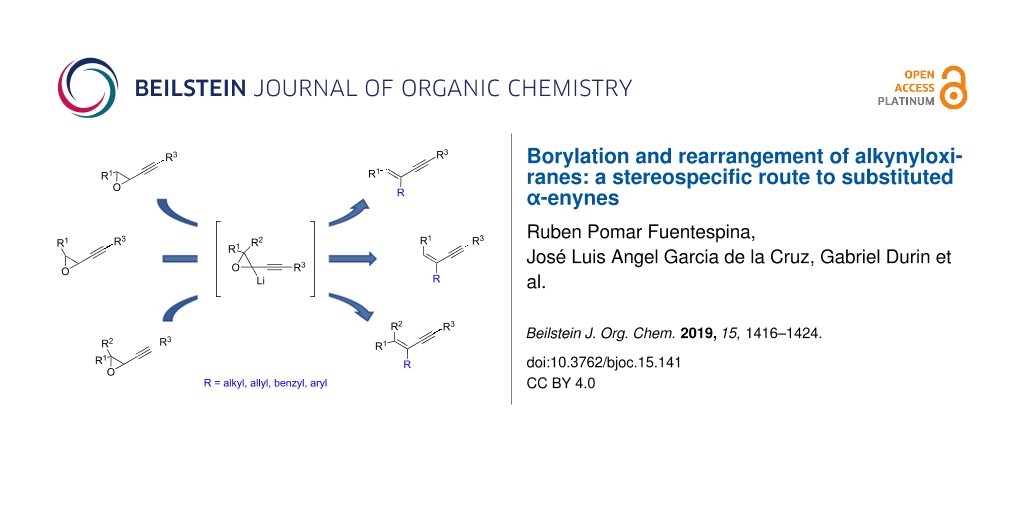
Abstract
1,3-Enynes are important building blocks in organic synthesis and also constitute the key motif in various bioactive natural products and functional materials. However, synthetic approaches to stereodefined substituted 1,3-enynes remain a challenge, as they are limited to Wittig and cross-coupling reactions. Herein, stereodefined 1,3-enynes, including tetrasubstituted ones, were straightforwardly synthesized from cis or trans-alkynylated oxiranes in good to excellent yields by a one-pot cascade process. The procedure relies on oxirane deprotonation, borylation and a stereospecific rearrangement of the so-formed alkynyloxiranyl borates. This stereospecific process overall transfers the cis or trans-stereochemistry of the starting alkynyloxiranes to the resulting 1,3-enynes.

Graphical Abstract
Introduction
Lithiated oxiranes are useful intermediates in organic synthesis [1,2]. Such species can undergo different reactions, driven either by their carbanionic or carbenic behavior [3,4], thus leading to various products. In contrast, the transmetalation of lithiated oxiranes and the evolution of the resulting species have been less explored. Utimoto’s group was probably the first who reported on the treatment of oxiranyllithium with diethylzinc or trimethylaluminium [5]. Shimizu et al. demonstrated that in situ formed trifluoromethyloxiranyllithium derivatives reacted with trimethylaluminium, dimethylzinc or organoboranes to yield tetrasubstituted alkenes [6,7]. Capitalizing on these precedents, Aggarwal et al. showed that oxiranes could be homologated to diols through lithiation, borylation, rearrangement and oxidation of the so-formed β-hydroxyboranes [8]. More recently, Blakemore et al. applied this sequence to sulfinyloxiranes [9]. Alternatively, Unemaya et al. and more recently Buchwald et al. described a Negishi cross coupling of aryl bromides or chlorides to α-CF3-oxiranyl zincates generated by lithiation of trifluoromethyloxirane and transmetalation with zinc chloride [10,11].
In this context and based on the pioneering work of Shimizu [6,7], and our experience on the metalation of alkynyloxiranes and reactivity studies of the so-formed alkynyloxirane anions [12,13], we explored the reaction of such anions with various boron derivatives in order to produce stereodefined α-enynes after Matteson-type rearrangement [14] and elimination [15] (Scheme 1). We also demonstrated the stereospecificity of these transformations [16].
![[1860-5397-15-141-i1]](/bjoc/content/inline/1860-5397-15-141-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Stereospecific formation of α-enynes from alkynyloxiranes.
Scheme 1: Stereospecific formation of α-enynes from alkynyloxiranes.
α-Enynes (1,3-enynes) are an interesting class of compounds in organic chemistry as these structures are found in various pharmacologically active natural products [17], and in functional materials [18]. α-Enynes are also important building blocks in organic syntheses or platforms for further synthesis [19]. Accordingly, significant efforts have been made to develop efficient syntheses of enynes [17-20]. The most common routes relied on Wittig-type reactions [21,22] and transition-metal-catalyzed coupling reactions of either terminal or organometallic alkynes with vinyl halides or of alkynyl halides with vinylic organometallics [20]. However, both routes may lead to mixtures of stereoisomers and synthetic approaches to stereodefined substituted α-enynes remain scarce and thus still represent a challenge in organic chemistry.
