

Abstract
The complexation study of cis-protected and bare palladium(II) components with a new tridentate ligand, i.e., pyridine-3,5-diylbis(methylene) dinicotinate (L1) is the focus of this work. Complexation of cis-Pd(tmeda)(NO3)2 with L1 at a 1:1 or 3:2 ratio produced [Pd(tmeda)(L1)](NO3)2 (1a). The reaction mixture obtained at 3:2 ratio upon prolonged heating, produced a small amount of [Pd3(tmeda)3(L1)2](NO3)6 (2a). Complexation of Pd(NO3)2 with L1 at a 1:2 or 3:4 ratios afforded [Pd(L1)2](NO3)2 (3a) and [(NO3)2@Pd3(L1)4](NO3)4 (4a), respectively. The encapsulated NO3– ions of 4a undergo anion exchange with halides (F–, Cl– and Br– but not with I–) to form [(X)2@Pd3(L1)4](NO3)4 5a–7a. The coordination behaviour of ligand L1 and some dynamic properties of these complexes are compared with a set of known complexes prepared using the regioisomeric ligand bis(pyridin-3-ylmethyl)pyridine-3,5-dicarboxylate (L2). Importantly, a ligand isomerism phenomenon is claimed by considering complexes prepared from L1 and L2.
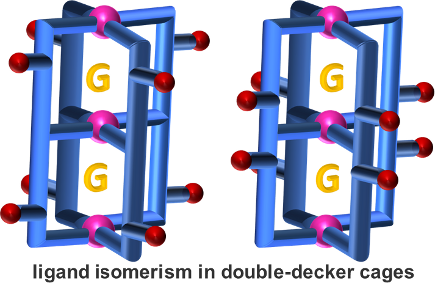
Graphical Abstract
Introduction
Coordination-driven self-assembly is a convenient strategy for the construction of supramolecules of desired dimensions via simple synthetic procedures. Well-defined metal–ligand coordination bonds enable the construction of designer targeted molecules with ease. The use of a palladium(II) component for complexation with a non-chelating bi- or polydentate ligand (usually N-donor ligands) is particularly advantageous for the construction of a variety of metallocages [1-5]. Complexation of cis-protected palladium(II), i.e., (PdL’) or bare palladium(II) with non-chelating bidentate ligands is known to afford a series of (PdL’)mLm or PdmL2m-type self-assembled coordination complexes [5]. Pd2L4-type cages are the simplest representatives among the PdmL2m-type complexes, yet most utilised [5,6]. The Pd2L4-type cages are well explored for the encapsulation of guests that are anionic [7-11], neutral [12-16], radical initiators [17], and drug molecules [18,19]. It is necessary to emphasize here that Pd2L4-type cages contain a cavity. McMorran and Steel reported the first Pd2L4-type cage and the anion binding ability of the cavity [20]. They also used a non-chelating tridentate ligand in an attempt to prepare a Pd3L4-type double-decker coordination cage that would possess two cavities, if formed. However, the plan could not be executed as one of the coordinating atoms of the ligand remained unutilized [21]. Instead of the desired Pd3L4 architecture, they observed a PdL2-type spirometallomacrocycle where bare palladium(II) is the juncture between two metallomacrocyclic rings. We report here a Pd3L4-type cage prepared from Pd(NO3)2 and pyridine-3,5-diylbis(methylene) dinicotinate (L1). We reported earlier the first Pd3L4-type double-decker coordination cage using a simple tridentate “E” shaped ester-based ligand bis(pyridin-3-ylmethyl)pyridine-3,5-dicarboxylate (L2) [22,23]. An additional feature in our design using L2 is the stoichiometrically controlled formation of PdL2-type spiro and Pd3L4-type double-decker complexes that is reversible under appropriate conditions. Subsequently, other research groups (Chand, Clever, Crowley and Yoshizawa groups) published Pd3L4-type cages [24]. This design has been further explored by Crowley et al. for the synthesis of a Pd4L4-type triple-decker cage [25]. The Clever research group reported a system in which two units of a double-decker cage are interlocked [26]. In this context, we revisited our earlier design of Pd3L4-type cages to prepare a corresponding analogues cages using the new ligand L1 (that is a positional isomer or regioisomer of L2) in order to exemplify ligand isomerism.
Ligand isomerism includes metal complexes (at least two) having the same molecular formula but are composed of different positional isomers (regioisomers) of the ligand. Positional isomers (regioisomers) of a non-chelating ligand system capable of forming palladium(II) complexes of same molecular formula is a rare phenomenon [27-34]. Such palladium(II) complexes should represent the phenomenon of “ligand isomerism”. We reported a family of Pd2L4-type complexes that fits under the definition of ligand isomerism [34]. In the pursuit of ligand isomerism in Pd3L4-type double-decker cages we intended to include our reported cage (prepared from palladium(II) and L2) [22,23] and construct a new isomeric Pd3L4-type complex. The complexation study of cis-protected and bare palladium(II) components with the new tridentate ligand, i.e., pyridine-3,5-diylbis(methylene) dinicotinate (L1) is the focus of this work. In addition, the dynamic behavior as well as anion binding abilities of selected complexes are also probed. Ligand L1 is a constitutional isomer of ligand L2 [22,23] and is expected to exhibit similarities but also some differences in complexation behavior with palladium(II) components. There are also some similarities and some differences in the related properties of these complexes.
Results and Discussion
Design and synthesis of ligand L1
The new ligand L1 was designed as a positional isomer (regioisomer) of the known ligand L2 (Figure 1). Each of these ligands has three pyridine moieties separated by two spacer moieties (–CH2OC(=O)–). Both ligands are semi-rigid/semi-flexible due to the spacers’ conformational mobility. The “E-shaped” conformation of the ligand that is suitable for the formation of the targeted Pd3L4-type complex is shown here for clarity of discussion. In a given ligand, two of the pyridine rings are substituted in the 3-position and are terminal and symmetrically disposed with respect to the central/internal 3,5-disubstituted pyridine ring. The spacers are identical in both ligands, however, their orientations are reversed in ligand L1 as compared to the known ligand L2. The primary intention of the design of L1 was to have a positional isomer (regioisomer) of the ligand L2.
![[1860-5397-15-109-1]](/bjoc/content/figures/1860-5397-15-109-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The ligands (i) L1 and (ii) L2 that are positional isomers (regioisomers).
Figure 1: The ligands (i) L1 and (ii) L2 that are positional isomers (regioisomers).
The tridentate ligand pyridine-3,5-diylbis(methylene) dinicotinate (L1, Figure 1) was prepared by condensation of pyridine-3,5-diyldimethanol [35] with nicotinoyl chloride hydrochloride in dry dichloromethane in the presence of triethylamine. The reaction mixture was stirred at room temperature for 24 h followed by aqueous work-up and column chromatography purification to afford pyridine-3,5-diylbis(methylene) dinicotinate (L1) as a white solid. The ligand was fully characterized by NMR spectroscopy and ESIMS techniques (Supporting Information File 1, Figures S1–S11). In addition, NOESY analysis was helpful in distinguishing the protons Ha and Hf.
