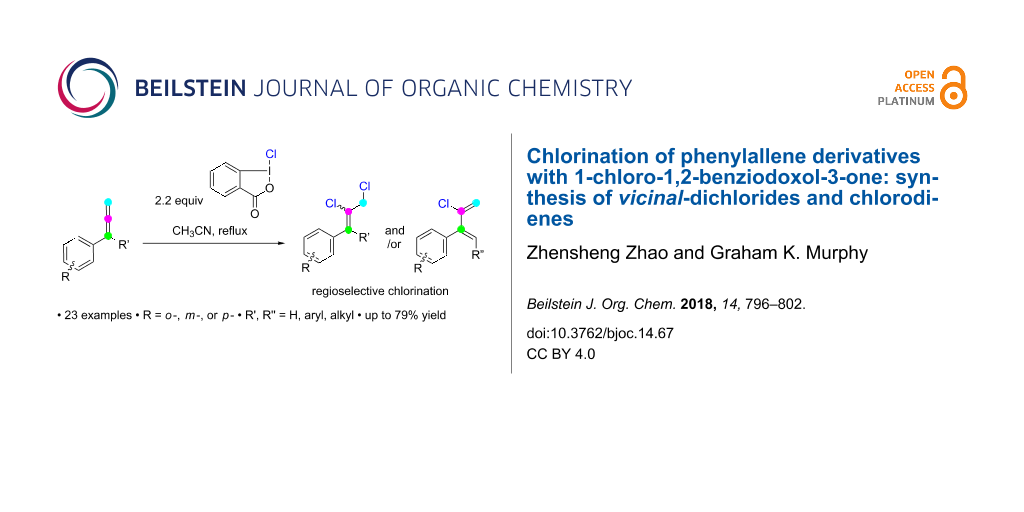
Abstract
Allyl and vinyl chlorides represent important structural motifs in organic chemistry. Herein is described the chemoselective and regioselective reaction of aryl- and α-substituted phenylallenes with the hypervalent iodine (HVI) reagent 1-chloro-1,2-benziodoxol-3-one. The reaction typically results in vicinal dichlorides, except with proton-containing α-alkyl substituents, which instead give chlorinated dienes as the major product. Experimental evidence suggests that a radical mechanism is involved.

Graphical Abstract
Introduction
Organochlorine compounds are vital as polymer precursors [1], as pharmaceuticals [2,3] and agrochemicals [4-6] and as functional materials [7,8]. And as there is an abundance of chlorine-containing natural products, the synthesis of chlorinated functional groups, such as allyl- and vinyl chlorides, can represent challenging obstacles that practitioners of natural product synthesis must surmount [9-12]. More commonly, allyl- and vinyl chlorides are highly sought-after intermediates for effecting allylations, and for use in transition metal-catalyzed carbon–carbon and carbon–heteroatom bond-forming reactions [13-28]. Given the versatility of allyl chloride and β-chlorostyrene groups, installing them in close proximity (as shown in 3) provides two handles for rapidly achieving high-density molecular complexity. Thus, the development of strategies for their synthesis is an important endeavour. We envisioned accomplishing this by developing a chemo- and regioselective vicinal-dichlorination of phenylallenes; however, no such chlorination reaction has yet been achieved [29-34].
Recent reports of reactions between hypervalent iodine reagents and phenylallenes have highlighted the possible product outcomes achievable through ionic and radical reaction pathways. For example, Liu and co-workers used Togni’s benziodoxolone reagent [35] in a radical-mediated vicinal 2,3-difunctionalization of allenes, which proceeded via CF3-radical adduct A (Scheme 1a) [36]. In contrast, Muñiz reported that with PhI(NTs2)2, an oxidative amination occurred via cation B, giving regioisomeric propargylamides upon elimination of the iodanyl adduct (Scheme 1b) [37]. Moriarty and Murphy, respectively, showed how reactions of arylallenes with either PhI(OH)OTs [38] or TolIF2 [39] provide α-disubstituted styrenes by sequences involving intermediates analogous to B, followed by a 1,2-phenyl shift (C to D, Scheme 1c). There has been no investigation of the chemistry between arylallenes and chlorinated hypervalent iodine reagents, and given the differing reactivities that might be achievable with (dichloroiodo)benzene [40] (1a) and chlorobenziodoxolone (1b) [30,41-45], there is potential for the selective generation of diverse, poly-chlorinated scaffolds. Reported here are the results of these investigations, and while indiscriminate chlorination of 2 was observed with 1a, 1b reacted chemo- and regioselectively to give 2,3-dichlorides (3) or chlorodienes (4).