Results and Discussion
Reaction set up
First we selected the simple 2-ethynyl-3-dimethyloxirane 1a to probe the feasibility of the reaction. In order to avoid unwanted reactions upon metalation [23], the ethynyl group was silylated and a bulky silyl group was installed to avoid any silyl migration [12]. As already shown [12,13], n-butyllithium proved to be very efficient, although stronger or complex bases are usually required to produce oxiranyl anions [1,2,24-28]. Upon deprotonation, the corresponding oxiranyllithium was sufficiently stable at −78 °C (see below) to be trapped by various phenylboronic esters 2. The resulting borate intermediate afforded the tetrasubstituted enyne 3 in good to moderate yields, depending on the boronic ester’s nature (Table 1). The catechol ester 2a only provided a modest yield of 3 (Table 1, entry 1), while the neopentylglycol ester 2b proved slightly better (Table 1, entry 2). The pinacol ester 2c turned out to be the most effective reagent for this transformation, giving around 70% of 3 after isolation and purification (Table 1, entry 3). These results showed that the steric bulk at the boron atom seems to facilitate the reaction. Indeed, the bulkier the boron substituent is, the more efficient is the process. Therefore, the substitution at the boron plays a key role, most probably at the boron rearrangement step (see below).
Table 1: Borylation and rearrangement of ethynyloxirane 1a.a,b.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-141-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Boronic ester |
Timec
(min) |
Yieldd
(%) |
|
| t1 | t2 | |||
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-141-i5.svg?max-width=637&scale=1.0)
2a |
30 | 20 | 33 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-141-i6.svg?max-width=637&scale=1.0)
2b |
30 | 20 | 43 |
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-141-i7.svg?max-width=637&scale=1.0)
2c |
30 | 20 | 66 |
aReaction conditions: 1) 1.5 equiv n-BuLi, THF, −78 °C, t1; 2) 1.5 equiv boronic ester 2, −78 °C, t2; 3) rt, 1 h; bTIPS: triisopropylsilyl); ct1 corresponds to the time of the lithiation step and t2 to the time of the borylation step; dyields of pure isolated products, unless otherwise stated.
Stereoselectivity, scope and limitations
Next, the stereoselectivity of this rearrangement was evaluated using cis and trans-disubstituted alkynyloxiranes (Table 2). For synthetic reasons [29,30], we selected cis-2,3-epoxy-5-triisopropylsilylpent-4-ynol, protected with the monomethoxytrityl group (MMTr) 1b and prepared its trans-isomer to compare the outcome of the reaction with both isomers.
Table 2: Lithiation, borylation and rearrangement of cis or trans-disubstituted ethynyloxiranes 1b.a,b.
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-141-i8.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Oxirane | R |
Temp.
(°C) |
Timec
(min) |
Enyne |
Yieldd
(%) |
|
| t1 | t2 | ||||||
| 1 | cis-1b | Ph | −78 | 30 | 20 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-141-i9.svg?max-width=637&scale=1.0)
(Z)-4 |
78 |
| 2 | trans-1b | " | −78 | 30 | 20 | (E)-4 + (Z)-4 4:1 | 80 |
| 3 | trans-1b | " | −92 | 60 | 90 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-141-i10.svg?max-width=637&scale=1.0)
(E)-4 |
70 |
| 4 | cis-1b | p-MeOPh | −78 | 30 | 20 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-141-i11.svg?max-width=637&scale=1.0)
(Z)-5 |
85 |
| 5 | trans-1b | " | −92 | 60 | 90 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-141-i12.svg?max-width=637&scale=1.0)
(E)-5 |
88 |
| 6 | cis-1b | p-ClPh | −78 | 30 | 20 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-141-i13.svg?max-width=637&scale=1.0)
(Z)-6 |
81 |
| 7 | trans-1b | " | −92 | 60 | 90 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-141-i14.svg?max-width=637&scale=1.0)
(E)-6 |
85 |
| 8 | cis-1b | Bn | −78 | 30 | 20 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-141-i15.svg?max-width=637&scale=1.0)
(Z)-7 |
62 |
| 9 | trans-1b | " | −92 | 60 | 90 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-141-i16.svg?max-width=637&scale=1.0)
(E)-7 |
65 |
| 10 | cis-1b | allyl | −78 | 30 | 20 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-141-i17.svg?max-width=637&scale=1.0)
(Z)-8 |
57 |
| 11 | trans-1b | " | −92 | 60 | 90 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-141-i18.svg?max-width=637&scale=1.0)
(E)-8 |
78 |
| 12 | cis-1b | n-butyl | −78 | 30 | 20 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-141-i19.svg?max-width=637&scale=1.0)
(Z)-9 |
85 |
| 13 | trans-1b | " | −92 | 60 | 90 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-141-i20.svg?max-width=637&scale=1.0)
(E)-9 |
90 |
aReactions were run under argon, c = 0.125 mol/L; bMMTr: monomethoxytrityl; ct1 corresponds to the time of the lithiation step and t2 to the time of the borylation step; dyields of pure isolated products, unless otherwise stated.