It is assumed that the electron density at the central pyridine ring in L1 (that is a lutidine derivative) should be higher than that at the central pyridine ring of L2 (that is a dinicotinate derivative) having electron-withdrawing carbonyl substituents. Also, the electron density at the terminal pyridine ring in L1 (that is a nicotinate derivative) should be lower than that at the terminal pyridine ring of L2 (that is a picolyl derivative). The electrostatic potential maps at the terminal and internal pyridine nitrogen calculated using DFT methods (Supporting Information File 1, Table S2), however, are found to be comparable. Nevertheless, it seemed interesting to check whether or not the subtle difference in the electron density at the pyridine N centers has any influence on the coordination behavior of the ligands.
Complexation of palladium(II) components with ligand L1
Complexation of cis-protected palladium(II) was carried out with the ligand L1 at two different metal-to-ligand ratios (1:1 and 3:2). We also carried out the complexation of bare palladium(II) with the ligand L1 at two different metal-to-ligand ratios (1:2 and 3:4). The complexation reactions performed in DMSO-d6 allowed the monitoring of complex formation and those performed in DMSO were used for isolation of the complex by precipitation methods. The resulting complexes at specified ratios of the reactants are depicted in Scheme 1 and the details of the complexation behavior are described below.
![[1860-5397-15-109-i1]](/bjoc/content/inline/1860-5397-15-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (i)/(ii) Complexation of Pd(tmeda)(Y)2 with the ligand L1 at 1:1 and 2:3 metal-to-ligand ratios, respectively; (iii)/(iv) complexation of Pd(Y)2 with the ligand L1 at 1:2 and 3:4 metal-to-ligand ratios, respectively. For (i)/(ii)/(iii): Y = NO3−, BF4−, ClO4−, or OTf− (2b was not formed).
Scheme 1: (i)/(ii) Complexation of Pd(tmeda)(Y)2 with the ligand L1 at 1:1 and 2:3 metal-to-ligand ratios, re...
Complexation of cis-protected palladium(II) with ligand L1 at 1:1 metal-to-ligand ratio
The addition of one equivalent of cis-Pd(tmeda)(NO3)2 to a clear solution of one equivalent of the ligand L1 in DMSO-d6 produced a turbid mixture. However, a clear yellow solution was obtained upon stirring the mixture at 90 °C for 5 min or at rt for 15 min. The reaction was repeated in DMSO and the (PdL’)L-type complex [Pd(tmeda)(L1)](NO3)2 (1a, Scheme 1(i)) was isolated from the reaction mixture by a precipitation method that is described in the experimental section. The 1H NMR spectrum of the solution showed formation of a single discrete complex (Figure 2(ii)). Counter-anion (BF4−, ClO4− and OTf−) variation was also carried out to successfully prepare a series of complexes [Pd(tmeda)(L1)](BF4)2 (1b); [Pd(tmeda)(L1)](ClO4)2 (1c); [Pd(tmeda)(L1)](OTf)2 (1d). The complexes 1a–d were prepared by mixing the corresponding metal component Pd(tmeda)(Y)2 with the ligand L1 where Y = NO3−, BF4−, ClO4− and OTf−. The metal components were prepared in situ by reacting Pd(tmeda)(Cl)2 with AgY in DMSO-d6 followed by separation of the precipitated AgCl.
The complex 1a was characterized by various NMR techniques (Supporting Information File 1, Figures S12–S16). The 1H NMR spectrum of compound 1a showed single set of peaks (Figure 2(ii)) characterized by complexation-induced downfield shifts of protons belonging to the terminal pyridines (Δδ = 0.77, 0.52, and 0.29 ppm for Ha, Hb, and Hc, respectively) as compared to the free ligand L1. The peak positions of Hf and Hg remained unchanged which indicated that the central pyridine ring is not involved in the complexation. The 1H NMR spectra of compounds 1b, 1c and 1d are very much comparable to that of 1a (Supporting Information File 1, Figure S17). One of the coordination sites of the ligand L1 thus remained unutilized in these mononuclear complexes.
![[1860-5397-15-109-2]](/bjoc/content/figures/1860-5397-15-109-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Partial 1H NMR spectra in DMSO-d6 for (i) L1, (ii) [Pd(tmeda)(L1)](NO3)2 (1a) and (iii) a mixture of [Pd(tmeda)(L1)](NO3)2 (1a) and [Pd3(tmeda)3(L1)2](NO3)6 (2a).
Figure 2: Partial 1H NMR spectra in DMSO-d6 for (i) L1, (ii) [Pd(tmeda)(L1)](NO3)2 (1a) and (iii) a mixture o...
The ESIMS data of the compounds 1b, 1c and 1d confirmed the formation of mononuclear complexes (Supporting Information File 1, Figures S18–S20). As an example, the ESIMS spectrum of compound 1b (Supporting Information File 1, Figure S18) showed isotopic peak patterns at m/z 658.14 and 285.57, respectively, which correspond to the cationic fragments [1b − BF4]+ and [1b − 2BF4]2+ that are formed due to the loss of one and two units of counter anions from 1b. The experimental and theoretical peak patterns were found to be in agreement. The data of 1c and 1d are given in Supporting Information File 1.
Complexation of cis-protected palladium(II) with ligand L1 at a 3:2 metal-to-ligand ratio
The addition of three equivalents of cis-Pd(tmeda)(NO3)2 to a clear solution of two equivalents of ligand L1 in DMSO-d6 produced a turbid mixture. However, a clear yellow solution was obtained upon stirring the mixture at 90 °C for 5 min. The progress of the complexation reaction was monitored by 1H NMR spectroscopy. We targeted a Pd3L’3L2-type complex [36], i.e., [Pd3(tmeda)3(L1)2](NO3)6 (2a, Scheme 1(ii)). However, the NMR spectrum showed the formation of [Pd(tmeda)(L1)](NO3)2 (1a) and uncomplexed cis-Pd(tmeda)2+. The reaction was allowed to continue where upon the integration ratios of the peaks corresponding to Hf and Hg were lower than expected and that of Hd was higher than expected. In addition, a new peak was observed at around 9.15 ppm (Figure 2(iii)). Careful analysis of the data led us to propose the formation of a minor proportion of 2a along with a major proportion of 1a and unutilized cis-Pd(tmeda)2+ remaining in solution. The peaks assigned to Hf and Hg of 2a are shifted downfield compared to those of Hf and Hg in 1a whereas other signals of 2a merged with the respective signals of 1a. The observed downfield shift of the Hf signal in 2a is complexation-induced and found to have a unique position. The downfield shift of the Hg signal in 2a could not be induced by complexation and is best described by considering an anisotropy effect of the nearby carbonyl groups the ligand strand. The Hg signal of 2a, however, merged with the Hd signal of 1a.
Subsequently, anion variation (BF4−, ClO4− and OTf−) was carried out in anticipation of the trinuclear products 2b–d (Scheme 1). Complexation of the metal component Pd(tmeda)(Y)2 with the ligand L1 were carried out where Y = NO3−, BF4−, ClO4− and OTf−. The metal components were prepared in situ by reacting Pd(tmeda)(Cl)2 with AgY in DMSO-d6 followed by separation of the precipitated AgCl. The 1H NMR spectra of the samples confirmed the formation of mononuclear complexes 1b–d at initial stages (Supporting Information File 1, Figures S22–S24). All reactions were allowed to continue and their progress was monitored by 1H NMR spectroscopy. The peak positions in the 1H NMR spectrum of the sample containing BF4− as counter anion remained unchanged but the same for the samples containing ClO4− and OTf− behaved in a manner very similar to the case of NO3−. A new peak was observed at around 9.10 ppm corresponding to Hf in each case. When integration ratio of Ha is taken as 1.0 the integration ratio of Hf for 1d and 2d in the mixture were found to be ≈0.8 and ≈0.2, respectively. It may be noted here that a comparable trinuclear complex of ligand L2, however, did not form [23].
