Abstract
Synthetic approaches towards N-acetylgalactosamine (GalNAc) have been attracting considerable interest since this compound is known for its pivotal role in cell–cell interaction and receptor induced cell signaling. Herein, we present a synthetic route in which two of the four stereogenic centers present in the target compound are derived from enantiopure tartaric acid being selectively converted to epoxy alcohols. The key step is the Pd-catalyzed, stereo- and regioselective epoxide opening and subsequent nucleophilic substitution of an azide functionality. This approach enables the synthesis of the naturally D- and unnaturally L-configured GalNAc, as well as both enantiomers of the largely unknown N-acetylidosamine (IdoNAc).

Graphical Abstract
Introduction
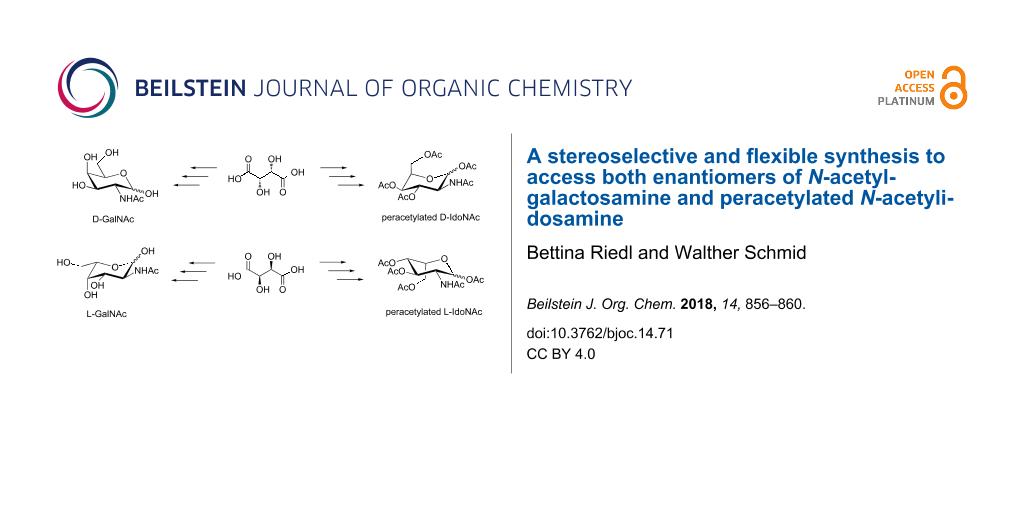
N-Acetylgalactosamine (GalNAc, Figure 1) and N-acetylidosamine (IdoNAc) belong to the group of 2-amino-2-deoxysugars, which can be found in a wide range of organisms as building blocks of, e.g., glycosaminoglycans, peptidoglycans or lipopolysaccharides [1]. As part of glycoproteins they are involved in numerous biological processes. On the one hand, these macromolecules influence the proper folding of newly synthesized proteins in the intracellular compartment of the endoplasmic reticulum (ER). On the other hand, glycoproteins serve as ligands for specific extracellular recognition processes toward, e.g., enzymes, lectins or antibodies [2]. In O-linked glycoproteins, also known as mucins, GalNAc becomes covalently α-linked to serine or threonine during post-translational modifications [3-5]. This glycoconjugate, known as T antigen nouvelle (Tn antigen), is further modified by the β(1→3) attachment of galactose leading to the Thomsen–Friedenreich (TF) antigen [6,7]. These two immunodeterminants are overexpressed on a high number of different cancer cells, located in the breast, lung, colon, liver, prostate and gastric tissues [8]. Furthermore, the TF-antigen region serves as the core region for the ABO and Lewis blood group determinants [9]. The high importance of GalNAc in diverse biological processes, together with its notable role in drug development, turns this compound into an interesting synthetic target. In contrast to the thoroughly investigated properties of GalNAc, other stereoisomers have not been probed for biological or pharmaceutical activity, yet. Highly flexible synthetic routes are required to access and probe the entire compound class of 2-amino-2-deoxysugars for further structure–activity relationship studies.