The cis-configured compound cis-1b readily reacted with pinacol phenylboronate 2c under the conditions set up above, providing enyne 4 as the sole product in high yield (Table 2, entry 1). NMR analysis revealed the Z-stereochemistry of this enyne [31]. In contrast, the trans-isomer trans-1b afforded a mixture of the 2 possible isomers, although in similar overall yield (Table 2, entry 2 vs 1). Decreasing the temperature from −78 to −92 °C for the deprotonation–borylation steps rewardingly allowed the exclusive formation of the E-enyne 4 as a single isomer (Table 2, entry 3).
After these encouraging results, the nature of the group transferred during the rearrangement was explored. Electron-withdrawing or electron-donating substituents on the phenyl group did not alter the stereochemical course of the reaction, while slightly improving the yields (Table 2, entries 4–7 vs 1 and 2). Benzyl or allyl groups could be transferred as well, although slightly lower yields were observed (Table 2, entries 8–11). Even an alkyl group could be transferred and interestingly, the best yields were achieved with this kind of group (Table 2, entries 12 and 13).
It is worth noting that the same stereochemical relationship between the starting oxirane and the enyne product was always obtained, whatever the group transferred from boron to the oxirane carbon. These results revealed the stereospecificity of the reaction and suggested that no epimerization of the oxiranyllithium intermediate occurred.
The present reaction was then further explored and extended to various ethynyloxiranes (Table 3). The non-functionalized cis or trans-3,4-epoxy-1-triisopropylsilyldec-1-ynes 1c stereospecifically gave the expected E or Z trisubstituted enynes 10 in moderate to good yields (Table 3, entries 1 and 2). It is worth mentioning that cis-1c contaminated with its trans-isomer (9:1 ratio) was converted to enyne 10 with the same E/Z ratio of 1:9. The cis or trans-phenyl analogues 1d were less reactive, as revealed by incomplete reactions at −92 °C and longer reaction times needed at −78 °C. Surprisingly, the trans isomer of 1d always provided a 4:1 mixture of the E/Z isomers of 11, while cis-1d gave the expected Z-isomer of 11 exclusively (Table 3, entries 3 and 4). The two isomers could, however, be separated by chromatography. Interestingly, the cis or trans-epoxydiynes 1e stereospecifically and solely provided the corresponding and expected E or Z-isomers of enyne 12 in quite good yields (Table 3, entries 5 and 6). However, in these reactions some degradation was observed, but the yields were satisfying considering the sensitive polyunsaturated nature of the starting compounds. The trisubstituted ethynyloxirane 1f proved also reactive, but longer reaction times were required, probably due to steric hindrance at some stage of the rearrangement (vide infra). Both isomers exclusively afforded the expected E or Z tetrasubstituted alkenes 13 in high yields (Table 3, entries 7 and 8).
Table 3: Lithiation, borylation and rearrangement of various cis or trans-ethynyloxiranes.a
![[Graphic 18]](/bjoc/content/inline/1860-5397-15-141-i21.svg?max-width=637&scale=1.0)
|
||||
| Entry | Oxirane | Temp. (°C) | Enyne | Yield (%)b |
| 1c |
![[Graphic 19]](/bjoc/content/inline/1860-5397-15-141-i22.svg?max-width=637&scale=1.0)
cis-1c |
−78 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-15-141-i23.svg?max-width=637&scale=1.0)
(Z)-10 |
55d |
| 2 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-15-141-i24.svg?max-width=637&scale=1.0)
trans-1c |
−92 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-15-141-i25.svg?max-width=637&scale=1.0)
(E)-10 |
56 |
| 3 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-15-141-i26.svg?max-width=637&scale=1.0)
cis-1d |
−78 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-15-141-i27.svg?max-width=637&scale=1.0)
(Z)-11 |
48 |
| 4 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-15-141-i28.svg?max-width=637&scale=1.0)
trans-1d |
−78 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-15-141-i29.svg?max-width=637&scale=1.0)
(E)-11 |
63e,f |
| 5 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-15-141-i30.svg?max-width=637&scale=1.0)
cis-1e |
−78 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-15-141-i31.svg?max-width=637&scale=1.0)
(Z)-12 |
59g |
| 6 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-15-141-i32.svg?max-width=637&scale=1.0)
trans-1e |
−92 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-15-141-i33.svg?max-width=637&scale=1.0)
(E)-12 |
57g |
| 7 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-15-141-i34.svg?max-width=637&scale=1.0)
cis-1f |
−78 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-15-141-i35.svg?max-width=637&scale=1.0)
(Z)-13 |
69h |
| 8 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-15-141-i36.svg?max-width=637&scale=1.0)
trans-1f |
−92 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-15-141-i37.svg?max-width=637&scale=1.0)
(E)-13 |
73h |
aReactions were run under argon, c = 0.125 mol/L; byields of pure isolated products, unless otherwise stated; cratio cis/trans 9:1; dratio Z/E 9:1; ea 4:1 mixture of stereoisomers was obtained; fthe yield refers to the isolated E-isomer; gsome degradation occurred; hafter addition of the boronic ester, the mixture was stirred overnight at room temperature.
