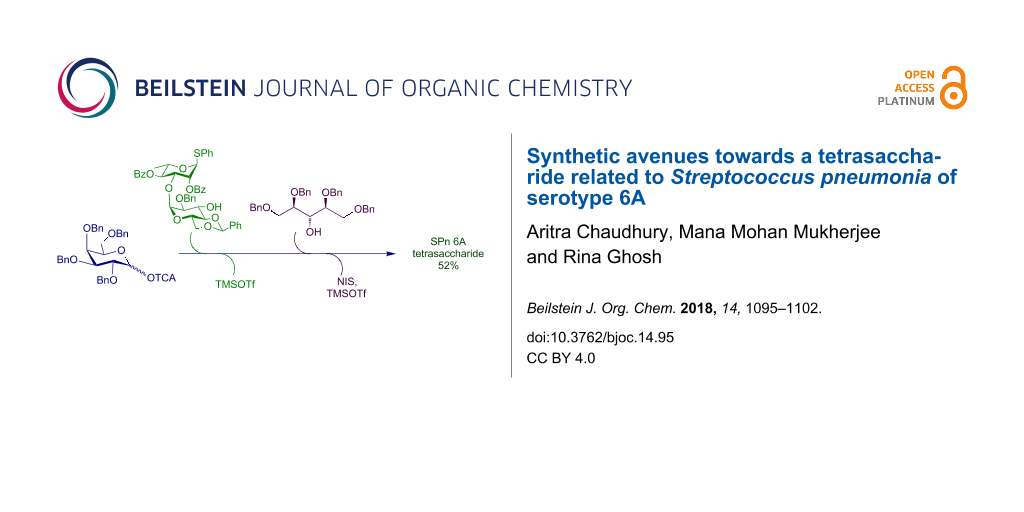
Abstract
Streptococcus pneumonia (SPn) is a Gram-positive bacterium which causes life threatening diseases. The bacteria protect themselves against non-specific host defence by an external polysaccharide (PS) capsule which bears a repeating unit, α-D-Galp(1->3)-α-D-Glcp(1->3)-α-L-Rhap(1->3)-D-Rib (SPn 6A). A closer look at the structure reveals the presence of α-linked galactose and glucose residues. The synthesis of these 1,2-cis glycosidic linkages are considered challenging particularly in the context of a one-pot oligosaccharide synthesis. We have synthesized the aforesaid tetrasaccharide (SPn 6A) based on both stepwise and sequential one-pot glycosylation reactions using easily accessible common building blocks; eventually similar overall yields were obtained in both cases.

Graphical Abstract
Introduction
Complex glycans serve as attractive targets for carbohydrate-based vaccines and therapeutics [1-3]. Streptococcus pneumonia (SPn) has been posing a serious threat in recent times. It is a major cause of pneumonia, bacteraemia, and meningitis in immune-compromised patients, elderly and children. A UNICEF/WHO survey has estimated that 920136 children died of pneumonia in 2015 accounting for 16% of all fatalities under the age of five [4]. Out of over 90 serotypes that have been reported for SPn [5,6], serogroup 6 has been ranked among the most important causes of invasive pneumonococcal diseases [7]. These facts have led to extensive research towards the establishment of polysaccharide structures associated with the SPn serogroup 6 [8,9] (Figure 1).
![[1860-5397-14-95-1]](/bjoc/content/figures/1860-5397-14-95-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The tetrasaccharides associated with the pneumonicoccal serogroup 6.
Figure 1: The tetrasaccharides associated with the pneumonicoccal serogroup 6.
Initially, it was thought that due to their similar carbohydrate core structures the antibodies elicited by SPn 6A would be effective against SPn 6B as well [10-13]. But recent studies have shown that serotype specific immune responses are elicited by the antibodies and that they cross react slowly [14]. As a result the importance of the presence of the capsular polysaccharide of SPn 6A in multicomponent vaccines like Pneumovax® has been recognized [15]. The low hydrolytic stability of the phosphodiester linkages in the clinical isolates of the SPn 6A polysaccharides poses a major drawback as it leads to low bioavailability [16]. Hence, the requirements of pure SPn 6A conjugate in proper amounts for future vaccine development can only be met via chemical synthesis. Therefore, a number of syntheses targeting the SPn 6A tetrasaccharide has been reported in literature. Initial reports of a linear synthesis were made by Vliegenthart et al. in the nineties [17-21]. After this, the Demchenko group improved these early reports with a convergent approach using glycosyl thioimidates as complementary glycosyl donors with respect to thioglycosides [22-25]. Herein, we wish to report synthetic routes to the SPn 6A tetrasaccharide via stepwise as well as one-pot sequential glycosylation strategies.