![[1860-5397-14-67-i1]](/bjoc/content/inline/1860-5397-14-67-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of substituted allenes with HVI reagents.
Scheme 1: Reactions of substituted allenes with HVI reagents.
Results and Discussion
We began our investigation of allene chlorination using p-tolylallene (2a), prepared from 4-methylstyrene through Doering–Moore–Skattebøl reaction [46], and iodane 1a. The reaction was carried out using a slight excess of 1a in acetonitrile, at both room temperature and at reflux, and upon consumption of the allene an inseparable mixture of chlorination products 3a and 3a’ were obtained. While the overall yield of the chlorinated products increased when under reflux conditions, very little change in chemoselectivity was observed (Scheme 2) [31]. As these results were consistent with those achievable by other allene chlorination reactions, it was not investigated further.
![[1860-5397-14-67-i2]](/bjoc/content/inline/1860-5397-14-67-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Chlorination of p-tolylallene (2a) with (dichloroiodo)benzene (1a).
Scheme 2: Chlorination of p-tolylallene (2a) with (dichloroiodo)benzene (1a).
We next investigated the chlorination of 2a with benziodoxolone 1b [47-49], which proved highly regioselective. An initial reaction with 2.2 equiv of 1b in acetonitrile at room temperature failed; however, repeating the reaction under reflux conditions gave 3a as a mixture of E/Z alkenes in 58% yield (Table 1, entries 1 and 2). The reaction was entirely selective for the terminal alkene, with none of 3a’ being observed. Toluene, chlorobenzene, DMF and DCE were also tested as reaction solvents, but none were superior to acetonitrile (Table 1, entries 3–6). A small improvement in yield was achieved by adding 2a dropwise over 30 minutes (Table 1, entry 7), and we ultimately found that adding 2a dropwise over one hour was optimal, giving 3a in 90% yield as a E:Z = 1:1.25 mixture (Table 1, entry 8). One final reaction was carried out using the related gem-dimethyl chlorobenziodoxole [49], but the yield of 3a decreased to 45% (Table 1, entry 9). This result is the first example of a selective chlorination reaction of phenylallenes, and as the regiochemical outcome parallels that observed by Liu (Scheme 1a), it is likely that radical pathways are involved [50].
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-67-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | solvent | temp. °C | yield (%) |
| 1 | CH3CN | rt | trace |
| 2 | CH3CN | 85 | 58% |
| 3 | toluene | 110 | 40%b |
| 4 | PhCl | 110 | trace |
| 5 | DMF | 110 | trace |
| 6 | DCE | 85 | NR |
| 7 | CH3CNc | 85 | 65% |
| 8 | CH3CNd | 85 | 90% |
| 9 | CH3CNe | 85 | 45% |
aGeneral conditions: Allene 2a (0.2 mmol, 1 equiv), 1b (0.44 mmol, 2.2 equiv) in 0.1 M solvent under reflux conditions for 2 h; isolated yield. b 1H NMR yield using HMDSO (hexamethyldisiloxane) as internal standard. cDropwise addition of 2a over 30 min. dDropwise addition of 2a over 1 h. e3,3-Dimethyl-1-chloro-1,2-benziodoxole used instead of 1b.
A series of aryl- and allenyl-substituted phenylallenes (2b–v) were then examined in the chlorination reaction. First, phenylallenes with various aryl substituents were investigated, and the p-tolyl and p-biphenyl derivatives gave the 1,2-dichlorides 3a and 3b in excellent yield, favouring the Z-alkene (Scheme 3). The 4-bromo and 2-, 3- or 4-chloro derivatives 2c–f led to 3c–f in only moderate yield, with the mass balance of chlorinated materials being made up by the regioisomeric vicinal-dichloroination products 3c’–f’ (compare with 3a’, Scheme 2) [30]. The p-anisyl derivative 2g was also viable in the reaction, giving 3g in 64% yield, as were the 1- and 2-naphthylallenes (2h and 2i), which gave the desired dichlorides 3h and 3i in 93% and 78% yield. In each case, preference for forming the Z-alkene was observed, with selectivities ranging from 1.2–4.3:1 Z:E. α-Substituents on the allenes were equally viable, as 1,1-diphenylallene gave 3j in 84% yield, and the related mono-methyl and mono-chloro derivatives 2k and 2l gave dichlorides 3k and 3l in 79% and 56% yield, respectively, with little preference observed for formation of either the Z or E alkene. Curiously, with 1,1-di(p-anisyl)allene (2m), only a trace of 3m was observed, and the reaction instead produced iodobenzoate 3m” in 57% yield. Presumably, this anomalous result arose due to the increased stability offered to an electron-deficient radical intermediate by the two methoxy groups, permitting a deviation in reaction outcome.