ESIMS data were collected in anticipation of detecting the trinuclear complexes (Supporting Information File 1, Figures S25 and S26). Isotopic peak patterns at m/z 391.55 corresponding to the fragment [2c − 4ClO4]4+ confirmed the existence of the trinuclear complex 2c. Similarly, isotopic peak patterns at m/z 2111.08 corresponding to the fragment [2d − OTf]+ confirmed the existence of trinuclear complex 2d. The experimental and theoretical patterns were found to be in agreement.
Complexation of bare palladium(II) with ligand L1 at a 1:2 metal-to-ligand ratio
The sample of Pd(NO3)2 used in this work was commercially acquired. A solution containing one equivalent of Pd(NO3)2 in DMSO-d6 was added to a separate solution containing two equivalents of ligand L1 in DMSO-d6. The 1H NMR spectrum of the resulting solution showed formation of a single discrete complex. The reaction was repeated in DMSO and the PdL2-type complex [Pd(L1)2](NO3)2 (3a, Scheme 1c) was isolated from the reaction mixture by precipitation as described in the experimental section. Counter-anion (BF4−, ClO4− and OTf−) variation was also carried out to successfully prepare a series of complexes [Pd(L1)2](BF4)2 (3b), [Pd(L1)2](ClO4)2 (3c), and [Pd(L1)2](OTf)2 (3d). These complexes were prepared by complexation of Pd(Y)2 with the ligand L1 where Y = BF4−, ClO4−, and OTf−. It is important to note that Pd(Y)2 solutions were prepared by reacting PdI2 with AgY and the precipitated AgI was removed by filtration. Following this procedure, the presence of iodide as impurity could not be ruled out but its presence was found to not influence the formation of the targeted complex. In contrast, the presence of chloride remaining as impurity when PdCl2 was reacted with AgY to prepare Pd(Y)2, contaminated Pd(Y)2 and produced upon complexation with L1 complexes 3a–d along with some other products. The choice of PdI2 is on the basis of our previous experience from related cages [23]. The complex 3a was characterized by various NMR techniques (Supporting Information File 1, Figures S27–S31). The 1H NMR spectrum of compound 3a showed a single set of peaks (Figure 3) featured with complexation-induced downfield shifts of protons belonging to the terminal pyridines (Δδ = 0.79, and 0.47 ppm for Ha, and Hb, respectively) as compared to the free ligand L1.
![[1860-5397-15-109-3]](/bjoc/content/figures/1860-5397-15-109-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Partial 1H NMR spectra in DMSO-d6 for (i) L1, (ii) [Pd(L1)2](NO3)2 (3a) and (iii) [(NO3)2@Pd3(L1)4](NO3)4 (4a).
Figure 3: Partial 1H NMR spectra in DMSO-d6 for (i) L1, (ii) [Pd(L1)2](NO3)2 (3a) and (iii) [(NO3)2@Pd3(L1)4]...
The peak position of Hf did not change indicating that the central pyridine is not involved in the complexation. The 1H NMR spectra of compounds 3b, 3c and 3d are very much comparable to that of 3a (Supporting Information File 1, Figure S32). One of the coordination sites of the ligand L1 thus remained unutilized in these mononuclear complexes.
The ESIMS spectrum of compound 3a (Supporting Information File 1, Figure S33) showed isotopic peak patterns at m/z 866.10 and 402.05, respectively, which corresponds to the cationic fragments [3a − NO3]+ and [3a − 2NO3]2+ that are formed due to the loss of one and two units of counter anions from 3a. The experimental and theoretical peak patterns were found to be in agreement. The ESIMS spectrum of 3b is provided in Supporting Information File 1, Figure S34.
Complexation of bare palladium(II) with ligand L1 at a 3:4 metal-to-ligand ratio
A solution containing three equivalents of commercially acquired Pd(NO3)2 in DMSO-d6 was added to the solution containing four equivalents of ligand L1 in DMSO-d6. The 1H NMR spectrum of the resulting solution recorded within 10 min showed a mixture of products. However, a single discrete complex was formed upon heating the solution at 90 °C for 5 min or upon stirring at rt for 20 min. A careful analysis of the 1H NMR spectrum revealed the existence of [Pd(L1)2](NO3)2 (3a) and [(NO3)2@Pd3(L1)4](NO3)4 (4a) in the mixture (Supporting Information File 1, Figure S35a). The mononuclear complex 3a is proposed here as a kinetically controlled product and the trinuclear 4a as the thermodynamic product. The reaction was repeated in DMSO and the Pd3L4-type complex 4a (Scheme 1d) was isolated from the reaction mixture by precipitation as described in the experimental section. Complexation of Pd(Y)2 (prepared from PdI2 and AgY, where Y = BF4−, ClO4−, and OTf−) with the ligand L1 at a 3:4 ratio resulted in the formation of only mononuclear complexes 3b–d (depicted in a later Scheme) and the unutilized proportion of Pd(Y)2 remained in solution. Thus, the counter ion nitrate has a determining role by acting as template for the cavities in the formation of the trinuclear complex 4a. It is proposed that the anion is essential to avoid charge repulsion between the metal centers in the ensuing cavities. However, the anions should be of fitting sizes only to get accommodated in the cavities so that discrete architectures are formed. Larger anions such as BF4−, ClO4− and OTf− are not accommodated in the cavities and not helpful as templates. The Pd(NO3)2 sample prepared from PdCl2 and AgNO3, contained chloride as impurity and resulted in a mixture of products along with the targeted 4a (Supporting Information File 1, Figure S35b). The products in the mixture were identified as [(Cl)(NO3)@Pd3(L1)4](NO3)4 (6a’) and [(Cl)2@Pd3(L1)4](NO3)4 (6a) and their proportion was found to depend on the amount of chloride as impurity. The influence of chloride on the product composition is discussed in a later section.
The complex 4a was characterized by various NMR techniques (Supporting Information File 1, Figures S37–S41). The 1H NMR spectrum of compound 4a showed a single set of peaks (Figure 3) featured with complexation-induced downfield shifts of protons belonging to the terminal as well as to the central pyridine rings (Δδ = 1.42, 0.82, 1.02 ppm for Ha, Hb, and Hf, respectively) as compared to the free ligand L1. The signal of Hf also got downfield-shifted indicating that the central pyridine ring is involved in the complexation.
The ESIMS spectrum of compound 4a confirmed the formation of a trinuclear complex (Supporting Information File 1, Figure S42). Isotopic peak patterns are found at m/z 982.04, 634.03 and 460.03, which correspond to the cationic fragments [4a − 2NO3]2+, [4a − 3NO3]3+ and [4a − 4NO3]4+ that are formed due to the loss of two, three and four units of counter anions from 4a. The experimental and theoretical peak patterns were found to be in agreement.