Results and Discussion
Keeping in mind our objective to synthesize the SPn 6A tetrasaccharide following stepwise as well as one-pot synthetic strategies based on common building blocks, a retrosynthetic analysis was made which led us to galactose-based donor 2 [26], ribitol-based acceptor 7 [22] and a gluco-rhamno-based disaccharide (3a/3b) to contemplate the synthesis of the tetrasaccharide derivative 1 (Figure 2).
![[1860-5397-14-95-2]](/bjoc/content/figures/1860-5397-14-95-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The disaccharides (3a/3b) can be synthesized from their parent monomeric units 6a/6b/6c and 5 [27]. To ensure high α-selection during the formation of the central gluco-rhamno disaccharide the benzylidene protected glucosyl donor (Figure 2) was selected, because the induction of the 1,2-cis selectivity in benzylidene-protected substrates via torsional/electronic effects have already been recognized [28].
The galactosyl trichloroacetimidate donor 2 was prepared following literature procedures [26]. On the other hand the D-glucosyl thioglycoside 8 was converted to the known benzylidene derivative 9 [29,30] according to our previously reported procedure. Benzylation of 9 under phase-transfer conditions led to 10 [31] in 49% yield (Scheme 1). Subsequently, 10 was subjected to naphthylmethylation/p-methoxybenzylation [32] with 2-(bromomethyl)naphthalene (NapBr)/p-methoxybenzyl chloride in DMF to afford 6a/11 [32] in 90% and 80% yields, respectively.
![[1860-5397-14-95-i1]](/bjoc/content/inline/1860-5397-14-95-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of D-glucosyl donor 6. Reaction conditions: a) NapBr/PMBCl, NaH, DMF, rt, 12 h, 90% (6a), 80% (11); b) TCCA, CH3COCH3/H2O (4:1), rt, 85% (12a), 89% (12b); c) CCl3CN, DBU, DCM, 0 °C, 93% (6b), 90% (6c).
Scheme 1: Preparation of D-glucosyl donor 6. Reaction conditions: a) NapBr/PMBCl, NaH, DMF, rt, 12 h, 90% (6a...
These derivatives were next subjected to thioglycoside hydrolysis using trichloroisocyanuric acid (TCCA) [33] in wet acetone which provided 12a/12b [34] in 85% and 89% yields, respectively. These were finally converted to their corresponding trichloroacetimidate 6b/6c [34] (Scheme 1) with yields of 93% and 90%, respectively.
The L-rhamnosyl thioglycoside 14 [29,30], prepared from L-rhamnose (13), was deacetylated quantitatively in the presence of Et3N/MeOH/H2O [35], and then stannylene-mediated selective naphthylmethylation at the O-3 position was carried out to give the known derivative 15 in 82% yield [36]. This was next benzoylated almost quantitatively to give 16. Finally DDQ-mediated deprotection of the naphthylmethyl group gave the acceptor 5 [27] in 83% yield (Scheme 2).
![[1860-5397-14-95-i2]](/bjoc/content/inline/1860-5397-14-95-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of L-rhamnosyl acceptor 5. Reaction conditions: a) Py, BzCl, rt, 12 h, 99%; b) DDQ, DCM/H2O (19:1), rt, 2 h, 83%.
Scheme 2: Preparation of L-rhamnosyl acceptor 5. Reaction conditions: a) Py, BzCl, rt, 12 h, 99%; b) DDQ, DCM...
For acceptor 7 (Figure 2) ribitol 17 was converted to its corresponding diisopropylidene derivative 18 [37] in the presence of dimethoxypropane and pTSA in acetone. Treatment with NapBr and NaH in DMF gave compound 19 in 93% yield. Subsequent deprotection of isopropylidene ketal with pTSA/MeOH (aq) and then benzylation furnished 20 in 95% yield over two steps. Deprotection of the naphthylmethyl group in the presence of DDQ in aqueous dichloromethane (19:1) gave the glycosyl acceptor 7 [22] in 85% yield (Scheme 3).
![[1860-5397-14-95-i3]](/bjoc/content/inline/1860-5397-14-95-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of ribitol acceptor 7. Reaction conditions: a) Me2C(OMe)2, pTSA, CH3COCH3, rt, 81%; b) NapBr, NaH, DMF, rt, 8 h, 93%; c) (i) pTSA, MeOH, 40 °C, 4 h, (ii) BnBr, NaH, DMF, rt, 12 h, 95% over 2 steps; d) DDQ, DCM/H2O (19:1), rt, 2 h, 85%.
Scheme 3: Preparation of ribitol acceptor 7. Reaction conditions: a) Me2C(OMe)2, pTSA, CH3COCH3, rt, 81%; b) ...