![[1860-5397-14-67-i3]](/bjoc/content/inline/1860-5397-14-67-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Chlorination of various aryl-substituted allenes. General conditions: Allene 2a (0.2 mmol, 1 equiv) was added dropwise over 1 h to a solution of 1b (0.44 mmol, 2.2 equiv) in 0.1 M CH3CN under reflux conditions, and the reaction stirred for 1 h; isolated yields. a 1H NMR yield using HMDSO as internal standard.
Scheme 3: Chlorination of various aryl-substituted allenes. General conditions: Allene 2a (0.2 mmol, 1 equiv)...
We next subjected α-alkyl-substituted phenylallenes 2n–v to the standard reaction conditions, which resulted in mixtures of exclusively the Z-dichloroalkenes (Z-3) and chlorodienes 4 (Scheme 4). When the para-Br and para-Cl phenylallenes bearing α-methyl groups (2n, 2o) were tested, the chlorodienes 4n and 4o were obtained in 31% and 38% yield, respectively, along with 23% of Z-3n and 34% of Z-3o. With α-methyl naphthylallene derivatives 2p and 2q, chlorodienes 4p and 4q were obtained in 58% and 65% yield, along with 37% and 20% of the 2,3-dichlorides. Substrates with α-ethyl (2r) and α-isopropyl (2s) substituents reacted similarly, giving chlorinated products in 63–65% yield. Lastly, vinylidene 2t gave chlorodiene 4t as the sole product in 74% yield.
![[1860-5397-14-67-i4]](/bjoc/content/inline/1860-5397-14-67-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Chlorination of various α-substituted phenylallene derivatives. General conditions: Allene 2a (0.2 mmol, 1 equiv) was added dropwise over 1 h to a solution of 1b (0.44 mmol, 2.2 equiv) in 0.1 M CH3CN under reflux conditions, and the reaction stirred for 1 h; isolated yields. a 1H NMR yield using HMDSO as internal standard.
Scheme 4: Chlorination of various α-substituted phenylallene derivatives. General conditions: Allene 2a (0.2 ...
As with alkoxy substrate 2m, the α-methylated substrates 2u and 2v possessing methoxy group(s) on the arene also deviated from the expected reaction course. These reactions failed to fully consume the starting materials 2u and 2v, even upon prolonged heating, which we discovered to be the result of 1b being also consumed through over-chlorination. 4-Methoxy derivative 2u gave trichloride 5u in 53% yield, with no trace of the expected dichloride 3u or chlorodiene 4u products observable by NMR (Scheme 5). The 3,4-dimethoxy substrate 2v gave trichlorides 5v and 6v in a combined 67% yield, or in 91% yield based on the loading of 1b (Scheme 5). These anomalous outcomes were again rationalized as resulting from the stabilization of radical intermediates gained upon methoxy substitution [51], which permitted further chlorination of either the methyl or arene groups.
![[1860-5397-14-67-i5]](/bjoc/content/inline/1860-5397-14-67-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Chlorination of methoxy-substituted α-methyl phenylallenes. General conditions: Allene 2a (0.2 mmol, 1 equiv) was added dropwise over 1 h to a solution of 1b (0.44 mmol, 2.2 equiv) in 0.1 M CH3CN under reflux conditions, and the reaction stirred overnight; isolated yields.
Scheme 5: Chlorination of methoxy-substituted α-methyl phenylallenes. General conditions: Allene 2a (0.2 mmol...
To gain insight into the reaction mechanism we carried out two key control experiments. First, to test for rearrangement processes that might not be elucidated through product analysis alone, deuterated biphenylallene [D2]-2b was subjected to the standard reaction conditions, and [D2]-3b (E/Z = 1:2) was obtained in 79% yield (Scheme 6 top, also see Supporting Information File 1). As there was no indication of deuterium scrambling observable by 1H or 2H NMR of the product mixture, it appeared that 1,2-phenyl shifts or other rearrangement processes were not involved in the reaction. A further reaction was carried out in the presence of the radical scavenger TEMPO (1.5 equiv), from which only a trace of 3b was recovered, along with 50% of 2b (Scheme 6 bottom). As the chlorination reactivity was suppressed, our hypothesis that these reactions involved radical intermediates was further supported.
![[1860-5397-14-67-i6]](/bjoc/content/inline/1860-5397-14-67-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Control reactions: (a) chlorination of deuterated biphenylallene [D2]-2b; (b) reaction with TEMPO.