Stereochemistry and mechanism
Based on the above results, we conclude that the alkynyloxirane-to-enyne reaction is stereospecific, maintaining the stereochemistry of the starting oxirane in the resulting enyne. This stereospecificity imposes the stereochemical integrity of the oxiranyllithium species formed upon lithiation, but also the existence of at least one concerted step during the reaction mechanism. This step most probably corresponds to the group transfer in organoboron derivatives, as the stereoselectivity of such transfer has been demonstrated [32,33].
Regarding the oxiranyllithium stereochemical integrity and stability, it has been demonstrated that solvent, oxirane substituent’s nature and of course temperature play key roles [34], in addition to the initial cis or trans stereochemistry, if present [35]. In our case, the oxiranyllithium derived from cis-alkynyloxiranes are configurationally stable as it was established by us [12,13]. However, we found here that in case of trans-alkynyloxiranes, a lower temperature was required to ensure stereospecificity (see entry 3 vs 2, Table 2). This observation suggests that the corresponding oxiranyllithium species could be labile. Trapping experiments with deuterated water were thus conducted at −92 and −78 °C with cis-1b or trans-1b, respectively. As expected, the cis-isomer gave the deuterated cis-derivative 1b-d with an excellent 92% yield, whatever the temperature was (Scheme 2, entry 1). However, the trans-isomer at −78 °C, produced a mixture of products (Scheme 2, entry 2). The expected deuterated trans-derivative was the major compound, the second product (2:1 ratio) was ascribed to ketone 14 after isolation. In addition, traces of the deuterated cis-derivative were detected by 1H NMR in the crude mixture, together with traces of other side-products. The formation of this set of products clearly evidenced the carbenic behavior of the involved oxiranyllithium (Scheme 2, entry 3) and revealed that the trans-alkynyloxiranes are unstable at −78 °C, but perfectly stable at −92 °C.
![[1860-5397-15-141-i2]](/bjoc/content/inline/1860-5397-15-141-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Trapping experiments of the oxiranyllithium derived from cis or trans-alkynyloxiranes 1b, and their interpretation.
Scheme 2: Trapping experiments of the oxiranyllithium derived from cis or trans-alkynyloxiranes 1b, and their...
As already proposed [36] and then demonstrated [12,13], deprotonation of oxiranes occurred upon oxygen-directed metalation. The resulting oxiranyllithium probably exists as a dimer in solution, as it has been nicely evidenced by structural characterization and NMR studies of the lithiated o-trifluoromethylstyrene oxide [37]. Such dimerization obviously stabilizes the oxiranyllithium species. Applied to oxiranyllithium derived from cis or trans-alkynyloxiranes, such dimer may easily form from the cis-oxirane (Scheme 2, entry 4), but not from the trans-isomer, or at least the large steric constrain present in it may hamper its stability. Indeed, the long oxirane C–O bond adjacent to lithium observed by X-ray diffraction in the lithiated o-trifluoromethylstyrene oxide [37] may be further elongated due to hindrance introduced by the trans-substituent adjacent to the lithium atom of the second oxirane in the dimer (Scheme 2, entry 5). Such effect may facilitate the formation of the corresponding carbenoid, especially with increasing temperature.
Once the oxiranyllithium species is formed (A in Scheme 3), oxiranylborate complexes B are obtained by the addition of aryl-, benzyl-, allyl- or alkylboronic esters. This was clearly evidenced by the fast disappearance of the coloration linked to the oxiranyllithium species upon the addition of the boronic ester. Unfortunately, all attempts to detect or isolate such ate complexes or related oxiranylboronic esters failed so far [38]. The oxiranylborate complex B then undergoes an intramolecular SN2-like process (Matteson-type rearrangement) [14] transferring the non-oxygenated boron substituent and promoting oxirane ring opening [39]. The group’s migratory aptitude cannot be directly compared to that known from cationic rearrangements, because the nature of the other boron substituents plays a key role in migration, as revealed by calculations [40-42]. From the results we so far gained, a migratory order could be assessed for the present reaction: Bu ≥ EDG-substitued Ar > EWG-substitued Ar ≥ Ar ≥ benzyl > allyl, and it appears that the larger the other substituents on the boron atom are, the easier the transfer is.
![[1860-5397-15-141-i3]](/bjoc/content/inline/1860-5397-15-141-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the rearrangement of alkynyloxiranes to α-enynes through metalation and borylation.
Scheme 3: Proposed mechanism for the rearrangement of alkynyloxiranes to α-enynes through metalation and bory...
A possible transition state B could be envisaged with the large pinacol substituent facing the small oxirane ring and thus placing the migrating group antiparallel to the adjacent oxiranyl C–O bond. This may explain the role of the diol substituent on the reaction efficacy (see Table 1). Upon migration and oxirane ring opening, a β-boron alkoxide C is formed, which probably isomerizes to a 1,2-oxaboretanide D, a known intermediate in bora-Wittig reactions [7], whose fragmentation then stereospecifically forms the observed enyne.
Conclusion
To summarize, we reported here on a convenient access to stereochemically defined α-enynes through lithiation, borylation and rearrangement of cis or trans-alkynyloxiranes. This cascade reaction is stereospecific, probably due to the stereocontrolled group transfer from boron to the adjacent oxiranyl carbon in an oxiranylborate complex intermediate, followed by an internal bora-Wittig reaction.