In order to construct the central disaccharide fragment 3 in high yield with 1,2-cis selectivity, several glycosylation reactions using glucosyl donors 6a/6b/6c/12a and rhamnosyl acceptor 5 were contemplated. None of the conditions, based on the use of thioglycoside 6a as the glycosyl donor and separately, BSP/Tf2O (Table 1, entry 1) or Ph2SO/Tf2O (Table 1, entry 2) as the corresponding activators, or based on 1-hydroxy donor 12a and Ph2SO/Tf2O (Table 1, entry 3), could furnish any desired result. After trying with the mentioned donors, and reagent combinations (Table 1), we switched over to utilize trichloroacetimidate donors (6b, Table 1, entries 5 and 6, and 6c, entry 4); the TMSOTf mediated glycosylation in DCM/Et2O solvent (Table 1, entry 6) was found to be effective in case of donor 6b and acceptor 5 which generated the desired disaccharide 3a in high yield and exclusive α-anomeric selectivity (evidenced from NMR). We presume that this near exclusivity in α-selection may be due to the synergistic effect from the 4,6-O-benzylidene group, which is a good promoter for 1,2-cis glycosylation in galactose-based systems [38], as well as the steric crowding caused by the bulky 3-O-naphthylmethyl group at the β-side of the ring. Having obtained the central disaccharide 3a in requisite yield and excellent stereochemical purity we now proceeded towards the synthesis of the trisaccharide fragment 21 (Scheme 4).
Table 1: Optimization of protocol for the synthesis of disaccharide 3.
| Entry | Donor | Acceptor | Conditions | Yielda |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-95-i6.svg?max-width=637&scale=1.0)
6a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-95-i7.svg?max-width=637&scale=1.0)
5 |
BSPb, Tf2O, DCM,
−60 °C→rt (A)c |
N.R.d |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-95-i8.svg?max-width=637&scale=1.0)
6a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-95-i9.svg?max-width=637&scale=1.0)
5 |
Ph2SO, TTBP, Tf2O, DCM,
−60 °C→−40 °C (B)c |
N.R.e |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-95-i10.svg?max-width=637&scale=1.0)
12a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-95-i11.svg?max-width=637&scale=1.0)
5 |
Ph2SO, TTBP, Tf2O, DCM,
−60 °C→−40 °C (B)c |
N.R.d |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-95-i12.svg?max-width=637&scale=1.0)
6c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-95-i13.svg?max-width=637&scale=1.0)
5 |
TMSOTf, DCM,
−30 °C (C)c |
N.R.f |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-95-i14.svg?max-width=637&scale=1.0)
6b |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-95-i15.svg?max-width=637&scale=1.0)
5 |
TMSOTf, DCM,
−10 °C (C)c |
3a, 54%
(α only) |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-95-i16.svg?max-width=637&scale=1.0)
6b |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-95-i17.svg?max-width=637&scale=1.0)
5 |
TMSOTf, DCM/Et2O (5:1),
−30 °C (C)c |
3a, 75%
(α only) |
aIsolated yields of products; bBSP = benzenesulfinylpiperidine; ccorresponding glycosylation procedure (see Supporting Information File 1); dstarting material was decomposed; edonor was decomposed but acceptor was recovered; fa complex mixture was formed from which the desired disaccharide could not be purified by column chromatography.
![[1860-5397-14-95-i4]](/bjoc/content/inline/1860-5397-14-95-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Stepwise synthesis of tetrasaccharide 1. Reaction conditions: a) TMSOTf, DCM/Et2O (5:1), 4 Å MS, −30 °C, 75%; b) DDQ, DCM/H2O (9:1), rt, 93%; c) TMSOTf, DCM/Et2O (4:1), 4 Å MS, −15 °C, 70%; d) NIS, TMSOTf, DCM, 4 Å MS, −20 °C, (procedure D, see Supporting Information File 1), 89%.
Scheme 4: Stepwise synthesis of tetrasaccharide 1. Reaction conditions: a) TMSOTf, DCM/Et2O (5:1), 4 Å MS, −3...
Compound 3a was treated with DDQ in dichloromethane to remove the 3-O-Nap protection group generating acceptor 4 in 93% yield. Glycosylation between donor 2 and acceptor 4 was achieved uneventfully in the presence of TMSOTf in dichloromethane/Et2O (4:1) to give the trisaccharide 21 in 70% yield. Successful glycosylation was also carried out between trisaccharide 21 and ribitol acceptor 7 in the presence of NIS and TMSOTf in dichloromethane at −20 °C to give the tetrasaccharide derivative 1 in 89% yield, thereby finishing the stepwise synthesis of SPn 6A tetrasaccharide 1 in the protected form (Scheme 4).