Scheme 6: Control reactions: (a) chlorination of deuterated biphenylallene [D2]-2b; (b) reaction with TEMPO.
When allene chlorination was carried out with 1a, the observed product distributions were consistent with the results previously obtained, suggesting that ionic processes were operative. Furthermore, since no evidence of propargyl chlorides or α-dichloromethylstyrenes were observed, it appears the chlorination of allenes with 1a proceeded without interruption of 1,2-phenyl shifts or iodane elimination, resulting in a reactivity pattern that differs from the related reagents TolIF2, PhI(OH)OTs or PhI(NTs2)2 (Scheme 1b and c). With 1b, however, the reactions were entirely selective for 2,3-dichlorination of the allene, which was consistent with the regiochemical outcome of reactions involving a trifluoromethyl radical (Scheme 1a). This, coupled with the results of Scheme 6, led us to propose a radical mechanism that was initiated by homolytic cleavage of the I–Cl bond of 1b at elevated temperature (Figure 1) [50]. Addition of the chlorine atom to the allene central carbon resulted in the highly stabilized radical intermediate E, which then abstracted a chlorine atom from a second equivalent of 1b, giving dichlorides 3. Or, in the case of α-alkyl groups, intermediate E was also subject to a competing hydrogen abstraction pathway, resulting in mixtures of 3 and chlorodienes 4.
![[1860-5397-14-67-1]](/bjoc/content/figures/1860-5397-14-67-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we report here an efficient new process for the chlorination of substituted phenylallene derivatives using the hypervalent iodine reagent 1-chloro-1,2-benziodoxol-3-one (1b). The reactions disclosed here represent the first report of a regioselective chlorination of phenylallenes, in which the 2,3-allene olefin undergoes selective vicinal dichlorination. Overall, the reactions were mild and operationally-simple, tolerant to a variety of different functional groups, and provided the products in typically good yield. The selectivity of the reaction is presumably derived from it being a radical, not ionic, process, which also enabled the formation of chlorodiene products with α-alkyl substituted allenes. This reaction offers a new strategy for accessing dichlorinated functional group building blocks not readily accessible with other reagents, and our continued work in this area will be disclosed in due course.
Supporting Information
| Supporting Information File 1: Experimental and characterization details, and NMR spectra of compounds. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Saunders, K. J. Poly(Vinyl Chloride) and Related Polymers. Organic Polymer Chemistry: An Introduction to the Organic Chemistry of Adhesives, Fibres, Paints, Plastics and Rubbers; Springer Netherlands: Dordrecht, 1988; pp 90–112. doi:10.1007/978-94-009-1195-6_4
Return to citation in text: [1] -
Hernandes, M. Z.; Cavalcanti, S. M. T.; Moreira, D. R. M.; Filgueira de Azevedo Junior, W.; Leite, A. C. L. Curr. Drug Targets 2010, 11, 303–314. doi:10.2174/138945010790711996
Return to citation in text: [1] -
Smith, B. R.; Eastman, C. M.; Njardarson, J. T. J. Med. Chem. 2014, 57, 9764–9773. doi:10.1021/jm501105n
Return to citation in text: [1] -
Jeschke, P. Pest Manage. Sci. 2010, 66, 10–27. doi:10.1002/ps.1829
Return to citation in text: [1] -
Herrera-Rodriguez, L. N.; Khan, F.; Robins, K. T.; Meyer, H. P. Chim. Oggi 2011, 29, 31–33.