![[1860-5397-14-71-1]](/bjoc/content/figures/1860-5397-14-71-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Four possible isomers reachable through the presented approach.
Figure 1: Four possible isomers reachable through the presented approach.
Several strategies for the synthesis of 2-amino sugars have been published so far. In one exemplary straightforward approach, GalNAc was prepared by inverting the stereogenic center at the C-4 position of N-acetylglucosamine (GlcNAc) [10]. However, the necessity of using a 2-amino sugar as starting material reduces the flexibility of this approach. Another strategy introduces the nitrogen at C-2 [11] via the 2-iodoxybenzoic acid-mediated generation of an oxazolidinone ring, selectively leading to C2–C3 syn-products [12]. A third approach features E-selective Julia olefination of (R)- or (S)-2,3-isopropylideneglyceraldehyde with a second chiral building block containing the amine function, followed by dihydroxylation [13,14]. Despite the compounds already available through these and other approaches, novel synthetic concepts to fill the remaining methodological gaps for accessing the missing isomers of 2-amino sugars, as well as new derivatives are still highly desired.
The here presented flexible approach towards GalNAc and IdoNAc from epoxythreitol 5 is a first step to answer this request. These molecules can be approached from tartaric acid (3), which already provides two of the four desired stereogenic centers. By extending compound 5 via a Horner–Wadsworth–Emmons (HWE) reaction [15] and subsequent stereoselective introduction of the corresponding nitrogen functionality, a route to four different 2-amino sugar stereoisomers has been elaborated.
Results and Discussion
A synthetic route was developed starting with both the D- and the L-epoxythreitol 5a and 5b. These isomers have been synthesized from D- (3a) and L-tartaric acid (3b), respectively, through an approach that has been published earlier by Iida et al. (Scheme 1) [16]. Here, tartaric acid predefines the configuration at C4 and C5 of the target compounds 5a and 5b. After the C4-chain of tartaric acid has been extended by two carbon atoms resulting in compounds 4a and 4b, two new stereocenters have been introduced by Sharpless epoxidation [17,18]. We applied this approach for the epoxidation of 4a with L-diethyl tartrate (L-DET) directing the reaction towards the D-galacto-configured epoxythreitol 5a. Alternatively, epoxidation of 4b with D-DET was used to access L-galacto-configured epoxythreitol 5b.
![[1860-5397-14-71-i1]](/bjoc/content/inline/1860-5397-14-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Sharpless epoxidation to gain D-galacto- 5a and L-galacto-configured epoxythreitol 5b.
Scheme 1: Sharpless epoxidation to gain D-galacto- 5a and L-galacto-configured epoxythreitol 5b.
A procedure by Miyashita et al. [19] for nucleophilic substitution of epoxides by an azide functionality was applied for the presented approach. Therefore, 5a and 5b had to be converted first to the unsaturated esters 6a and 6b (Scheme 2) [20,21]. These compounds were reacted with trimethylsilyl azide (TMSN3) under Pd0 catalysis. With this protocol, the azide was introduced under double inversion of the stereocenter on C4, resulting in the desired cis-selective epoxide opening and therefore, galacto-configured azido alcohols 7a and 7b (≥95% de) [22]. Initial limitations in reaction size and yield could have been overcome by switching the solvent from THF, as described in the Miyashita protocol, to EtOH. This improvement can be explained by a protonation of TMSN3 causing the in situ generation of highly reactive HN3 (Scheme 3). Additionally, no acidic work-up, as described by Miyashita et al., was necessary to gain the free alcohol on C5, which corroborates the proposed mechanism.
![[1860-5397-14-71-i2]](/bjoc/content/inline/1860-5397-14-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: a) i) (COCl)2, DMSO, Et3N, DCM, ii) triethyl phosphonoacetate, NaH, DCM; b) TMSN3, Pd(PPh3)4, EtOH; c) i) DOWEX H+, MeOH; ii) O3, dimethyl sulfide, DCM/MeOH; iii) Ac2O, DMAP, pyridine; d) H2, Pd/C, Ac2O; e) MeOH/H2O/Et3N (10:10:1).