Having standardized a stepwise synthesis of the tetrasaccharide 1 in the protected form we then turned our attention to devise a one-pot protocol to achieve the same derivative. The one-pot synthesis of this target is particularly challenging because of the presence of the two 1,2-cis glycosidic linkages which are likely to make product isolation particularly difficult at the end of the glycosylative protocol. Assuming equal preference of formation for each and every possible diastereomer across the three glycosylation steps a mixture of 8 different diastereomers may be formed if the four monomers are sequentially added in a (1 + 1 + 1 + 1) one-pot strategy. However, the number of possibilities may be reduced to 4 isomers by using a participating group on the rhamnose residue to induce near exclusive α-selectivity in the last step. Further reduction can be ensured by incorporating a (1 + 2 + 1) approach where the number of possible diastereomers becomes 2. So, we selected this (1 + 2 + 1) glycosylation for our synthesis.
Two different strategies were attempted in this direction. Recently, Mong et al. have reported high α-selectivity in the formation of glucan and galactan under non-participating conditions from the O-2 protecting group [39,40]. With this method, we tried to couple donor p-tolyl 2,3,4,6-tetra-O-benzyl-1-thio-β-D-galactopyranoside (22) with acceptor 4 using a NIS/TMSOTf combination in the presence of DMF acting as a modulating solvent (inset, Scheme 5). Unfortunately, when this strategy was applied to our case it could not produce a viable result. So we switch to the conventional orthogonal strategy for a one-pot synthesis of the targeted tetrasaccharide (Scheme 5).
![[1860-5397-14-95-i5]](/bjoc/content/inline/1860-5397-14-95-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: One-pot synthesis of tetrasaccharide 1. Reaction conditions: a) TMSOTf, 4 Å MS, DCM/Et2O (4:1), N2, −15 °C, 1 h; b) NIS, TMSOTf, −10 °C, 45 min; c) NaOMe, MeOH, rt; d) H2, Pd/C, EtOH/EtOAc/AcOH, rt.
Scheme 5: One-pot synthesis of tetrasaccharide 1. Reaction conditions: a) TMSOTf, 4 Å MS, DCM/Et2O (4:1), N2,...
The disaccharide acceptor 4 was glycosylated with galactosyl trichloroacetimidate donor 2 at −15 °C using 30 mol % of TMSOTf. After full consumption of the starting materials (TLC), into the same pot the second acceptor 7 followed by NIS were added. The reaction mixture was allowed to reach −10 °C before another 30 mol % of TMSOTf were added; TLC after 45 minutes showed complete consumption of the starting materials. Thus the targeted tetrasaccharide derivative was prepared via a three component, one-pot sequential glycosylation technique in 52% yield (Scheme 5). It is to be noted that the temperature had to be raised to −10 °C from −20 °C in the second step of the one-pot protocol. This was necessary to improve the overall yield of the final product. The tetrasaccharide derivative 1 was next deprotected under Zemplén conditions [41], followed by hydrogenation with H2/Pd-C in EtOH/EtOAc/AcOH solvent to give the deprotected tetrasaccharide 23 in 85% yield over two steps.
1H NMR in D2O of the target tetrasaccharide 23 showed the anomeric protons of the galactose, glucose, and rhamnose residues from the non-reducing end appearing at δ 5.32 (d, J = 3.5 Hz), δ 5.02 (d, J = 3 Hz), and δ 4.93 (bs), respectively. 13C NMR along with the HSQC in the same solvent revealed that the chemical shifts of the anomeric carbons of the same units from the non-reducing end are at δ 99.2 (1JC1-H1 = 167.7 Hz), 95.4 (1JC1-H1 = 169.3 Hz) and 100.1 (1JC1-H1 = 169.2 Hz), respectively. The values are indicative of α-stereochemistry at all the anomeric centers [42]. Moreover, the chemical shifts were found to be in fair agreement with the reported C-1 chemical shifts at δ 99.5, 95.6, and 100.3, exhibited in D2O corresponding to the anomeric centers of compound 23 [22].
Conclusion
Summarizing our work we have achieved stepwise and sequential one-pot syntheses of the tetrasaccharide repeating unit of SPn 6A via an orthogonal glycosylation strategy using commonly used trichloroacetimidate and thioglycoside donors. The challenging 1,2-cis linkages could be prepared with a yield and a selectivity which were high enough to allow the one-pot synthesis.