DFT studies of the complexes
The energy-minimized structures of [Pd(tmeda)(L1)]2+, [Pd3(tmeda)3(L1)2]6+, [Pd(L1)2]2+, and [(NO3)2@Pd3(L1)4]4+ are shown in Figure 4 (see Supporting Information File 1 for details). Geometry optimization and calculation of frequencies were performed using Gaussian 09 software package at the B3LYP/6-31G* level of theory [37]. Since the complex [Pd3(tmeda)3(L1)2]6+ could not be prepared exclusively, we looked into the energetics of the system. The overall Gibbs free energies (∆G) and the enthalpies (∆H) for the formation of the trinuclear complex [Pd3(tmeda)3(L1)2]6+ considering its formation from 1 equivalent of [Pd(tmeda)(NO3)2] and 2 equivalents of [Pd(tmeda)(L1)]2+ were found to be not feasible (616.349 kcal mol−1) and endothermic (+537.727 kcal mol−1), respectively (see Figure S71 and Table S3 in Supporting Information File 1). However, a small amount of the trinuclear complex was formed experimentally. Probably, the counter anions stayed in the hemi-cage part of the trinuclear structure making it somewhat feasible. A detailed investigation of solvent and counter anion might help.
![[1860-5397-15-109-4]](/bjoc/content/figures/1860-5397-15-109-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Energy-minimized structures of (i) [Pd(tmeda)(L1)]2+, (ii) [Pd3(tmeda)3(L1)2]6+, (iii) [Pd(L1)2]2+, and (iv) [(NO3)2@Pd3(L1)4]4+. Hydrogen atoms are omitted for clarity, red, blue, grey and cyan colors represent oxygen, nitrogen, carbon and palladium, respectively.
Figure 4: Energy-minimized structures of (i) [Pd(tmeda)(L1)]2+, (ii) [Pd3(tmeda)3(L1)2]6+, (iii) [Pd(L1)2]2+,...
Complex-to-complex transformations: 3a versus 4a
The in situ prepared mononuclear complex [Pd(L1)2](NO3)2 (3a) was found to be stable at room temperature for days in DMSO-d6 (Supporting Information File 1, Figure S43) but not upon heating. The 1H NMR spectrum recorded after heating the solution at 90 °C for 24 h revealed decomplexation and signals for the free ligand were observed (Supporting Information File 1, Figure S44). In addition, the solution turned dark and dark particles were observed. Upon cooling the solution, the free ligand should have undergone complexation to form 3a. However, no complexation was observed and it is assumed that palladium(II) got reduced to palladium(0). In another experiment, Pd(NO3)2 was added to a solution of 3a at a 2:1 ratio where upon complex-to-complex conversion was observed at room temperature or upon heating to afford 4a (Scheme 2(i)). With the appropriate amount of Pd(NO3)2 a complete formation of 4a was observed within 20 min at rt or 5 min at 90 °C (Supporting Information File 1, Figures S45 and S46).
![[1860-5397-15-109-i2]](/bjoc/content/inline/1860-5397-15-109-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reorganization of (i) a mixture of Pd(NO3)2 and 3a at a 2:1 ratio leading to 4a with a complete conversion and (ii) a mixture of L1 and 4a at a 2:1 ratio leading to 3a but with a partial conversion.
Scheme 2: Reorganization of (i) a mixture of Pd(NO3)2 and 3a at a 2:1 ratio leading to 4a with a complete con...
On the other hand, the in situ prepared trinuclear complex 4a was found to be stable at room temperature as well as at 90 °C for days (Supporting Information File 1, Figures S47 and S48). The free ligand L1 was added to a solution of 4a in DMSO-d6 at room temperature and the sample was monitored by 1H NMR spectroscopy. The calculated amount of ligand was added to the solution of 4a (at 2:1 ratio) to match the stoichiometric requirement for the formation of 3a. Although the formation of 3a was observed, it remained only as a minor product, and the added ligand L1 was partially consumed. Thus, the unbound ligand remained in its free state along with 4a. No further change was observed after 30 min (Supporting Information File 1, Figure S49). Heating of the reaction mixture did not help in further pushing the conversion towards the formation of 3a (Supporting Information File 1, Figure S50). Prolonged heating could not help because the complex 3a is unstable under such conditions. This provided additional support on the higher stability of 4a as compared to 3a.
Halide binding by the cavities of a double-decker cage
The trinuclear complex [(NO3)2@Pd3(L1)4](NO3)4 (4a) was prepared by mixing Pd(NO3)2 with ligand L1 at a 3:4 ratio (Scheme 3(i), also Scheme 1(iv)). Complexation of metal components like Pd(BF4)2, Pd(ClO4)2 or Pd(OTf)2 with L1 at a 3:4 ratio did not afford the analogous trinuclear complexes; rather the corresponding mononuclear complexes were formed and the uncomplexed palladium(II) remained in solution (Scheme 3(ii)).
![[1860-5397-15-109-i3]](/bjoc/content/inline/1860-5397-15-109-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Halide (F−, Cl− and Br− but not I−) encapsulation by the cavities of the double-decker cage.
Scheme 3: Halide (F−, Cl− and Br− but not I−) encapsulation by the cavities of the double-decker cage.
Each of the two cavities of cage 4a is loaded with one NO3–. This phenomenon of NO3– encapsulation by a related isomeric cage was established by us earlier [22,23]. Halide recognition by the complex 4a through anion exchange was studied by portionwise addition of freshly prepared solutions of tetra-n-butylammonium halide, i.e., TBA(X) (where X stands for F−, Cl−, Br− and I−) in four separate experiments using DMSO-d6 as the solvent. The anion exchange processes were monitored by 1H NMR spectroscopy of the samples (Supporting Information File 1, Figures S51–S54). The addition of a portion of TBACl to the complex 4a resulted in a mixture of 4a, [(Cl)(NO3)@Pd3(L1)4](NO3)4 (6a’) and [(Cl)2@Pd3(L1)4](NO3)4 (6a). With further addition of TBACl, the proportion of 6a increased at the cost of 4a and 6a’ to finally yield compound 6a as the only product. Similarly, the addition of TBABr initially produced a mixture of 4a, [(Br)(NO3)@Pd3(L1)4](NO3)4 (7a’) and [(Br)2@Pd3(L1)4](NO3)4 (7a) with complex 7a as the exclusive final product. In the case of TBAF, initially there was no change observable except a slight broadening of the signals for Hb and Hc. Further addition of TBAF led to the formation of [(F)2@Pd3(L1)4](NO3)4 (5a) along with minor impurities. However, the addition of TBAI to 4a showed no changes and hence iodide encapsulation did not happen. The 1H NMR spectra for the mixtures of products formed at intermediate and final stages are provided in Supporting Information File 1 (Figures S51–S54) and those of 4a, 5a, 6a and 7a are shown in Figure 5. The positions of the signals in the 1H NMR spectra of these anion-encapsulated complexes are influenced by coordination of ligand L1 with palladium(II) and interaction of the encapsulated anion with the endohedrally oriented hydrogens of the cages.
![[1860-5397-15-109-5]](/bjoc/content/figures/1860-5397-15-109-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Partial 1H NMR spectra at 400 MHz in DMSO-d6 for (i) [(NO3)2@Pd3(L1)4](NO3)4 (4a), (ii) [(F)2@Pd3(L1)4](NO3)4 (5a), (iii) [(Cl)2@Pd3(L1)4](NO3)4 (6a) and (iv) [(Br)2@Pd3(L1)4](NO3)4 (7a).
Figure 5: Partial 1H NMR spectra at 400 MHz in DMSO-d6 for (i) [(NO3)2@Pd3(L1)4](NO3)4 (4a), (ii) [(F)2@Pd3(L1...