Scheme 2: Reagents and conditions: a) i) (COCl)2, DMSO, Et3N, DCM, ii) triethyl phosphonoacetate, NaH, DCM; b...
![[1860-5397-14-71-i3]](/bjoc/content/inline/1860-5397-14-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism of the Pd-catalyzed azide substitution of 6a in protic solvent.
Scheme 3: Proposed mechanism of the Pd-catalyzed azide substitution of 6a in protic solvent.
The protecting groups were cleaved by the use of acidic ion exchange (DOWEX H+). Subsequent ozonolysis caused the formation of the aldehyde by the oxidation of the methylene functionality. Consequently, intramolecular hemiacetal formation resulted immediately in the cyclization of the unprotected azido sugar, which was peracetylated to yield compounds 8a and 8b. The azide function was reduced with H2 and Pd/C in acetic anhydride to gain the fully acetylated amino sugars 2a and 2b. In the final step, the compounds were deprotected using a mixture of MeOH/H2O/Et3N to yield D-GalNAc (1a) and L-GalNAc (1b).
As proof of concept, this approach was additionally adapted for the synthesis of peracetylated D-N-acetylidosamine 2c. Therefore, the epoxidation of 4a was performed with D-DET instead of L-DET, resulting in D-epoxythreitol 5c (Scheme 4). Performing the presented, optimized reaction sequence resulted in the formation of peracetylated D-IdoNAc 2c.
![[1860-5397-14-71-i4]](/bjoc/content/inline/1860-5397-14-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Approach towards peracetylated D-IdoNAc 2c, reactions and conditions: a) Ti(OiPr)4, t-BuOOH, D-DET, DCM; b) i) (COCl)2, DMSO, Et3N, DCM, ii) triethyl phosphonoacetate, NaH, DCM; c) TMSN3, Pd(PPh3)4, EtOH; d) i) DOWEX H+, MeOH; ii) O3, dimethyl sulfide, DCM/MeOH; iii) Ac2O, DMAP, pyridine; e) H2, Pd/C, Ac2O.
Scheme 4: Approach towards peracetylated D-IdoNAc 2c, reactions and conditions: a) Ti(OiPr)4, t-BuOOH, D-DET,...
Conclusion
The presented synthetic route was successfully used to prepare both, D- and L-GalNAc, from non-carbohydrate starting materials over 9 steps with overall yields of 22% and 12%, respectively. Our approach combines the Sharpless epoxidation and Pd-catalyzed azide substitution, both, highly stereoselective protocols resulting in an advantageous flexibility. Therefore, this route can also be adapted for the synthesis of the rarely known D- and L-IdoNAc. As a proof of concept, the synthesis of the peracetylated D-IdoNAc was performed (Scheme 4, 8 steps, 11%). Furthermore, this approach opens up new perspectives for additional modifications: meso-tartaric acid as possible starting material or different epoxide opening protocols, as well as, the selective isotopic labelling are only a few to mention. This convertible synthetic route brings us a step closer to access the whole library of 2-amino-2-deoxysugars, as well as a variety of derivatives, for further studies.
Supporting Information
| Supporting Information File 1: Experimental procedures, as well as 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Salton, M. R. J. Annu. Rev. Biochem. 1965, 34, 143–174. doi:10.1146/annurev.bi.34.070165.001043
Return to citation in text: [1] -
Shylaja, M.; Seshadri, H. S. Biochem. Educ. 1989, 17, 170–178. doi:10.1016/0307-4412(89)90136-2
Return to citation in text: [1] -
Varki, A.; Cummings, R. D.; Esko, J. D.; Stanley, P.; Hart, G. W.; Aebi, M.; Darvill, A. G.; Kinoshita, T.; Packer, N. H.; Prestegard, J. H.; Schnaar, R. L.; Seeberger, P. H., Eds. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, 2017.