Supporting Information
| Supporting Information File 1: Experimental details for the preparation of compounds 1, 3a, 4, 5, 6a, 6b, 7, 12a, 19, 20, 21, and 23 and the corresponding characterization data. | ||
| Format: PDF | Size: 581.8 KB | Download |
| Supporting Information File 2: 1H and 13C NMR of compounds 1, 3a, 4, 5, 6a, 6b, 7, 12a, 19, 20, 21, and 23 and 2D NMR (COSY, HSQC and HMBC) of compound 23. | ||
| Format: PDF | Size: 3.1 MB | Download |
References
-
Kuberan, B.; Linhardt, R. J. Curr. Org. Chem. 2000, 4, 653. doi:10.2174/1385272003376111
Return to citation in text: [1] -
Danishefsky, S. J.; Allen, J. R. Angew. Chem., Int. Ed. 2000, 39, 836. doi:10.1002/(SICI)1521-3773(20000303)39:5<836::AID-ANIE836>3.0.CO;2-I
Return to citation in text: [1] -
Wong, C.-H., Ed. Carbohydrate-Based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/3527602437
Return to citation in text: [1] -
http://www.who.int/mediacentre/factsheets/fs331/en/ (accessed Sept 15, 2016).
Return to citation in text: [1] -
Kamerling, J. P. In Streptococcus pneumonia, Molecular Biology and Mechanism of Disease; Tomasz, A., Ed.; Mary Ann Liebert: Larchmont, New York, NY, 2000.
Return to citation in text: [1] -
Park, I. H.; Pritchard, D. G.; Cartee, R.; Brandao, A.; Brandileone, M. C. C.; Nahm, M. H. J. Clin. Microbiol. 2007, 45, 1225. doi:10.1128/JCM.02199-06
Return to citation in text: [1] -
Robinson, D. A.; Briles, D. E.; Crain, M. J.; Hollingshead, S. K. J. Bacteriol. 2002, 184, 6367. doi:10.1128/JB.184.22.6367-6375.2002
Return to citation in text: [1] -
Rebers, P. A.; Heidelberger, M. J. Am. Chem. Soc. 1961, 83, 3056. doi:10.1021/ja01475a021
Return to citation in text: [1] -
Robbins, J. B.; Lee, C. J.; Rastogi, S. C.; Schiffman, G.; Henrichsen, J. Infect. Immun. 1979, 26, 1116.
Return to citation in text: [1] -
Robbins, J. B.; Austrian, R.; Lee, C.-J.; Rastogi, S. C.; Schiffman, G.; Henrichsen, J.; Makela, P. H.; Broome, C. V.; Facklam, R. R.; Tiesjema, R. H.; Parke, J. C., Jr. J. Infect. Dis. 1983, 148, 1136. doi:10.1093/infdis/148.6.1136
Return to citation in text: [1] -
Alonsodevelasco, E.; Verheul, A. F. M.; Verhoef, J.; Snippe, H. Microbiol. Rev. 1995, 59, 591.
Return to citation in text: [1] -
Moreau, M.; Schulz, D. J. J. Carbohydr. Chem. 2000, 19, 419. doi:10.1080/07328300008544091
Return to citation in text: [1] -
Ledwith, M. Curr. Opin. Pediatr. 2001, 13, 70. doi:10.1097/00008480-200102000-00013
Return to citation in text: [1] -
Väkeväinen, M.; Eklund, C.; Eskola, J.; Käyhty, H. J. Infect. Dis. 2001, 184, 789. doi:10.1086/322984
Return to citation in text: [1] -
Eschaniz-Aviles, I. G.; Solorzano-Santos, F. Salud Publica Mexico 2001, 43, 352.
Return to citation in text: [1] -
Zon, G.; Szu, S. C.; Egan, W.; Robbins, J. D.; Robbins, J. B. Infect. Immun. 1982, 37, 89.
Return to citation in text: [1] -
Slaghek, T. M.; van Vliet, M. J.; Maas, A. A. M.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1989, 195, 75. doi:10.1016/0008-6215(89)85090-6
Return to citation in text: [1] -
Slaghek, T. M.; van Oijen, A. H.; Maas, A. A. M.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1990, 207, 237. doi:10.1016/0008-6215(90)84051-U
Return to citation in text: [1] -
Thijssen, M. J. L.; van Rijswijk, M. N.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1998, 306, 93. doi:10.1016/S0008-6215(97)00271-1
Return to citation in text: [1] -