Is iodide not capable of replacing the preexisting nitrate in a competition or iodide is not suited at all for the cavity irrespective of any competition? The following argument might answer this question. The complexation reaction shown in steps (ii) of Scheme 3 suggest that the presence of BF4−, ClO4− or OTf− could not support the formation of the double-decker cage even though the required amount of palladium(II) was available. The addition of TBAI to any of these solutions containing Pd(Y)2 and 3b, 3c or 3d, respectively, did not lead to double-decker cages indicates that I− is not suited for the cavity. However, addition of TBANO3, TBAF, TBACl and TBABr produced the corresponding anion encapsulated double-decker cages as shown in steps (iv), (v) and (vi) of Scheme 3. Representative 1H NMR spectra for the conversion of 3b to corresponding products 4b, 5b, 6b and 7b are shown in Supporting Information File 1 (Figure S55).
The 1H NMR spectral analysis of the two nitrate anions incorporating compound 4a discussed in an earlier section revealed downfield shifts of particular signals as compared to the free ligand L1 and the Δδ values were 1.42, 0.82 and 1.02 ppm for the signals of Ha, Hb, and Hf, respectively. A similar comparison for the (i) two F− encapsulated compound 5a: Δδ = 1.75, 0.99 and 1.78 ppm for Ha, Hb, and Hf, respectively), (ii) the two Cl− encapsulated compound 6a: (Δδ = 1.91, 1.01 and 1.69 ppm for Ha, Hb and Hf, respectively), and (iii) the two Br− encapsulated compound 7a: (Δδ = 2.24, 1.02 and 1.74 ppm for Ha, Hb and Hf, respectively) are in line with the expectation. Although fluoride (F–) could replace NO3– in 4a to afford complex 5a, the complex 5a was found to be unstable and it decomposed within a few hours. Thus the 1H NMR and ESIMS spectrum of 5a were recorded from freshly prepared samples. The complexes 4a, 6a and 7a are quite stable and no decomposition was observed. Detailed characterization data of 4a–7a form a variety of NMR techniques are provided in Supporting Information File 1, Figures S56–S66.
The molecular compositions of the halide encapsulated complexes were confirmed by recording ESIMS data for the systems. The double halide encapsulated complexes 6a and 7a were detected. Also, one of the mixed halide–nitrate encapsulated complexes, i.e., 7a’ was also detected.
The ESIMS spectrum of compound 6a (Supporting Information File 1, Figure S67) showed isotopic peak pattern at m/z = 956.02 which corresponds to the cationic fragment [6a − 2NO3]2+ that was formed due to the loss of two counter anions from 6a. The ESIMS spectrum of compound 7a (Supporting Information File 1, Figure S68) showed isotopic peak patterns at m/z = 645.99 and at 468.99 which correspond to the cationic fragments [7a − 3NO3]3+ and [7a − 4NO3]4+ that are formed due to the loss of three and four units of counter anions from 7a. The ESIMS spectrum of compound 7a’ (Supporting Information File 1, Figure S69) showed isotopic peak pattern at m/z = 991.51 which corresponds to the cationic fragment [7a’ − 2NO3]2+ that is formed due to the loss of two units of counter anions from 7a’. The experimental and theoretical peak patterns were found to be in agreement.
Coordination complexes of L1 versus L2: ligand isomerism phenomenon
As discussed in the introduction section “The definition of ligand isomerism includes metal complexes (at least two) having the same molecular formula but are composed of different structural isomers of the ligand.” The complexes prepared in this work namely [Pd(tmeda)(L1)](NO3)2 (1a), [Pd(L1)2](NO3)2 (3a), [(NO3)2@Pd3(L1)4](NO3)4 (4a), and [(X)2@Pd3(L1)4](NO3)4 5a–7a fulfill the definition of ligand isomerism when compared with the reported complexes [Pd(tmeda)(L2)](NO3)2 (8a), [Pd(L2)2](NO3)2 (10a), [(NO3)2@Pd3(L2)4](NO3)4 (11a), and [(X)2@Pd3(L2)4](NO3)4 12a–14a, respectively. We have demonstrated ligand isomerism in Pd2L4-type cages [29]. The present work demonstrates ligand isomerism in some complexes and more interestingly for the Pd3L4-type double-decker coordination cages for the first time.
Palladium(II)-based self-assembled complexes of ligands L1 and L2: a comparison
The complexation behavior of L1 and L2 are broadly comparable. However, a closer look revealed certain differences. While complexation of cis-Pd(tmeda)(NO3)2 with L1 produced the trinuclear complex [Pd3(tmeda)3(L1)2](NO3)6 (2a), the ligand L2 did not afford the targeted [Pd3(tmeda)3(L1)2](NO3)6 (9a). The complex [Pd(L1)2](NO3)2 (3a) was unstable when heated in DMSO medium whereas the corresponding complex [Pd(L2)2](NO3)2 (10a) was stable under comparable conditions. The addition of two equivalents of L1 to a solution of [(NO3)2@Pd3(L1)4](NO3)4 (4a) produced only a small amount of [Pd(L1)2](NO3)2 (3a) and the added ligand remained in solution. On the other hand, addition of the required amount of L2 to a solution of [(NO3)2@Pd3(L2)4](NO3)4 (11a) resulted in complete transformation to complex Pd(L2)2](NO3)2 (10a). The F− encapsulated complex [(F)2@Pd3(L1)4](NO3)4 (5a) decomposed within a few hours whereas [(F)2@Pd3(L2)4](NO3)4 (12a) was stable for a few hours. These differences are ascribed to the positional exchanged functionalities in the ligands L1 and L2. Probably, the coordination ability of the central pyridine ring is better than that of the terminal pyridine rings in case of L1. However, in the mononuclear complexes of L1 the central pyridine remained uncomplexed, which may be due to the formation of metallomacrocyclic rings. This behavior is not observed in the case of mononuclear complexes of L2. Thus, the mononuclear complexes of L1 are reluctant to form (e.g., from 4a and L1) and prone to decomposition. As far as trinuclear complex formation is concerned the central pyridine ring of L1 is in a relatively favorable situation, thus the complex 4a could form and 3a was a kinetic product.
Conclusion
A set of mononuclear and trinuclear complexes were prepared through complexation of cis-protected palladium(II) and bare palladium(II) components with the new tridentate ligand L1. A variety of counter anions were employed to broaden the scope of the choice of metal components. Mononuclear complexes with PdL’L composition could be prepared easily, however, Pd3L’3L2-type trinuclear complexes were obtained in only small amounts. Also, mononuclear complexes of PdL2 and trinuclear complexes of Pd3L4-type compositions were prepared easily. The choice of the counter anion did not influence the formation of mononuclear complexes whereas the counter anion displayed a template role for the formation of trinuclear complexes, especially for Pd3L4-type complexes. The anions helped to screen the charge repulsion between the palladium(II) ions. The complexation behavior of palladium(II) components with the ligand L2 have been reported earlier [23]. The similarities and differences in the complexation behaviors of the ligands L1 and L2 were highlighted. A qualitative comparison indicated that ligands L1 and L2 are well suited for the formation of trinuclear only and mononuclear/trinuclear complexes, respectively. The Pd3L’3L2-type complexes could be prepared, though in small proportions, using ligand L1 but not L2. The ligands L1 and L2 are positional isomers (regioisomers) hence many of their complexes could be rightfully considered under ligand isomerism in coordination complexes.