Return to citation in text: [1] -
Hussain, M. R. M.; Hassan, M.; Afzal, I.; Afzal, A. Egypt. J. Med. Hum. Genet. 2012, 13, 1–9. doi:10.1016/j.ejmhg.2011.07.006
Return to citation in text: [1] -
Kobata, A. Eur. J. Biochem. 1992, 209, 483–501. doi:10.1111/j.1432-1033.1992.tb17313.x
Return to citation in text: [1] -
Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology; Varki, A.; Cummings, R. D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2009; pp 115–128.
Return to citation in text: [1] -
Haslam, S. M.; Julien, S.; Burchell, J. M.; Monk, C. R.; Ceroni, A.; Garden, O. A.; Dell, A. Immunol. Cell Biol. 2008, 86, 564–573. doi:10.1038/icb.2008.54
Return to citation in text: [1] -
Chia, J.; Goh, G.; Bard, F. Biochim. Biophys. Acta 2016, 1860, 1623–1639. doi:10.1016/j.bbagen.2016.03.008
Return to citation in text: [1] -
Hara, A.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. Molecules 2014, 19, 414–437. doi:10.3390/molecules19010414
Return to citation in text: [1] -
Araki, S.; Ito, H.; Butsugan, Y. J. Org. Chem. 1988, 53, 1831–1833. doi:10.1021/jo00243a052
Return to citation in text: [1] -
Mirabella, S.; Cardona, F.; Goti, A. Org. Biomol. Chem. 2016, 14, 5186–5204. doi:10.1039/C6OB00649C
Return to citation in text: [1] -
Nicolaou, K. C.; Baran, P. S.; Zhong, Y.-L.; Vega, J. A. Angew. Chem., Int. Ed. 2000, 39, 2525–2529. doi:10.1002/1521-3773(20000717)39:14<2525::AID-ANIE2525>3.0.CO;2-1
Return to citation in text: [1] -
Ermolenko, L.; Sasaki, N. A.; Potier, P. Tetrahedron Lett. 1999, 40, 5187–5190. doi:10.1016/S0040-4039(99)00978-8
Return to citation in text: [1] -
Ermolenko, L.; Sasaki, N. A.; Potier, P. J. Chem. Soc., Perkin Trans. 1 2000, 2465–2473. doi:10.1039/b001721n
Return to citation in text: [1] -
Wadsworth, W. S.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042
Return to citation in text: [1] -
Iida, H.; Yamazaki, N.; Kibayashi, C. J. Org. Chem. 1987, 52, 3337–3342. doi:10.1021/jo00391a031
Return to citation in text: [1] -
Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974–5976. doi:10.1021/ja00538a077
Return to citation in text: [1] -
Ko, S. Y.; Lee, A. W. M.; Masamune, S.; Reed, L. A., III; Sharpless, K. B.; Walker, F. J. Science 1983, 220, 949–951. doi:10.1126/science.220.4600.949
Return to citation in text: [1] -
Miyashita, M.; Mizutani, T.; Tadano, G.; Iwata, Y.; Miyazawa, M.; Tanino, K. Angew. Chem., Int. Ed. 2005, 44, 5094–5097. doi:10.1002/anie.200500838
Return to citation in text: [1] -
Ghosh, A. K.; Wang, Y. J. Org. Chem. 1999, 64, 2789–2795. doi:10.1021/jo9822378
Return to citation in text: [1] -
Sarabia, F.; Martín-Gálvez, F.; García-Ruiz, C.; Sánchez-Ruiz, A.; Vivar-García, C. J. Org. Chem. 2013, 78, 5239–5253. doi:10.1021/jo4003272
Return to citation in text: [1] -
Albler, C.; Hollaus, R.; Kählig, H.; Schmid, W. Beilstein J. Org. Chem. 2014, 10, 2230–2234. doi:10.3762/bjoc.10.231
Return to citation in text: [1]