Thijssen, M. J. L.; Bijkerk, M. H. G.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1998, 306, 111. doi:10.1016/S0008-6215(97)10013-1
Return to citation in text: [1] -
Slaghek, T. M.; Maas, A. A. M.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1991, 211, 25. doi:10.1016/0008-6215(91)84143-3
Return to citation in text: [1] -
Parameswar, A. R.; Pornsuriyasak, P.; Lubanowski, N. A.; Demchenko, A. V. Tetrahedron 2007, 63, 10083. doi:10.1016/j.tet.2007.07.036
Return to citation in text: [1] [2] [3] [4] -
Demchenko, A. V.; Rousson, E.; Boons, G.-J. Tetrahedron Lett. 1999, 40, 6523. doi:10.1016/S0040-4039(99)01203-4
Return to citation in text: [1] -
Parameswar, A.; Hasty, S. J.; Demchenko, A. V. Carbohydr. Res. 2008, 343, 1707. doi:10.1016/j.carres.2008.03.035
Return to citation in text: [1] -
Parameswar, A. R.; Park, I. H.; Saksena, R.; Kováč, P.; Nahm, M. H.; Demchenko, A. V. ChemBioChem 2009, 10, 2893. doi:10.1002/cbic.200900587
Return to citation in text: [1] -
Schmidt, R. R.; Michel, J.; Roos, M. Liebigs Ann. Chem. 1984, 1343. doi:10.1002/jlac.198419840710
Return to citation in text: [1] [2] -
Rao, Y.; Boons, G.-J. Angew. Chem., Int. Ed. 2007, 46, 6148. doi:10.1002/anie.200701750
Return to citation in text: [1] [2] -
Weingart, R.; Schmidt, R. R. Tetrahedron Lett. 2000, 41, 8753. doi:10.1016/S0040-4039(00)01497-0
Return to citation in text: [1] -
Mukherjee, M. M.; Basu, N.; Chaudhury, A.; Ghosh, R. RSC Adv. 2016, 6, 109301. doi:10.1039/C6RA23198E
Return to citation in text: [1] [2] -
Basu, N.; Maity, S. K.; Roy, S.; Singha, S.; Ghosh, R. Carbohydr. Res. 2011, 346, 534. doi:10.1016/j.carres.2011.01.003
Return to citation in text: [1] [2] -
Garegg, P. J.; Kvarnström, I.; Niklasson, A.; Niklasson, G.; Svensson, S. C. T. J. Carbohydr. Chem. 1993, 12, 933. doi:10.1080/07328309308020107
Return to citation in text: [1] -
Müller, M.; Schmidt, R. R. Eur. J. Org. Chem. 2001, 2055. doi:10.1002/1099-0690(200106)2001:11<2055::AID-EJOC2055>3.0.CO;2-N
Return to citation in text: [1] [2] -
Basu, N.; Maity, S. K.; Chaudhury, A.; Ghosh, R. Carbohydr. Res. 2013, 369, 10. doi:10.1016/j.carres.2013.01.001
Return to citation in text: [1] -
Sumida, Y.; Kokubo, S.; Kunisada, S.; Miyamoto, S. Immunostimulants containing polysaccharides comprising β-1,3-glucan as main chain and β-1,3-glucan side chains attached via β1→6 bond, and antitumor agents containing them. Jpn. Kokai Tokkyo Koho JP2005225775 A, Aug 25, 2005.
Return to citation in text: [1] [2] -
Meier, L.; Monteiro, G. C.; Baldissera, R. A. M.; Sá, M. M. J. Braz. Chem. Soc. 2010, 21, 859. doi:10.1590/S0103-50532010000500013
Return to citation in text: [1] -
Crich, D.; Vinogradova, O. J. Org. Chem. 2007, 72, 3581. doi:10.1021/jo062411p
Return to citation in text: [1] -
Xie, Z.-F.; Suemune, H.; Sakai, K. Tetrahedron: Asymmetry 1993, 4, 973. doi:10.1016/S0957-4166(00)80142-1
Return to citation in text: [1] -
Crich, D. Acc. Chem. Res. 2010, 43, 1144. doi:10.1021/ar100035r
Return to citation in text: [1] -
Lu, S.-R.; Lai, Y.-H.; Chen, J.-H.; Liu, C.-Y.; Mong, K.-K. T. Angew. Chem., Int. Ed. 2011, 50, 7315. doi:10.1002/anie.201100076
Return to citation in text: [1] -
Liu, C.-Y. I.; Mulani, S.; Mong, K.-K. T. Adv. Synth. Catal. 2012, 354, 3299. doi:10.1002/adsc.201200396
Return to citation in text: [1] -
Zemplén, G. Ber. Dtsch. Chem. Ges. 1926, 59, 1254. doi:10.1002/cber.19260590626