Experimental
Synthesis of ligand L1: A mixture of pyridine-3,5-diyldimethanol (282.6 mg, 2.03 mmol) and nicotinoyl chloride hydrochloride (500.0 mg, 4.06 mmol) in dry DCM (50 mL) was placed in a 100 mL round-bottomed flask. The flask was placed in an ice bath to cool the mixture followed by the dropwise addition of triethylamine (2 mL). Then, the reaction mixture was stirred at room temperature for 24 h followed by the addition of a saturated aqueous solution of sodium bicarbonate. The organic layer was separated and the solvent was evaporated using a rotavapor. The crude product was purified by column chromatography using EtOAc/hexane 8:2 to afford the product as white solid (507.3 mg, isolated yield 71%) after evaporation of the solvent and drying under vacuum. Mp 124 °C; 1H NMR (500 MHz, DMSO-d6, 300 K) δ 9.13 (dd, J1 = 2.8 Hz, J2 = 1.5 Hz, 1H, Ha), 8.83 (dd, J1 = 6.5 Hz, J2 = 3.2 Hz, 1H, Hb), 8.72 (d, J = 2.0 Hz, 1H, Hf), 8.34–8.32 (m, 1H, Hd), 8.09 (t, J = 2.0 Hz, 1H, Hg), 7.58–7.55 (m, 1H, Hc), 5.47 (s, 2H, He); 13C NMR (100 MHz, DMSO-d6, 300 K) δ 164.59, 153.88, 150.12, 149.10, 137.01, 135.62, 131.46, 125.37, 123.97, 64.13; ESIMS (m/z): 372.098 [M + Na]+.
[Pd(tmeda)(L1)](NO3)2 (1a): To a solution of cis-Pd(tmeda)(NO3)2 (10.3 mg, 0.03 mmol), in 3 mL of DMSO ligand L1 (10.5 mg, 0.03 mmol) was added. The reaction mixture was stirred at room temperature for 10 min to obtain a clear yellow solution. The product was precipitated by the addition of ethyl acetate (10 mL), separated by centrifugation, washed with acetone (4 mL) and dried under vacuum to afford complex 1a (16.8 mg, isolated yield 80%). 1H NMR (500 MHz, DMSO-d6, 300 K) δ 9.90 (d, J = 1.5 Hz, 1H, Ha), 9.35 (dd, J1 = 1.0 Hz, J2 = 5.8 Hz, 1H, Hb), 8.67 (s, 1H, Hf), 8.54–8.52 (m, 1H, Hd), 8.18 (s, 1H, Hg), 7.85 (dd, J1 = 7.9 Hz, J2 = 5.8 Hz, 1H, Hc), 5.91–5.29 (dd, J1 = 13.8 Hz, J2 = 13.8 Hz, 2H, He); 13C NMR (125 MHz, DMSO-d6, 300 K) δ 164.50, 154.60, 151.91, 147.21, 140.92, 133.03, 132.24, 129.41, 127.31, 63.41, 62.31.
[Pd(L1)2](NO3)2 (3a): To a solution of ligand L1 (20.9 mg, 0.06 mmol) in 3 mL of DMSO, Pd(NO3)2 (6.9 mg, 0.03 mmol) was added and the reaction mixture was stirred for 10 min at room temperature to give a clear yellow solution. The product was precipitated by the addition of 10 mL of ethyl acetate. The pale yellow precipitate was separated by centrifugation, washed with acetone and dried under vacuum to afford complex 3a (18.6 mg, isolated yield 66%). 1H NMR (500 MHz, DMSO-d6, 300 K) δ 9.92 (s, 1H, Ha), 9.30 (d, J = 5.3 Hz, 1H, Hb), 8.75 (d, J = 1.3 Hz, 1H, Hf), 8.55–8.53 (m, 1H, Hd), 8.31 (s, 1H, Hg), 7.82 (dd, J1 = 7.9 Hz, J2 = 5.8 Hz, 1H, Hc), 5.63 (s, 1H, He); 13C NMR (125 MHz, DMSO-d6, 300 K) δ 162.43, 154.57, 151.59, 147.83, 141.30, 133.97, 132.31 129.57, 127.38, 63.86; ESIMS (m/z): 866.10 [3a − 1NO3]+; 402.05 [3a − 2NO3]2+.
[(NO3)2@Pd3(L1)4](NO3)4 (4a): To a solution of ligand L1 (14.0 mg, 0.04 mmol) in 2 mL of DMSO, Pd(NO3)2 (7.0 mg, 0.03 mmol) in 1 mL of DMSO was added and the reaction mixture was stirred for 5 min at 90 °C to give a clear yellow solution. The product was precipitated by the addition of 10 mL of ethyl acetate. The pale yellow precipitate was separated by centrifugation, washed with acetone and dried under vacuum to afford the complex 4a (15.6 mg, isolated yield 75%). 1H NMR (500 MHz, DMSO-d6, 300 K) δ 10.54 (d, J = 1.4 Hz, 1H, Ha), 9.74 (s, 1H, Hf), 9.66 (d, J = 5.0 Hz, 1H, Hb), 8.58 (d, J = 8.0 Hz, 1H, Hd), 8.26 (s, 1H, Hg), 7.95–7.93 (m, 1H, Hc), 5.45 (s, 1H, He); 13C NMR (125 MHz, DMSO-d6, 300 K) δ 162.40, 155.05, 153.19, 149.91, 141.88, 138.39, 133.83, 128.66, 127.68, 64.84; ESIMS (m/z): 982.04 [4a − 2NO3]2+; 634.03 [4a − 3NO3]3+; 460.03 [4a − 4NO3]4+.
Supporting Information
| Supporting Information File 1: Experimental procedures, NMR, ESIMS data, and theoretical study. | ||
| Format: PDF | Size: 8.4 MB | Download |
Acknowledgements
D. K. C. thanks the Science and Engineering Research Board (SERB), the Department of Science and Technology, and the Government of India (Project No. EMR/2017/002262) for financial support. S. S. and B. S. R. thank CSIR and UGC, India for research fellowships, respectively. We thank IIT Madras for infrastructure and facilities.