Return to citation in text: [1] -
Jeschke, P. The Unique Role of Halogen Substituents in the Design of Modern Crop Protection Compounds. In Modern Methods in Crop Protection Research; Jeschke, P.; Krämer, W.; Schirmer, U., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp 73–128. doi:10.1002/9783527655908.ch4
Return to citation in text: [1] -
Tang, M. L.; Bao, Z. Chem. Mater. 2011, 23, 446–455. doi:10.1021/cm102182x
Return to citation in text: [1] -
Hiszpanski, A. M.; Saathoff, J. D.; Shaw, L.; Wang, H.; Kraya, L.; Lüttich, F.; Brady, M. A.; Chabinyc, M. L.; Kahn, A.; Clancy, P.; Loo, Y.-L. Chem. Mater. 2015, 27, 1892–1900. doi:10.1021/acs.chemmater.5b00329
Return to citation in text: [1] -
Gribble, G. W. Environ. Sci. Technol. 1994, 28, 310A–319A. doi:10.1021/es00056a712
Return to citation in text: [1] -
Gribble, G. W. Heterocycles 2012, 84, 157–207. doi:10.3987/REV-11-SR(P)5
Return to citation in text: [1] -
Wang, B.-G.; Gloer, J. B.; Ji, N.-Y.; Zhao, J.-C. Chem. Rev. 2013, 113, 3632–3685. doi:10.1021/cr9002215
Return to citation in text: [1] -
Gribble, G. W. Acc. Chem. Res. 1998, 31, 141–152. doi:10.1021/ar9701777
Return to citation in text: [1] -
Wallner, O. A.; Szabó, K. J. J. Org. Chem. 2003, 68, 2934–2943. doi:10.1021/jo026767m
Return to citation in text: [1] -
Gosmini, C.; Bégouin, J.-M.; Moncomble, A. Chem. Commun. 2008, 3221–3233. doi:10.1039/b805142a
Return to citation in text: [1] -
Nakamura, H.; Shim, J.-G.; Yamamoto, Y. J. Am. Chem. Soc. 1997, 119, 8113–8114. doi:10.1021/ja971599e
Return to citation in text: [1] -
Solin, N.; Narayan, S.; Szabó, K. J. J. Org. Chem. 2001, 66, 1686–1693. doi:10.1021/jo001374d
Return to citation in text: [1] -
Solin, N.; Narayan, S.; Szabó, K. J. Org. Lett. 2001, 3, 909–912. doi:10.1021/ol015545s
Return to citation in text: [1] -
Tsuji, J. Pd(0)-Catalyzed Reactions of Allylic Compounds via π-Allylpalladium Complexes. Palladium Reagents and Catalysts; John Wiley and Sons, Ltd.: Chichester, UK, 2005; pp 431–517. doi:10.1002/0470021209.ch4
Return to citation in text: [1] -
Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h
Return to citation in text: [1] -
Barluenga, J.; Fernández, M. A.; Aznar, F.; Valdés, C. Chem. Commun. 2004, 1400–1401. doi:10.1039/B403655G
Return to citation in text: [1] -
Reddy, C.; Reddy, V.; Urgaonkar, S.; Verkade, J. G. Org. Lett. 2005, 7, 4427–4430. doi:10.1021/ol051612x
Return to citation in text: [1] -
Ma, S.; Jiang, X.; Cheng, X.; Hou, H. Adv. Synth. Catal. 2006, 348, 2114–2124. doi:10.1002/adsc.200600234
Return to citation in text: [1] -
Lemay, A. B.; Vulic, K. S.; Ogilvie, W. W. J. Org. Chem. 2006, 71, 3615–3618. doi:10.1021/jo060144h
Return to citation in text: [1] -
Geary, L. M.; Hultin, P. G. J. Org. Chem. 2010, 75, 6354–6371. doi:10.1021/jo1014678
Return to citation in text: [1] -
Rossi, R.; Bellina, F.; Lessi, M. Tetrahedron 2011, 67, 6969–7025. doi:10.1016/j.tet.2011.06.001
Return to citation in text: [1] -
Thakur, A.; Zhang, K.; Louie, J. Chem. Commun. 2012, 48, 203–205. doi:10.1039/C1CC15990A
Return to citation in text: [1] -
Han, F.-S. Chem. Soc. Rev. 2013, 42, 5270–5298. doi:10.1039/c3cs35521g
Return to citation in text: [1] -
Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564–12649. doi:10.1021/acs.chemrev.6b00512
Return to citation in text: [1] -
Boyes, A. L.; Wild, M. Tetrahedron Lett. 1998, 39, 6725–6728. doi:10.1016/S0040-4039(98)01410-5
Return to citation in text: [1] -
Laali, K. K.; Nandi, G. C.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 2401–2405. doi:10.1016/j.tetlet.2014.02.110
Return to citation in text: [1] [2] [3] -
Bäckvall, J.-E.; Jonasson, C. Tetrahedron Lett. 1997, 38, 291–294. doi:10.1016/S0040-4039(96)02295-2
Return to citation in text: [1] [2] -
Mueller, W. H.; Butler, P. E.; Griesbaum, K. J. Org. Chem. 1967, 32, 2651–2654. doi:10.1021/jo01283a073
Return to citation in text: [1] -
Poutsma, M. L. J. Org. Chem. 1968, 33, 4080–4084. doi:10.1021/jo01275a016
Return to citation in text: [1] -
Lasne, M. C.; Thuillie, A. Bull. Soc. Chim. Fr. 1974, 249–252.