| 1. | Salton, M. R. J. Annu. Rev. Biochem. 1965, 34, 143–174. doi:10.1146/annurev.bi.34.070165.001043 |
| 8. | Chia, J.; Goh, G.; Bard, F. Biochim. Biophys. Acta 2016, 1860, 1623–1639. doi:10.1016/j.bbagen.2016.03.008 |
| 20. | Ghosh, A. K.; Wang, Y. J. Org. Chem. 1999, 64, 2789–2795. doi:10.1021/jo9822378 |
| 21. | Sarabia, F.; Martín-Gálvez, F.; García-Ruiz, C.; Sánchez-Ruiz, A.; Vivar-García, C. J. Org. Chem. 2013, 78, 5239–5253. doi:10.1021/jo4003272 |
| 6. | Brockhausen, I.; Schachter, H.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology; Varki, A.; Cummings, R. D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, 2009; pp 115–128. |
| 7. | Haslam, S. M.; Julien, S.; Burchell, J. M.; Monk, C. R.; Ceroni, A.; Garden, O. A.; Dell, A. Immunol. Cell Biol. 2008, 86, 564–573. doi:10.1038/icb.2008.54 |
| 22. | Albler, C.; Hollaus, R.; Kählig, H.; Schmid, W. Beilstein J. Org. Chem. 2014, 10, 2230–2234. doi:10.3762/bjoc.10.231 |
| 3. | Varki, A.; Cummings, R. D.; Esko, J. D.; Stanley, P.; Hart, G. W.; Aebi, M.; Darvill, A. G.; Kinoshita, T.; Packer, N. H.; Prestegard, J. H.; Schnaar, R. L.; Seeberger, P. H., Eds. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, New York, 2017. |
| 4. | Hussain, M. R. M.; Hassan, M.; Afzal, I.; Afzal, A. Egypt. J. Med. Hum. Genet. 2012, 13, 1–9. doi:10.1016/j.ejmhg.2011.07.006 |
| 5. | Kobata, A. Eur. J. Biochem. 1992, 209, 483–501. doi:10.1111/j.1432-1033.1992.tb17313.x |
| 17. | Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974–5976. doi:10.1021/ja00538a077 |
| 18. | Ko, S. Y.; Lee, A. W. M.; Masamune, S.; Reed, L. A., III; Sharpless, K. B.; Walker, F. J. Science 1983, 220, 949–951. doi:10.1126/science.220.4600.949 |
| 2. | Shylaja, M.; Seshadri, H. S. Biochem. Educ. 1989, 17, 170–178. doi:10.1016/0307-4412(89)90136-2 |
| 19. | Miyashita, M.; Mizutani, T.; Tadano, G.; Iwata, Y.; Miyazawa, M.; Tanino, K. Angew. Chem., Int. Ed. 2005, 44, 5094–5097. doi:10.1002/anie.200500838 |
| 12. | Nicolaou, K. C.; Baran, P. S.; Zhong, Y.-L.; Vega, J. A. Angew. Chem., Int. Ed. 2000, 39, 2525–2529. doi:10.1002/1521-3773(20000717)39:14<2525::AID-ANIE2525>3.0.CO;2-1 |
| 15. | Wadsworth, W. S.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042 |
| 11. | Mirabella, S.; Cardona, F.; Goti, A. Org. Biomol. Chem. 2016, 14, 5186–5204. doi:10.1039/C6OB00649C |
| 16. | Iida, H.; Yamazaki, N.; Kibayashi, C. J. Org. Chem. 1987, 52, 3337–3342. doi:10.1021/jo00391a031 |
| 10. | Araki, S.; Ito, H.; Butsugan, Y. J. Org. Chem. 1988, 53, 1831–1833. doi:10.1021/jo00243a052 |
| 9. | Hara, A.; Imamura, A.; Ando, H.; Ishida, H.; Kiso, M. Molecules 2014, 19, 414–437. doi:10.3390/molecules19010414 |
| 13. | Ermolenko, L.; Sasaki, N. A.; Potier, P. Tetrahedron Lett. 1999, 40, 5187–5190. doi:10.1016/S0040-4039(99)00978-8 |
| 14. | Ermolenko, L.; Sasaki, N. A.; Potier, P. J. Chem. Soc., Perkin Trans. 1 2000, 2465–2473. doi:10.1039/b001721n |
© 2018 Riedl and Schmid; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)