Return to citation in text: [1] -
Bock, K.; Pedersen, C. J. Chem. Soc., Perkin Trans. 2 1974, 293. doi:10.1039/p29740000293
Return to citation in text: [1]
| 37. | Xie, Z.-F.; Suemune, H.; Sakai, K. Tetrahedron: Asymmetry 1993, 4, 973. doi:10.1016/S0957-4166(00)80142-1 |
| 22. | Parameswar, A. R.; Pornsuriyasak, P.; Lubanowski, N. A.; Demchenko, A. V. Tetrahedron 2007, 63, 10083. doi:10.1016/j.tet.2007.07.036 |
| 1. | Kuberan, B.; Linhardt, R. J. Curr. Org. Chem. 2000, 4, 653. doi:10.2174/1385272003376111 |
| 2. | Danishefsky, S. J.; Allen, J. R. Angew. Chem., Int. Ed. 2000, 39, 836. doi:10.1002/(SICI)1521-3773(20000303)39:5<836::AID-ANIE836>3.0.CO;2-I |
| 3. | Wong, C.-H., Ed. Carbohydrate-Based Drug Discovery; Wiley-VCH: Weinheim, Germany, 2003. doi:10.1002/3527602437 |
| 8. | Rebers, P. A.; Heidelberger, M. J. Am. Chem. Soc. 1961, 83, 3056. doi:10.1021/ja01475a021 |
| 9. | Robbins, J. B.; Lee, C. J.; Rastogi, S. C.; Schiffman, G.; Henrichsen, J. Infect. Immun. 1979, 26, 1116. |
| 28. | Weingart, R.; Schmidt, R. R. Tetrahedron Lett. 2000, 41, 8753. doi:10.1016/S0040-4039(00)01497-0 |
| 7. | Robinson, D. A.; Briles, D. E.; Crain, M. J.; Hollingshead, S. K. J. Bacteriol. 2002, 184, 6367. doi:10.1128/JB.184.22.6367-6375.2002 |
| 26. | Schmidt, R. R.; Michel, J.; Roos, M. Liebigs Ann. Chem. 1984, 1343. doi:10.1002/jlac.198419840710 |
| 5. | Kamerling, J. P. In Streptococcus pneumonia, Molecular Biology and Mechanism of Disease; Tomasz, A., Ed.; Mary Ann Liebert: Larchmont, New York, NY, 2000. |
| 6. | Park, I. H.; Pritchard, D. G.; Cartee, R.; Brandao, A.; Brandileone, M. C. C.; Nahm, M. H. J. Clin. Microbiol. 2007, 45, 1225. doi:10.1128/JCM.02199-06 |
| 22. | Parameswar, A. R.; Pornsuriyasak, P.; Lubanowski, N. A.; Demchenko, A. V. Tetrahedron 2007, 63, 10083. doi:10.1016/j.tet.2007.07.036 |
| 27. | Rao, Y.; Boons, G.-J. Angew. Chem., Int. Ed. 2007, 46, 6148. doi:10.1002/anie.200701750 |
| 16. | Zon, G.; Szu, S. C.; Egan, W.; Robbins, J. D.; Robbins, J. B. Infect. Immun. 1982, 37, 89. |
| 22. | Parameswar, A. R.; Pornsuriyasak, P.; Lubanowski, N. A.; Demchenko, A. V. Tetrahedron 2007, 63, 10083. doi:10.1016/j.tet.2007.07.036 |
| 23. | Demchenko, A. V.; Rousson, E.; Boons, G.-J. Tetrahedron Lett. 1999, 40, 6523. doi:10.1016/S0040-4039(99)01203-4 |
| 24. | Parameswar, A.; Hasty, S. J.; Demchenko, A. V. Carbohydr. Res. 2008, 343, 1707. doi:10.1016/j.carres.2008.03.035 |
| 25. | Parameswar, A. R.; Park, I. H.; Saksena, R.; Kováč, P.; Nahm, M. H.; Demchenko, A. V. ChemBioChem 2009, 10, 2893. doi:10.1002/cbic.200900587 |
| 42. | Bock, K.; Pedersen, C. J. Chem. Soc., Perkin Trans. 2 1974, 293. doi:10.1039/p29740000293 |
| 15. | Eschaniz-Aviles, I. G.; Solorzano-Santos, F. Salud Publica Mexico 2001, 43, 352. |
| 26. | Schmidt, R. R.; Michel, J.; Roos, M. Liebigs Ann. Chem. 1984, 1343. doi:10.1002/jlac.198419840710 |
| 22. | Parameswar, A. R.; Pornsuriyasak, P.; Lubanowski, N. A.; Demchenko, A. V. Tetrahedron 2007, 63, 10083. doi:10.1016/j.tet.2007.07.036 |
| 14. | Väkeväinen, M.; Eklund, C.; Eskola, J.; Käyhty, H. J. Infect. Dis. 2001, 184, 789. doi:10.1086/322984 |
| 39. | Lu, S.-R.; Lai, Y.-H.; Chen, J.-H.; Liu, C.-Y.; Mong, K.-K. T. Angew. Chem., Int. Ed. 2011, 50, 7315. doi:10.1002/anie.201100076 |
| 40. | Liu, C.-Y. I.; Mulani, S.; Mong, K.-K. T. Adv. Synth. Catal. 2012, 354, 3299. doi:10.1002/adsc.201200396 |