Return to citation in text: [1] -
Eisenberger, P.; Gischig, S.; Togni, A. Chem. – Eur. J. 2006, 12, 2579–2586. doi:10.1002/chem.200501052
Return to citation in text: [1] -
Zhu, N.; Wang, F.; Chen, P.; Ye, J.; Liu, G. Org. Lett. 2015, 17, 3580–3583. doi:10.1021/acs.orglett.5b01677
Return to citation in text: [1] -
Purkait, N.; Okumura, S.; Souto, J. A.; Muñiz, K. Org. Lett. 2014, 16, 4750–4753. doi:10.1021/ol502179z
Return to citation in text: [1] -
Moriarty, R. M.; Hopkins, T. E.; Vaid, R. K.; Vaid, B. K.; Levy, S. G. Synthesis 1992, 847–849. doi:10.1055/s-1992-26244
Return to citation in text: [1] -
Zhao, Z.; Racicot, L.; Murphy, G. K. Angew. Chem., Int. Ed. 2017, 56, 11620–11623. doi:10.1002/anie.201706798
Return to citation in text: [1] -
Knight, D. W.; Russell, G. A. Phenyliodine(III) Dichloride. e-EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons, 2001.
Return to citation in text: [1] -
Xue, C.; Jiang, X.; Fu, C.; Ma, S. Chem. Commun. 2013, 49, 5651–5653. doi:10.1039/c3cc42014k
Return to citation in text: [1] -
Egami, H.; Yoneda, T.; Uku, M.; Ide, T.; Kawato, Y.; Hamashima, Y. J. Org. Chem. 2016, 81, 4020–4030. doi:10.1021/acs.joc.6b00295
Return to citation in text: [1] -
Wang, M.; Zhang, Y.; Wang, T.; Wang, C.; Xue, D.; Xiao, J. Org. Lett. 2016, 18, 1976–1979. doi:10.1021/acs.orglett.6b00547
Return to citation in text: [1] -
Yang, X.; She, Y.; Chong, Y.; Zhai, H.; Zhu, H.; Chen, B.; Huang, G.; Yan, R. Adv. Synth. Catal. 2016, 358, 3130–3134. doi:10.1002/adsc.201600304
Return to citation in text: [1] -
Parvathaneni, S. P.; Perumgani, P. C. Asian J. Org. Chem. 2018, 7, 324–327. doi:10.1002/ajoc.201700620
Return to citation in text: [1] -
Wang, Z. Doering-Moore-Skattebøl Reaction. Comprehensive Organic Name Reactions and Reagents; John Wiley and Sons, 2010. doi:10.1002/9780470638859.conrr199
Return to citation in text: [1] -
Willgerodt, C. J. Prakt. Chem. 1894, 49, 466–482. doi:10.1002/prac.18940490134
Return to citation in text: [1] -
Keefer, R. M.; Andrews, L. J. J. Am. Chem. Soc. 1959, 81, 2374–2379. doi:10.1021/ja01519a026
Return to citation in text: [1] -
Amey, R. L.; Martin, J. C. J. Org. Chem. 1979, 44, 1779–1784. doi:10.1021/jo01325a007
Return to citation in text: [1] [2] -
Li, X.-Q.; Zhang, C. Synthesis 2009, 1163–1169. doi:10.1055/s-0028-1087850
Return to citation in text: [1] [2] -
Tokumura, K.; Ozaki, T.; Nosaka, H.; Saigusa, Y.; Itoh, M. J. Am. Chem. Soc. 1991, 113, 4974–4980. doi:10.1021/ja00013a037
Return to citation in text: [1]
| 47. | Willgerodt, C. J. Prakt. Chem. 1894, 49, 466–482. doi:10.1002/prac.18940490134 |
| 48. | Keefer, R. M.; Andrews, L. J. J. Am. Chem. Soc. 1959, 81, 2374–2379. doi:10.1021/ja01519a026 |
| 49. | Amey, R. L.; Martin, J. C. J. Org. Chem. 1979, 44, 1779–1784. doi:10.1021/jo01325a007 |
| 49. | Amey, R. L.; Martin, J. C. J. Org. Chem. 1979, 44, 1779–1784. doi:10.1021/jo01325a007 |
| 1. | Saunders, K. J. Poly(Vinyl Chloride) and Related Polymers. Organic Polymer Chemistry: An Introduction to the Organic Chemistry of Adhesives, Fibres, Paints, Plastics and Rubbers; Springer Netherlands: Dordrecht, 1988; pp 90–112. doi:10.1007/978-94-009-1195-6_4 |