This method is particularly appealing, providing with good to high yields a wide diversity of functionalized E or Z tri- or tetrasubstituted α-enynes.
References
-
Capriati, V.; Florio, S.; Luisi, R. Chem. Rev. 2008, 108, 1918–1942. doi:10.1021/cr0683921
Return to citation in text: [1] [2] -
Florio, S., Ed. Oxiranyl; aziridinyl, anions as reactive intermediates in synthetic organic chemistry. Tetrahedron 2003, 59, 9683–9864. doi:10.1016/j.tet.2003.09.015
Return to citation in text: [1] [2] -
Chemla, F.; Vrancken, E. Reactivity of oxiranes with organolithioum reagents. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2004; pp 1165–1242. doi:10.1002/047002111x.ch18
Return to citation in text: [1] -
Braun, M. Lithium Carbenoids. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2004; pp 829–900. doi:10.1002/047002111x.ch13
Return to citation in text: [1] -
Taniguchi, M.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1991, 32, 2783–2786. doi:10.1016/0040-4039(91)85085-j
Return to citation in text: [1] -
Shimizu, M.; Fujimoto, T.; Minezaki, H.; Hata, T.; Hiyama, T. J. Am. Chem. Soc. 2001, 123, 6947–6948. doi:10.1021/ja016077k
Return to citation in text: [1] [2] -
Shimizu, M.; Fujimoto, T.; Liu, X.; Minezaki, H.; Hata, T.; Hiyama, T. Tetrahedron 2003, 59, 9811–9823. doi:10.1016/j.tet.2003.09.098
Return to citation in text: [1] [2] [3] -
Vedrenne, E.; Wallner, O. A.; Vitale, M.; Schmidt, F.; Aggarwal, V. K. Org. Lett. 2009, 11, 165–168. doi:10.1021/ol802651b
Return to citation in text: [1] -
Alwedi, E.; Zakharov, L. N.; Blakemore, P. R. Eur. J. Org. Chem. 2014, 6643–6648. doi:10.1002/ejoc.201403105
Return to citation in text: [1] -
Yamauchi, Y.; Kawate, T.; Katagiri, T.; Uneyama, K. Tetrahedron 2003, 59, 9839–9847. doi:10.1016/j.tet.2003.09.019
Return to citation in text: [1] -
Zhang, H.; Buchwald, S. L. J. Am. Chem. Soc. 2017, 139, 11590–11594. doi:10.1021/jacs.7b06630
Return to citation in text: [1] -
Klein, S.; Zhang, J. H.; Holler, M.; Weibel, J.-M.; Pale, P. Tetrahedron 2003, 59, 9793–9802. doi:10.1016/j.tet.2003.10.012
Return to citation in text: [1] [2] [3] [4] [5] -
Grandjean, D.; Pale, P.; Chuche, J. Tetrahedron: Asymmetry 1993, 4, 1991–1994. doi:10.1016/s0957-4166(00)82247-8
Return to citation in text: [1] [2] [3] [4] -
Matteson, D. S. Chem. Rev. 1989, 89, 1535–1551. doi:10.1021/cr00097a009
Return to citation in text: [1] [2] -
Kawashima, T.; Yamashita, N.; Okazaki, R. J. Am. Chem. Soc. 1995, 117, 6142–6143. doi:10.1021/ja00127a036
Return to citation in text: [1] -
Bojaryn, K.; Fritsch, S.; Hirschhäuser, C. Org. Lett. 2019, 21, 2218–2222. doi:10.1021/acs.orglett.9b00517
See for a recently reported similar but iterative process producing alkenes of increasing substitution.
Return to citation in text: [1] -
Halbes-Letinois, U.; Pale, P. Palladium-catalyzed synthesis of enynes. In Leading Edge Organometallic Chemistry Research; Cato, M. A., Ed.; Nova Science Publishers, Inc., 2006; pp 93–131.