References
-
Cook, T. R.; Stang, P. J. Chem. Rev. 2015, 115, 7001–7045. doi:10.1021/cr5005666
Return to citation in text: [1] -
Fujita, M.; Ogura, K. Bull. Chem. Soc. Jpn. 1996, 69, 1471–1482. doi:10.1246/bcsj.69.1471
Return to citation in text: [1] -
Fujita, M. Chem. Soc. Rev. 1998, 27, 417–425. doi:10.1039/a827417z
Return to citation in text: [1] -
Chambron, J.-C.; Sauvage, J.-P. New J. Chem. 2013, 37, 49–57. doi:10.1039/c2nj40555e
Return to citation in text: [1] -
Debata, N. B.; Tripathy, D.; Chand, D. K. Coord. Chem. Rev. 2012, 256, 1831–1945. doi:10.1016/j.ccr.2012.04.001
Return to citation in text: [1] [2] [3] -
Schmidt, A.; Casini, A.; Kühn, F. E. Coord. Chem. Rev. 2014, 275, 19–36. doi:10.1016/j.ccr.2014.03.037
Return to citation in text: [1] -
Chand, D. K.; Biradha, K.; Fujita, M. Chem. Commun. 2001, 1652–1653. doi:10.1039/b104853h
Return to citation in text: [1] -
Clever, G. H.; Tashiro, S.; Shionoya, M. Angew. Chem., Int. Ed. 2009, 48, 7010–7012. doi:10.1002/anie.200902717
Return to citation in text: [1] -
Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373
Return to citation in text: [1] -
Zhu, R.; Lübben, J.; Dittrich, B.; Clever, G. H. Angew. Chem., Int. Ed. 2015, 54, 2796–2800. doi:10.1002/anie.201408068
Return to citation in text: [1] -
Clever, G. H.; Shionoya, M. Chem. – Eur. J. 2010, 16, 11792–11796. doi:10.1002/chem.201002013
Return to citation in text: [1] -
Kishi, N.; Li, Z.; Yoza, K.; Akita, M.; Yoshizawa, M. J. Am. Chem. Soc. 2011, 133, 11438–11441. doi:10.1021/ja2037029
Return to citation in text: [1] -
Yamashina, M.; Yuki, T.; Sei, Y.; Akita, M.; Yoshizawa, M. Chem. – Eur. J. 2015, 21, 4200–4204. doi:10.1002/chem.201406445
Return to citation in text: [1] -
Liao, P.; Langloss, B. W.; Johnson, A. M.; Knudsen, E. R.; Tham, F. S.; Julian, R. R.; Hooley, R. J. Chem. Commun. 2010, 46, 4932–4934. doi:10.1039/c0cc00234h
Return to citation in text: [1] -
Kishi, N.; Li, Z.; Sei, Y.; Akita, M.; Yoza, K.; Siegel, J. S.; Yoshizawa, M. Chem. – Eur. J. 2013, 19, 6313–6320. doi:10.1002/chem.201204010
Return to citation in text: [1] -
Bandi, S.; Chand, D. K. Chem. – Eur. J. 2016, 22, 10330–10335. doi:10.1002/chem.201602039
Return to citation in text: [1] -
Yamashina, M.; Sei, Y.; Akita, M.; Yoshizawa, M. Nat. Commun. 2014, 5, No. 4662. doi:10.1038/ncomms5662
Return to citation in text: [1] -
Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/c2sc00899h
Return to citation in text: [1] -
Ganta, S.; Chand, D. K. Inorg. Chem. 2018, 57, 3634–3645. doi:10.1021/acs.inorgchem.7b02239
Return to citation in text: [1] -
McMorran, D. A.; Steel, P. J. Angew. Chem., Int. Ed. 1998, 37, 3295–3297. doi:10.1002/(sici)1521-3773(19981217)37:23<3295::aid-anie3295>3.0.co;2-5
Return to citation in text: [1] -
McMorran, D. A.; Steel, P. J. Supramol. Chem. 2002, 14, 79–85. doi:10.1080/10610270290006600
Return to citation in text: [1] -
Bandi, S.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Chem. – Eur. J. 2014, 20, 13122–13126. doi:10.1002/chem.201403808
Return to citation in text: [1] [2] [3] [4] -
Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Vasdev, R. A. S.; Preston, D.; Crowley, J. D. Chem. – Asian J. 2017, 12, 2513–2523. doi:10.1002/asia.201700948
Return to citation in text: [1] -
Preston, D.; Lewis, J. E. M.; Crowley, J. D. J. Am. Chem. Soc. 2017, 139, 2379–2386. doi:10.1021/jacs.6b11982
Return to citation in text: [1] -
Zhu, R.; Regeni, I.; Holstein, J. J.; Dittrich, B.; Simon, M.; Prévost, S.; Gradzielski, M.; Clever, G. H. Angew. Chem., Int. Ed. 2018, 57, 13652–13656. doi:10.1002/anie.201806047
Return to citation in text: [1] -
Fujita, M.; Yu, S.-Y.; Kusukawa, T.; Funaki, H.; Ogura, K.; Yamaguchi, K. Angew. Chem., Int. Ed. 1998, 37, 2082–2085. doi:10.1002/(sici)1521-3773(19980817)37:15<2082::aid-anie2082>3.0.co;2-0
Return to citation in text: [1] -
Fujita, M.; Oguro, D.; Miyazawa, M.; Oka, H.; Yamaguchi, K.; Ogura, K. Nature 1995, 378, 469–471. doi:10.1038/378469a0
Return to citation in text: [1] -
Qin, Z.; Jennings, M. C.; Puddephatt, R. J. Inorg. Chem. 2003, 42, 1956–1965. doi:10.1021/ic020322z
Return to citation in text: [1] [2] -
Troff, R. W.; Hovorka, R.; Weilandt, T.; Lützen, A.; Cetina, M.; Nieger, M.; Lentz, D.; Rissanen, K.; Schalley, C. A. Dalton Trans. 2012, 41, 8410–8420. doi:10.1039/c2dt30190c
Return to citation in text: [1] -
Harris, K.; Sun, Q.-F.; Sato, S.; Fujita, M. J. Am. Chem. Soc. 2013, 135, 12497–12499. doi:10.1021/ja4043609
Return to citation in text: [1] -
Yu, H.-J.; Liu, Z.-M.; Pan, M.; Wu, K.; Wei, Z.-W.; Xu, Y.-W.; Fan, Y.-N.; Wang, H.-P.; Su, C.-Y. Eur. J. Inorg. Chem. 2018, 80–85. doi:10.1002/ejic.201701319
Return to citation in text: [1] -
Preston, D.; McNeill, S. M.; Lewis, J. E. M.; Giles, G. I.; Crowley, J. D. Dalton Trans. 2016, 45, 8050–8060. doi:10.1039/c6dt00133e
Return to citation in text: [1] -
Dasary, H.; Jagan, R.; Chand, D. K. Inorg. Chem. 2018, 57, 12222–12231. doi:10.1021/acs.inorgchem.8b01884
Return to citation in text: [1] [2] -
Mateescu, M.; Nuss, I.; Southan, A.; Messenger, H.; Wegner, S. V.; Kupka, J.; Bach, M.; Tovar, G. M. E.; Boehm, H.; Laschat, S. Synthesis 2014, 46, 1243–1253. doi:10.1055/s-0033-1338614
Return to citation in text: [1] -
Dasary, H.; Jagan, R.; Chand, D. K. Chem. – Eur. J. 2015, 21, 1499–1507. doi:10.1002/chem.201405255
Return to citation in text: [1] -
Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1]
| 1. | Cook, T. R.; Stang, P. J. Chem. Rev. 2015, 115, 7001–7045. doi:10.1021/cr5005666 |
| 2. | Fujita, M.; Ogura, K. Bull. Chem. Soc. Jpn. 1996, 69, 1471–1482. doi:10.1246/bcsj.69.1471 |
| 3. | Fujita, M. Chem. Soc. Rev. 1998, 27, 417–425. doi:10.1039/a827417z |