| 9. | Gribble, G. W. Environ. Sci. Technol. 1994, 28, 310A–319A. doi:10.1021/es00056a712 |
| 10. | Gribble, G. W. Heterocycles 2012, 84, 157–207. doi:10.3987/REV-11-SR(P)5 |
| 11. | Wang, B.-G.; Gloer, J. B.; Ji, N.-Y.; Zhao, J.-C. Chem. Rev. 2013, 113, 3632–3685. doi:10.1021/cr9002215 |
| 12. | Gribble, G. W. Acc. Chem. Res. 1998, 31, 141–152. doi:10.1021/ar9701777 |
| 46. | Wang, Z. Doering-Moore-Skattebøl Reaction. Comprehensive Organic Name Reactions and Reagents; John Wiley and Sons, 2010. doi:10.1002/9780470638859.conrr199 |
| 7. | Tang, M. L.; Bao, Z. Chem. Mater. 2011, 23, 446–455. doi:10.1021/cm102182x |
| 8. | Hiszpanski, A. M.; Saathoff, J. D.; Shaw, L.; Wang, H.; Kraya, L.; Lüttich, F.; Brady, M. A.; Chabinyc, M. L.; Kahn, A.; Clancy, P.; Loo, Y.-L. Chem. Mater. 2015, 27, 1892–1900. doi:10.1021/acs.chemmater.5b00329 |
| 31. | Bäckvall, J.-E.; Jonasson, C. Tetrahedron Lett. 1997, 38, 291–294. doi:10.1016/S0040-4039(96)02295-2 |
| 4. | Jeschke, P. Pest Manage. Sci. 2010, 66, 10–27. doi:10.1002/ps.1829 |
| 5. | Herrera-Rodriguez, L. N.; Khan, F.; Robins, K. T.; Meyer, H. P. Chim. Oggi 2011, 29, 31–33. |
| 6. | Jeschke, P. The Unique Role of Halogen Substituents in the Design of Modern Crop Protection Compounds. In Modern Methods in Crop Protection Research; Jeschke, P.; Krämer, W.; Schirmer, U., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp 73–128. doi:10.1002/9783527655908.ch4 |
| 40. | Knight, D. W.; Russell, G. A. Phenyliodine(III) Dichloride. e-EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley and Sons, 2001. |
| 2. | Hernandes, M. Z.; Cavalcanti, S. M. T.; Moreira, D. R. M.; Filgueira de Azevedo Junior, W.; Leite, A. C. L. Curr. Drug Targets 2010, 11, 303–314. doi:10.2174/138945010790711996 |
| 3. | Smith, B. R.; Eastman, C. M.; Njardarson, J. T. J. Med. Chem. 2014, 57, 9764–9773. doi:10.1021/jm501105n |
| 30. | Laali, K. K.; Nandi, G. C.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 2401–2405. doi:10.1016/j.tetlet.2014.02.110 |
| 41. | Xue, C.; Jiang, X.; Fu, C.; Ma, S. Chem. Commun. 2013, 49, 5651–5653. doi:10.1039/c3cc42014k |
| 42. | Egami, H.; Yoneda, T.; Uku, M.; Ide, T.; Kawato, Y.; Hamashima, Y. J. Org. Chem. 2016, 81, 4020–4030. doi:10.1021/acs.joc.6b00295 |
| 43. | Wang, M.; Zhang, Y.; Wang, T.; Wang, C.; Xue, D.; Xiao, J. Org. Lett. 2016, 18, 1976–1979. doi:10.1021/acs.orglett.6b00547 |
| 44. | Yang, X.; She, Y.; Chong, Y.; Zhai, H.; Zhu, H.; Chen, B.; Huang, G.; Yan, R. Adv. Synth. Catal. 2016, 358, 3130–3134. doi:10.1002/adsc.201600304 |
| 45. | Parvathaneni, S. P.; Perumgani, P. C. Asian J. Org. Chem. 2018, 7, 324–327. doi:10.1002/ajoc.201700620 |
| 36. | Zhu, N.; Wang, F.; Chen, P.; Ye, J.; Liu, G. Org. Lett. 2015, 17, 3580–3583. doi:10.1021/acs.orglett.5b01677 |
| 38. | Moriarty, R. M.; Hopkins, T. E.; Vaid, R. K.; Vaid, B. K.; Levy, S. G. Synthesis 1992, 847–849. doi:10.1055/s-1992-26244 |