Return to citation in text: [1] [2] -
Mori, M. Adv. Synth. Catal. 2007, 349, 121–135. doi:10.1002/adsc.200600484
Return to citation in text: [1] [2] -
Raviola, C.; Protti, S.; Ravelli, D.; Fagnoni, M. Chem. Soc. Rev. 2016, 45, 4364–4390. doi:10.1039/c6cs00128a
Return to citation in text: [1] [2] -
Zhou, Y.; Zhang, Y.; Wang, J. Org. Biomol. Chem. 2016, 14, 6638–6650. doi:10.1039/c6ob00944a
Return to citation in text: [1] [2] -
Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042
Return to citation in text: [1] -
Karatholuvhu, M. S.; Fuchs, P. L. J. Am. Chem. Soc. 2004, 126, 14314–14315. doi:10.1021/ja045112v
Return to citation in text: [1] -
Denichoux, A.; Ferreira, F.; Chemla, F. Org. Lett. 2004, 6, 3509–3512. doi:10.1021/ol048630j
Return to citation in text: [1] -
Eisch, J. J.; Galle, J. E. J. Am. Chem. Soc. 1976, 98, 4646–4648. doi:10.1021/ja00431a052
Return to citation in text: [1] -
Eisch, J. J.; Galle, J. E. J. Org. Chem. 1976, 41, 2615–2621. doi:10.1021/jo00877a022
Return to citation in text: [1] -
Eisch, J. J.; Galle, J. E. J. Organomet. Chem. 1976, 121, C10–C14. doi:10.1016/s0022-328x(00)85519-9
Return to citation in text: [1] -
Eisch, J. J.; Galle, J. E. J. Organomet. Chem. 1988, 341, 293–313. doi:10.1016/0022-328x(88)89085-5
Return to citation in text: [1] -
Burford, C.; Cooke, F.; Roy, G.; Magnus, P. Tetrahedron 1983, 39, 867–876. doi:10.1016/s0040-4020(01)88585-9
Return to citation in text: [1] -
Halbes-Létinois, U.; Pale, P. J. Organomet. Chem. 2003, 687, 420–424. doi:10.1016/j.jorganchem.2003.07.003
Return to citation in text: [1] -
Bertus, P.; Zhang, J.-H.; Sir, G.; Weibel, J.-M.; Pale, P. Tetrahedron Lett. 2003, 44, 3391–3395. doi:10.1016/s0040-4039(03)00565-3
Return to citation in text: [1] -
Hoffmann, M.; Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. RSC Adv. 2015, 5, 37138–37148. doi:10.1039/c5ra03228h
Return to citation in text: [1] -
Midland, M. M.; Zolopa, A. R.; Halterman, R. L. J. Am. Chem. Soc. 1979, 101, 248–249. doi:10.1021/ja00495a056
Return to citation in text: [1] -
Matteson, D. S. Stereodirected Synthesis with Organoboranes; Reactivity and Structure Concepts in Organic Chemistry; Springer Berlin: Berlin, Germany, 1995. doi:10.1007/978-3-642-79709-5
Return to citation in text: [1] -
Capriati, V.; Florio, S.; Luisi, R. Eur. J. Org. Chem. 2014, 5397–5417. doi:10.1002/ejoc.201402744
Return to citation in text: [1] -
Abbotto, A.; Capriati, V.; Degennaro, L.; Florio, S.; Luisi, R.; Pierrot, M.; Salomone, A. J. Org. Chem. 2001, 66, 3049–3058. doi:10.1021/jo0057607
Return to citation in text: [1] -
Molander, G. A.; Mautner, K. J. Org. Chem. 1989, 54, 4042–4050. doi:10.1021/jo00278a013
Return to citation in text: [1] -
Salomone, A.; Perna, F. M.; Falcicchio, A.; Nilsson Lill, S. O.; Moliterni, A.; Michel, R.; Florio, S.; Stalke, D.; Capriati, V. Chem. Sci. 2014, 5, 528–538. doi:10.1039/c3sc52099d
Return to citation in text: [1] [2] -
For example, addition of pinacolboron derivatives carrying a leaving group did not lead to the expected pinacol oxiranylboron product, but to decomposition.
Return to citation in text: [1] -
It is worth noticing here that the lower stereoselectivity observed at −78 °C with trans-1b could be explained by trapping of either the corresponding trans and cis-oxyranyllithium and stereospecific migration, or the carbenoid, which apparently formed under such conditions, and further non-selective migration.
Return to citation in text: [1] -
Bottoni, A.; Lombardo, M.; Neri, A.; Trombini, C. J. Org. Chem. 2003, 68, 3397–3405. doi:10.1021/jo026733e
Return to citation in text: [1] -
Stoddard, J. M.; Shea, K. J. Chem. Commun. 2004, 830–831. doi:10.1039/b311292f
Return to citation in text: [1] -
Robiette, R.; Fang, G. Y.; Harvey, J. N.; Aggarwal, V. K. Chem. Commun. 2006, 741–743. doi:10.1039/b514987h
Return to citation in text: [1]