| 4. | Chambron, J.-C.; Sauvage, J.-P. New J. Chem. 2013, 37, 49–57. doi:10.1039/c2nj40555e |
| 5. | Debata, N. B.; Tripathy, D.; Chand, D. K. Coord. Chem. Rev. 2012, 256, 1831–1945. doi:10.1016/j.ccr.2012.04.001 |
| 12. | Kishi, N.; Li, Z.; Yoza, K.; Akita, M.; Yoshizawa, M. J. Am. Chem. Soc. 2011, 133, 11438–11441. doi:10.1021/ja2037029 |
| 13. | Yamashina, M.; Yuki, T.; Sei, Y.; Akita, M.; Yoshizawa, M. Chem. – Eur. J. 2015, 21, 4200–4204. doi:10.1002/chem.201406445 |
| 14. | Liao, P.; Langloss, B. W.; Johnson, A. M.; Knudsen, E. R.; Tham, F. S.; Julian, R. R.; Hooley, R. J. Chem. Commun. 2010, 46, 4932–4934. doi:10.1039/c0cc00234h |
| 15. | Kishi, N.; Li, Z.; Sei, Y.; Akita, M.; Yoza, K.; Siegel, J. S.; Yoshizawa, M. Chem. – Eur. J. 2013, 19, 6313–6320. doi:10.1002/chem.201204010 |
| 16. | Bandi, S.; Chand, D. K. Chem. – Eur. J. 2016, 22, 10330–10335. doi:10.1002/chem.201602039 |
| 34. | Dasary, H.; Jagan, R.; Chand, D. K. Inorg. Chem. 2018, 57, 12222–12231. doi:10.1021/acs.inorgchem.8b01884 |
| 7. | Chand, D. K.; Biradha, K.; Fujita, M. Chem. Commun. 2001, 1652–1653. doi:10.1039/b104853h |
| 8. | Clever, G. H.; Tashiro, S.; Shionoya, M. Angew. Chem., Int. Ed. 2009, 48, 7010–7012. doi:10.1002/anie.200902717 |
| 9. | Han, M.; Michel, R.; He, B.; Chen, Y.-S.; Stalke, D.; John, M.; Clever, G. H. Angew. Chem., Int. Ed. 2013, 52, 1319–1323. doi:10.1002/anie.201207373 |
| 10. | Zhu, R.; Lübben, J.; Dittrich, B.; Clever, G. H. Angew. Chem., Int. Ed. 2015, 54, 2796–2800. doi:10.1002/anie.201408068 |
| 11. | Clever, G. H.; Shionoya, M. Chem. – Eur. J. 2010, 16, 11792–11796. doi:10.1002/chem.201002013 |
| 22. | Bandi, S.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Chem. – Eur. J. 2014, 20, 13122–13126. doi:10.1002/chem.201403808 |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
| 5. | Debata, N. B.; Tripathy, D.; Chand, D. K. Coord. Chem. Rev. 2012, 256, 1831–1945. doi:10.1016/j.ccr.2012.04.001 |
| 6. | Schmidt, A.; Casini, A.; Kühn, F. E. Coord. Chem. Rev. 2014, 275, 19–36. doi:10.1016/j.ccr.2014.03.037 |
| 26. | Zhu, R.; Regeni, I.; Holstein, J. J.; Dittrich, B.; Simon, M.; Prévost, S.; Gradzielski, M.; Clever, G. H. Angew. Chem., Int. Ed. 2018, 57, 13652–13656. doi:10.1002/anie.201806047 |
| 5. | Debata, N. B.; Tripathy, D.; Chand, D. K. Coord. Chem. Rev. 2012, 256, 1831–1945. doi:10.1016/j.ccr.2012.04.001 |
| 27. | Fujita, M.; Yu, S.-Y.; Kusukawa, T.; Funaki, H.; Ogura, K.; Yamaguchi, K. Angew. Chem., Int. Ed. 1998, 37, 2082–2085. doi:10.1002/(sici)1521-3773(19980817)37:15<2082::aid-anie2082>3.0.co;2-0 |
| 28. | Fujita, M.; Oguro, D.; Miyazawa, M.; Oka, H.; Yamaguchi, K.; Ogura, K. Nature 1995, 378, 469–471. doi:10.1038/378469a0 |
| 29. | Qin, Z.; Jennings, M. C.; Puddephatt, R. J. Inorg. Chem. 2003, 42, 1956–1965. doi:10.1021/ic020322z |
| 30. | Troff, R. W.; Hovorka, R.; Weilandt, T.; Lützen, A.; Cetina, M.; Nieger, M.; Lentz, D.; Rissanen, K.; Schalley, C. A. Dalton Trans. 2012, 41, 8410–8420. doi:10.1039/c2dt30190c |
| 31. | Harris, K.; Sun, Q.-F.; Sato, S.; Fujita, M. J. Am. Chem. Soc. 2013, 135, 12497–12499. doi:10.1021/ja4043609 |
| 32. | Yu, H.-J.; Liu, Z.-M.; Pan, M.; Wu, K.; Wei, Z.-W.; Xu, Y.-W.; Fan, Y.-N.; Wang, H.-P.; Su, C.-Y. Eur. J. Inorg. Chem. 2018, 80–85. doi:10.1002/ejic.201701319 |
| 33. | Preston, D.; McNeill, S. M.; Lewis, J. E. M.; Giles, G. I.; Crowley, J. D. Dalton Trans. 2016, 45, 8050–8060. doi:10.1039/c6dt00133e |
| 34. | Dasary, H.; Jagan, R.; Chand, D. K. Inorg. Chem. 2018, 57, 12222–12231. doi:10.1021/acs.inorgchem.8b01884 |
| 21. | McMorran, D. A.; Steel, P. J. Supramol. Chem. 2002, 14, 79–85. doi:10.1080/10610270290006600 |
| 24. | Vasdev, R. A. S.; Preston, D.; Crowley, J. D. Chem. – Asian J. 2017, 12, 2513–2523. doi:10.1002/asia.201700948 |
| 20. | McMorran, D. A.; Steel, P. J. Angew. Chem., Int. Ed. 1998, 37, 3295–3297. doi:10.1002/(sici)1521-3773(19981217)37:23<3295::aid-anie3295>3.0.co;2-5 |
| 25. | Preston, D.; Lewis, J. E. M.; Crowley, J. D. J. Am. Chem. Soc. 2017, 139, 2379–2386. doi:10.1021/jacs.6b11982 |
| 18. | Lewis, J. E. M.; Gavey, E. L.; Cameron, S. A.; Crowley, J. D. Chem. Sci. 2012, 3, 778–784. doi:10.1039/c2sc00899h |
| 19. | Ganta, S.; Chand, D. K. Inorg. Chem. 2018, 57, 3634–3645. doi:10.1021/acs.inorgchem.7b02239 |
| 17. | Yamashina, M.; Sei, Y.; Akita, M.; Yoshizawa, M. Nat. Commun. 2014, 5, No. 4662. doi:10.1038/ncomms5662 |
| 22. | Bandi, S.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Chem. – Eur. J. 2014, 20, 13122–13126. doi:10.1002/chem.201403808 |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
| 36. | Dasary, H.; Jagan, R.; Chand, D. K. Chem. – Eur. J. 2015, 21, 1499–1507. doi:10.1002/chem.201405255 |
| 22. | Bandi, S.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Chem. – Eur. J. 2014, 20, 13122–13126. doi:10.1002/chem.201403808 |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
| 35. | Mateescu, M.; Nuss, I.; Southan, A.; Messenger, H.; Wegner, S. V.; Kupka, J.; Bach, M.; Tovar, G. M. E.; Boehm, H.; Laschat, S. Synthesis 2014, 46, 1243–1253. doi:10.1055/s-0033-1338614 |
| 29. | Qin, Z.; Jennings, M. C.; Puddephatt, R. J. Inorg. Chem. 2003, 42, 1956–1965. doi:10.1021/ic020322z |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
| 22. | Bandi, S.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Chem. – Eur. J. 2014, 20, 13122–13126. doi:10.1002/chem.201403808 |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
| 23. | Bandi, S.; Samantray, S.; Chakravarthy, R. D.; Pal, A. K.; Hanan, G. S.; Chand, D. K. Eur. J. Inorg. Chem. 2016, 2816–2827. doi:10.1002/ejic.201600259 |
© 2019 Samantray et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