| 35. | Eisenberger, P.; Gischig, S.; Togni, A. Chem. – Eur. J. 2006, 12, 2579–2586. doi:10.1002/chem.200501052 |
| 39. | Zhao, Z.; Racicot, L.; Murphy, G. K. Angew. Chem., Int. Ed. 2017, 56, 11620–11623. doi:10.1002/anie.201706798 |
| 29. | Boyes, A. L.; Wild, M. Tetrahedron Lett. 1998, 39, 6725–6728. doi:10.1016/S0040-4039(98)01410-5 |
| 30. | Laali, K. K.; Nandi, G. C.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 2401–2405. doi:10.1016/j.tetlet.2014.02.110 |
| 31. | Bäckvall, J.-E.; Jonasson, C. Tetrahedron Lett. 1997, 38, 291–294. doi:10.1016/S0040-4039(96)02295-2 |
| 32. | Mueller, W. H.; Butler, P. E.; Griesbaum, K. J. Org. Chem. 1967, 32, 2651–2654. doi:10.1021/jo01283a073 |
| 33. | Poutsma, M. L. J. Org. Chem. 1968, 33, 4080–4084. doi:10.1021/jo01275a016 |
| 34. | Lasne, M. C.; Thuillie, A. Bull. Soc. Chim. Fr. 1974, 249–252. |
| 30. | Laali, K. K.; Nandi, G. C.; Bunge, S. D. Tetrahedron Lett. 2014, 55, 2401–2405. doi:10.1016/j.tetlet.2014.02.110 |
| 13. | Wallner, O. A.; Szabó, K. J. J. Org. Chem. 2003, 68, 2934–2943. doi:10.1021/jo026767m |
| 14. | Gosmini, C.; Bégouin, J.-M.; Moncomble, A. Chem. Commun. 2008, 3221–3233. doi:10.1039/b805142a |
| 15. | Nakamura, H.; Shim, J.-G.; Yamamoto, Y. J. Am. Chem. Soc. 1997, 119, 8113–8114. doi:10.1021/ja971599e |
| 16. | Solin, N.; Narayan, S.; Szabó, K. J. J. Org. Chem. 2001, 66, 1686–1693. doi:10.1021/jo001374d |
| 17. | Solin, N.; Narayan, S.; Szabó, K. J. Org. Lett. 2001, 3, 909–912. doi:10.1021/ol015545s |
| 18. | Tsuji, J. Pd(0)-Catalyzed Reactions of Allylic Compounds via π-Allylpalladium Complexes. Palladium Reagents and Catalysts; John Wiley and Sons, Ltd.: Chichester, UK, 2005; pp 431–517. doi:10.1002/0470021209.ch4 |
| 19. | Dounay, A. B.; Overman, L. E. Chem. Rev. 2003, 103, 2945–2964. doi:10.1021/cr020039h |
| 20. | Barluenga, J.; Fernández, M. A.; Aznar, F.; Valdés, C. Chem. Commun. 2004, 1400–1401. doi:10.1039/B403655G |
| 21. | Reddy, C.; Reddy, V.; Urgaonkar, S.; Verkade, J. G. Org. Lett. 2005, 7, 4427–4430. doi:10.1021/ol051612x |
| 22. | Ma, S.; Jiang, X.; Cheng, X.; Hou, H. Adv. Synth. Catal. 2006, 348, 2114–2124. doi:10.1002/adsc.200600234 |
| 23. | Lemay, A. B.; Vulic, K. S.; Ogilvie, W. W. J. Org. Chem. 2006, 71, 3615–3618. doi:10.1021/jo060144h |
| 24. | Geary, L. M.; Hultin, P. G. J. Org. Chem. 2010, 75, 6354–6371. doi:10.1021/jo1014678 |
| 25. | Rossi, R.; Bellina, F.; Lessi, M. Tetrahedron 2011, 67, 6969–7025. doi:10.1016/j.tet.2011.06.001 |
| 26. | Thakur, A.; Zhang, K.; Louie, J. Chem. Commun. 2012, 48, 203–205. doi:10.1039/C1CC15990A |
| 27. | Han, F.-S. Chem. Soc. Rev. 2013, 42, 5270–5298. doi:10.1039/c3cs35521g |
| 28. | Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564–12649. doi:10.1021/acs.chemrev.6b00512 |
| 37. | Purkait, N.; Okumura, S.; Souto, J. A.; Muñiz, K. Org. Lett. 2014, 16, 4750–4753. doi:10.1021/ol502179z |
| 51. | Tokumura, K.; Ozaki, T.; Nosaka, H.; Saigusa, Y.; Itoh, M. J. Am. Chem. Soc. 1991, 113, 4974–4980. doi:10.1021/ja00013a037 |
© 2018 Zhao and Murphy; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)