| 12. | Klein, S.; Zhang, J. H.; Holler, M.; Weibel, J.-M.; Pale, P. Tetrahedron 2003, 59, 9793–9802. doi:10.1016/j.tet.2003.10.012 |
| 13. | Grandjean, D.; Pale, P.; Chuche, J. Tetrahedron: Asymmetry 1993, 4, 1991–1994. doi:10.1016/s0957-4166(00)82247-8 |
| 36. | Molander, G. A.; Mautner, K. J. Org. Chem. 1989, 54, 4042–4050. doi:10.1021/jo00278a013 |
| 12. | Klein, S.; Zhang, J. H.; Holler, M.; Weibel, J.-M.; Pale, P. Tetrahedron 2003, 59, 9793–9802. doi:10.1016/j.tet.2003.10.012 |
| 13. | Grandjean, D.; Pale, P.; Chuche, J. Tetrahedron: Asymmetry 1993, 4, 1991–1994. doi:10.1016/s0957-4166(00)82247-8 |
| 1. | Capriati, V.; Florio, S.; Luisi, R. Chem. Rev. 2008, 108, 1918–1942. doi:10.1021/cr0683921 |
| 2. | Florio, S., Ed. Oxiranyl; aziridinyl, anions as reactive intermediates in synthetic organic chemistry. Tetrahedron 2003, 59, 9683–9864. doi:10.1016/j.tet.2003.09.015 |
| 8. | Vedrenne, E.; Wallner, O. A.; Vitale, M.; Schmidt, F.; Aggarwal, V. K. Org. Lett. 2009, 11, 165–168. doi:10.1021/ol802651b |
| 19. | Raviola, C.; Protti, S.; Ravelli, D.; Fagnoni, M. Chem. Soc. Rev. 2016, 45, 4364–4390. doi:10.1039/c6cs00128a |
| 7. | Shimizu, M.; Fujimoto, T.; Liu, X.; Minezaki, H.; Hata, T.; Hiyama, T. Tetrahedron 2003, 59, 9811–9823. doi:10.1016/j.tet.2003.09.098 |
| 6. | Shimizu, M.; Fujimoto, T.; Minezaki, H.; Hata, T.; Hiyama, T. J. Am. Chem. Soc. 2001, 123, 6947–6948. doi:10.1021/ja016077k |
| 7. | Shimizu, M.; Fujimoto, T.; Liu, X.; Minezaki, H.; Hata, T.; Hiyama, T. Tetrahedron 2003, 59, 9811–9823. doi:10.1016/j.tet.2003.09.098 |
| 17. | Halbes-Letinois, U.; Pale, P. Palladium-catalyzed synthesis of enynes. In Leading Edge Organometallic Chemistry Research; Cato, M. A., Ed.; Nova Science Publishers, Inc., 2006; pp 93–131. |
| 18. | Mori, M. Adv. Synth. Catal. 2007, 349, 121–135. doi:10.1002/adsc.200600484 |
| 19. | Raviola, C.; Protti, S.; Ravelli, D.; Fagnoni, M. Chem. Soc. Rev. 2016, 45, 4364–4390. doi:10.1039/c6cs00128a |
| 20. | Zhou, Y.; Zhang, Y.; Wang, J. Org. Biomol. Chem. 2016, 14, 6638–6650. doi:10.1039/c6ob00944a |
| 5. | Taniguchi, M.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1991, 32, 2783–2786. doi:10.1016/0040-4039(91)85085-j |
| 17. | Halbes-Letinois, U.; Pale, P. Palladium-catalyzed synthesis of enynes. In Leading Edge Organometallic Chemistry Research; Cato, M. A., Ed.; Nova Science Publishers, Inc., 2006; pp 93–131. |
| 39. | It is worth noticing here that the lower stereoselectivity observed at −78 °C with trans-1b could be explained by trapping of either the corresponding trans and cis-oxyranyllithium and stereospecific migration, or the carbenoid, which apparently formed under such conditions, and further non-selective migration. |
| 3. | Chemla, F.; Vrancken, E. Reactivity of oxiranes with organolithioum reagents. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2004; pp 1165–1242. doi:10.1002/047002111x.ch18 |
| 4. | Braun, M. Lithium Carbenoids. In The Chemistry of Organolithium Compounds; Rappoport, Z.; Marek, I., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2004; pp 829–900. doi:10.1002/047002111x.ch13 |
| 40. | Bottoni, A.; Lombardo, M.; Neri, A.; Trombini, C. J. Org. Chem. 2003, 68, 3397–3405. doi:10.1021/jo026733e |
| 41. | Stoddard, J. M.; Shea, K. J. Chem. Commun. 2004, 830–831. doi:10.1039/b311292f |
| 42. | Robiette, R.; Fang, G. Y.; Harvey, J. N.; Aggarwal, V. K. Chem. Commun. 2006, 741–743. doi:10.1039/b514987h |
| 12. | Klein, S.; Zhang, J. H.; Holler, M.; Weibel, J.-M.; Pale, P. Tetrahedron 2003, 59, 9793–9802. doi:10.1016/j.tet.2003.10.012 |
| 13. | Grandjean, D.; Pale, P.; Chuche, J. Tetrahedron: Asymmetry 1993, 4, 1991–1994. doi:10.1016/s0957-4166(00)82247-8 |
| 15. | Kawashima, T.; Yamashita, N.; Okazaki, R. J. Am. Chem. Soc. 1995, 117, 6142–6143. doi:10.1021/ja00127a036 |
| 38. | For example, addition of pinacolboron derivatives carrying a leaving group did not lead to the expected pinacol oxiranylboron product, but to decomposition. |