| 10. | Robbins, J. B.; Austrian, R.; Lee, C.-J.; Rastogi, S. C.; Schiffman, G.; Henrichsen, J.; Makela, P. H.; Broome, C. V.; Facklam, R. R.; Tiesjema, R. H.; Parke, J. C., Jr. J. Infect. Dis. 1983, 148, 1136. doi:10.1093/infdis/148.6.1136 |
| 11. | Alonsodevelasco, E.; Verheul, A. F. M.; Verhoef, J.; Snippe, H. Microbiol. Rev. 1995, 59, 591. |
| 12. | Moreau, M.; Schulz, D. J. J. Carbohydr. Chem. 2000, 19, 419. doi:10.1080/07328300008544091 |
| 13. | Ledwith, M. Curr. Opin. Pediatr. 2001, 13, 70. doi:10.1097/00008480-200102000-00013 |
| 17. | Slaghek, T. M.; van Vliet, M. J.; Maas, A. A. M.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1989, 195, 75. doi:10.1016/0008-6215(89)85090-6 |
| 18. | Slaghek, T. M.; van Oijen, A. H.; Maas, A. A. M.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1990, 207, 237. doi:10.1016/0008-6215(90)84051-U |
| 19. | Thijssen, M. J. L.; van Rijswijk, M. N.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1998, 306, 93. doi:10.1016/S0008-6215(97)00271-1 |
| 20. | Thijssen, M. J. L.; Bijkerk, M. H. G.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1998, 306, 111. doi:10.1016/S0008-6215(97)10013-1 |
| 21. | Slaghek, T. M.; Maas, A. A. M.; Kamerling, J. P.; Vliegenthart, J. F. G. Carbohydr. Res. 1991, 211, 25. doi:10.1016/0008-6215(91)84143-3 |
| 41. | Zemplén, G. Ber. Dtsch. Chem. Ges. 1926, 59, 1254. doi:10.1002/cber.19260590626 |
| 32. | Müller, M.; Schmidt, R. R. Eur. J. Org. Chem. 2001, 2055. doi:10.1002/1099-0690(200106)2001:11<2055::AID-EJOC2055>3.0.CO;2-N |
| 29. | Mukherjee, M. M.; Basu, N.; Chaudhury, A.; Ghosh, R. RSC Adv. 2016, 6, 109301. doi:10.1039/C6RA23198E |
| 30. | Basu, N.; Maity, S. K.; Roy, S.; Singha, S.; Ghosh, R. Carbohydr. Res. 2011, 346, 534. doi:10.1016/j.carres.2011.01.003 |
| 31. | Garegg, P. J.; Kvarnström, I.; Niklasson, A.; Niklasson, G.; Svensson, S. C. T. J. Carbohydr. Chem. 1993, 12, 933. doi:10.1080/07328309308020107 |
| 36. | Crich, D.; Vinogradova, O. J. Org. Chem. 2007, 72, 3581. doi:10.1021/jo062411p |
| 27. | Rao, Y.; Boons, G.-J. Angew. Chem., Int. Ed. 2007, 46, 6148. doi:10.1002/anie.200701750 |
| 29. | Mukherjee, M. M.; Basu, N.; Chaudhury, A.; Ghosh, R. RSC Adv. 2016, 6, 109301. doi:10.1039/C6RA23198E |
| 30. | Basu, N.; Maity, S. K.; Roy, S.; Singha, S.; Ghosh, R. Carbohydr. Res. 2011, 346, 534. doi:10.1016/j.carres.2011.01.003 |
| 35. | Meier, L.; Monteiro, G. C.; Baldissera, R. A. M.; Sá, M. M. J. Braz. Chem. Soc. 2010, 21, 859. doi:10.1590/S0103-50532010000500013 |
| 34. | Sumida, Y.; Kokubo, S.; Kunisada, S.; Miyamoto, S. Immunostimulants containing polysaccharides comprising β-1,3-glucan as main chain and β-1,3-glucan side chains attached via β1→6 bond, and antitumor agents containing them. Jpn. Kokai Tokkyo Koho JP2005225775 A, Aug 25, 2005. |
| 34. | Sumida, Y.; Kokubo, S.; Kunisada, S.; Miyamoto, S. Immunostimulants containing polysaccharides comprising β-1,3-glucan as main chain and β-1,3-glucan side chains attached via β1→6 bond, and antitumor agents containing them. Jpn. Kokai Tokkyo Koho JP2005225775 A, Aug 25, 2005. |
| 32. | Müller, M.; Schmidt, R. R. Eur. J. Org. Chem. 2001, 2055. doi:10.1002/1099-0690(200106)2001:11<2055::AID-EJOC2055>3.0.CO;2-N |
| 33. | Basu, N.; Maity, S. K.; Chaudhury, A.; Ghosh, R. Carbohydr. Res. 2013, 369, 10. doi:10.1016/j.carres.2013.01.001 |
© 2018 Chaudhury et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)