| 6. | Shimizu, M.; Fujimoto, T.; Minezaki, H.; Hata, T.; Hiyama, T. J. Am. Chem. Soc. 2001, 123, 6947–6948. doi:10.1021/ja016077k |
| 7. | Shimizu, M.; Fujimoto, T.; Liu, X.; Minezaki, H.; Hata, T.; Hiyama, T. Tetrahedron 2003, 59, 9811–9823. doi:10.1016/j.tet.2003.09.098 |
| 16. |
Bojaryn, K.; Fritsch, S.; Hirschhäuser, C. Org. Lett. 2019, 21, 2218–2222. doi:10.1021/acs.orglett.9b00517
See for a recently reported similar but iterative process producing alkenes of increasing substitution. |
| 10. | Yamauchi, Y.; Kawate, T.; Katagiri, T.; Uneyama, K. Tetrahedron 2003, 59, 9839–9847. doi:10.1016/j.tet.2003.09.019 |
| 11. | Zhang, H.; Buchwald, S. L. J. Am. Chem. Soc. 2017, 139, 11590–11594. doi:10.1021/jacs.7b06630 |
| 37. | Salomone, A.; Perna, F. M.; Falcicchio, A.; Nilsson Lill, S. O.; Moliterni, A.; Michel, R.; Florio, S.; Stalke, D.; Capriati, V. Chem. Sci. 2014, 5, 528–538. doi:10.1039/c3sc52099d |
| 9. | Alwedi, E.; Zakharov, L. N.; Blakemore, P. R. Eur. J. Org. Chem. 2014, 6643–6648. doi:10.1002/ejoc.201403105 |
| 37. | Salomone, A.; Perna, F. M.; Falcicchio, A.; Nilsson Lill, S. O.; Moliterni, A.; Michel, R.; Florio, S.; Stalke, D.; Capriati, V. Chem. Sci. 2014, 5, 528–538. doi:10.1039/c3sc52099d |
| 23. | Denichoux, A.; Ferreira, F.; Chemla, F. Org. Lett. 2004, 6, 3509–3512. doi:10.1021/ol048630j |
| 21. | Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1961, 83, 1733–1738. doi:10.1021/ja01468a042 |
| 22. | Karatholuvhu, M. S.; Fuchs, P. L. J. Am. Chem. Soc. 2004, 126, 14314–14315. doi:10.1021/ja045112v |
| 20. | Zhou, Y.; Zhang, Y.; Wang, J. Org. Biomol. Chem. 2016, 14, 6638–6650. doi:10.1039/c6ob00944a |
| 34. | Capriati, V.; Florio, S.; Luisi, R. Eur. J. Org. Chem. 2014, 5397–5417. doi:10.1002/ejoc.201402744 |
| 35. | Abbotto, A.; Capriati, V.; Degennaro, L.; Florio, S.; Luisi, R.; Pierrot, M.; Salomone, A. J. Org. Chem. 2001, 66, 3049–3058. doi:10.1021/jo0057607 |
| 31. | Hoffmann, M.; Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. RSC Adv. 2015, 5, 37138–37148. doi:10.1039/c5ra03228h |
| 32. | Midland, M. M.; Zolopa, A. R.; Halterman, R. L. J. Am. Chem. Soc. 1979, 101, 248–249. doi:10.1021/ja00495a056 |
| 33. | Matteson, D. S. Stereodirected Synthesis with Organoboranes; Reactivity and Structure Concepts in Organic Chemistry; Springer Berlin: Berlin, Germany, 1995. doi:10.1007/978-3-642-79709-5 |
| 1. | Capriati, V.; Florio, S.; Luisi, R. Chem. Rev. 2008, 108, 1918–1942. doi:10.1021/cr0683921 |
| 2. | Florio, S., Ed. Oxiranyl; aziridinyl, anions as reactive intermediates in synthetic organic chemistry. Tetrahedron 2003, 59, 9683–9864. doi:10.1016/j.tet.2003.09.015 |
| 24. | Eisch, J. J.; Galle, J. E. J. Am. Chem. Soc. 1976, 98, 4646–4648. doi:10.1021/ja00431a052 |
| 25. | Eisch, J. J.; Galle, J. E. J. Org. Chem. 1976, 41, 2615–2621. doi:10.1021/jo00877a022 |
| 26. | Eisch, J. J.; Galle, J. E. J. Organomet. Chem. 1976, 121, C10–C14. doi:10.1016/s0022-328x(00)85519-9 |
| 27. | Eisch, J. J.; Galle, J. E. J. Organomet. Chem. 1988, 341, 293–313. doi:10.1016/0022-328x(88)89085-5 |
| 28. | Burford, C.; Cooke, F.; Roy, G.; Magnus, P. Tetrahedron 1983, 39, 867–876. doi:10.1016/s0040-4020(01)88585-9 |
| 29. | Halbes-Létinois, U.; Pale, P. J. Organomet. Chem. 2003, 687, 420–424. doi:10.1016/j.jorganchem.2003.07.003 |
| 30. | Bertus, P.; Zhang, J.-H.; Sir, G.; Weibel, J.-M.; Pale, P. Tetrahedron Lett. 2003, 44, 3391–3395. doi:10.1016/s0040-4039(03)00565-3 |
| 12. | Klein, S.; Zhang, J. H.; Holler, M.; Weibel, J.-M.; Pale, P. Tetrahedron 2003, 59, 9793–9802. doi:10.1016/j.tet.2003.10.012 |
| 12. | Klein, S.; Zhang, J. H.; Holler, M.; Weibel, J.-M.; Pale, P. Tetrahedron 2003, 59, 9793–9802. doi:10.1016/j.tet.2003.10.012 |
| 13. | Grandjean, D.; Pale, P.; Chuche, J. Tetrahedron: Asymmetry 1993, 4, 1991–1994. doi:10.1016/s0957-4166(00)82247-8 |
© 2019 Fuentespina et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)