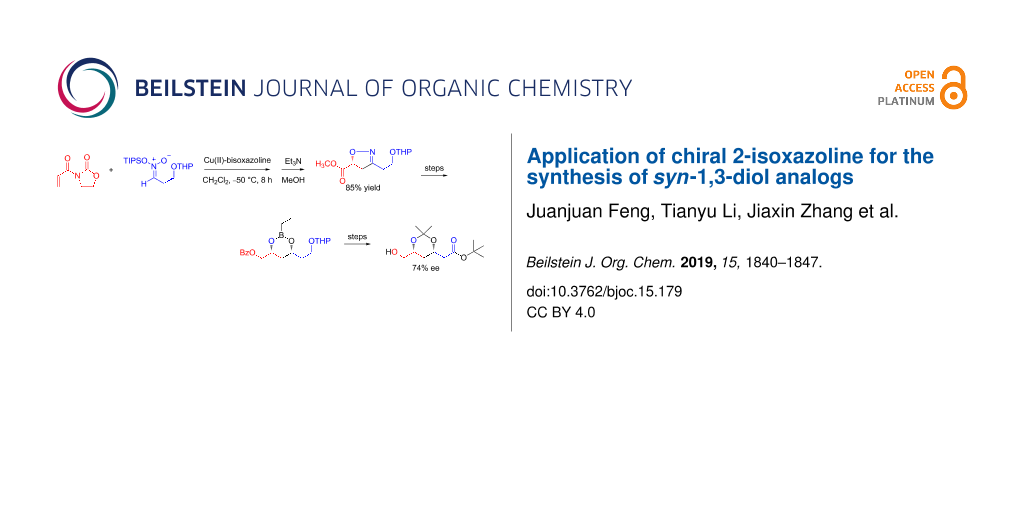
Abstract
The asymmetric cycloaddition of TIPS nitronate catalyzed by “Cu(II)-bisoxazoline” gave the 2-isoxazoline product in 95% yield, which was converted into tert-butyl (3S,5R)-6-hydroxy-3,5-O-isopropylidene-3,5-dihydroxyhexanoate in 14 steps through a β-hydroxy ketone.

Graphical Abstract
Introduction
The chiral 1,3-diol structure is widespread in a broad spectrum of natural products [1,2]. (3R)-β-Hydroxy-δ-lactone or its open-ring equivalent (3R)-syn-3,5-dihydroxypentanoic acid, is a common structure in naturally occurring mevastatin (or compactin), lovastatin or closely related statins, and synthetic statins. Either the syn or anti-1,3-diol could be prepared from enantiomerically pure β-hydroxy ketones through β-hydroxy-directed carbonyl reduction following Evans’ [3] or Prasad’s [4-11] method. The Narasaka–Prasad reduction of a δ-hydroxy-β-keto esters derived from β-hydroxy esters [12-23] is widely used to prepare tert-butyl (3R)-3,5-O-isopropylidene-3,5-dihydroxyhexanoate (Scheme 1a) [24-37], which is a building block for synthetic statins [38-41], though enzymatic syntheses [42-48] of the chiral β-hydroxy-δ-lactone moiety or its equivalents, pioneered by Wong [42], is equally competitive. Here, we report the preparations of tert-butyl (3S,5R)-6-hydroxy-3,5-O-isopropylidene-3,5-dihydroxyhexanoate and related syn-1,3-diol analogs from a chiral 2-isoxazoline (Scheme 1b). This work is part of our continuous efforts in asymmetric syntheses and applications of chiral 2-isoxazolines [49-51].
![[1860-5397-15-179-i1]](/bjoc/content/inline/1860-5397-15-179-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Accesses to tert-butyl 3,5-O-isopropylidene-3,5-dihydroxyhexanoates. (a) Previous methods using Claisen condensation. (b) Our new method using cycloaddition.
Scheme 1: Accesses to tert-butyl 3,5-O-isopropylidene-3,5-dihydroxyhexanoates. (a) Previous methods using Cla...
Results and Discussion
Our synthesis commenced with a chiral 3,5-disubstituted-2-isoxazoline 3 or 4, which were prepared from silyl nitronate through an asymmetric 1,3-dipolar cycloaddition developed in our lab (Table 1) [49]. The synthesis of the triisopropylsilyl nitronate was initially attempted starting with 3-nitropropionic acid methyl ester but no desired product was observed. However, switching to 3-nitropropanol, protected as the THP ether, succeeded to prepare the required triisopropylsilyl nitronate. Then, the catalytic asymmetric cycloaddition gave the 2-isoxazolidine cycloadduct 1 in a high yield. In the light of our previous ligand screening results [49], two bisoxazolines with an isopropyl (ligand B) or tert-butyl group (ligand A) were tested. Optimization of the conditions established that 26 mol % of ligand B together with 20 mol % Cu(OTf)2 in anhydrous CH2Cl2 catalyzed the cycloaddition between N-acryloyl-1,3-oxazolidin-2-one and the silyl nitronate at −50 °C to give 1 in 95% isolated yield, which subsequently generated 3,5-disubstituted isoxazoline 4 in 80% ee. Decreasing the amount of the chiral Lewis acid catalyst led to a decrease of both the ee and the yield. Desilylation of the 2-isoxazolidine 1 was effected in CHCl3 using catalytic amounts of p-toluenesulfonic acid (PTSA). Though the yield of the in situ-generated 2-isoxazoline 2 bearing the 1,3-oxazolidin-2-one auxiliary was perfect, purification of 2 by silica gel chromatography was problematic due to decomposition. No pure product was isolated from crude 2 by chromatography on silica gel. Decomposition occurred to a compound similar to 2, in which the 3-substituent was CH2OH [49]. To overcome this problem, the crude reaction mixture containing 2 and PTSA was concentrated before excess Et3N was added followed by CH3OH as the solvent. These operations removed the 1,3-oxazolidin-2-one auxiliary while preserving the THP group, and afforded the corresponding methyl ester 3 (Table 1), which was stable and could be subjected to silica gel chromatography. Compound 4 was used to determine the stereoselectivity of the cycloaddition step as well as for oxidation.
Table 1: Optimization of the conditions for the asymmetric cycloaddition.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-179-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Ligand | x mol % Cu | T (°C) | Yield (%) (1) | ee (%) (4) |
| 1 | A | 10 | −50 | 25 | 18 |
| 2 | A | 20 | −50 | 75 | 72 |
| 3 | B | 10 | −50 | 70 | 66 |
| 4 | B | 20 | −50 | 95 | 80 |
| 5 | B | 20 | −40 | 78 | 78 |
| 6 | B | 20 | −60 | 84 | 80 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-179-i7.svg?max-width=637&scale=1.18182)
Oxidation of the 2-isoxazoline 4 with Jones’ reagent gave a complicated mixture, in which the desired carboxylic acid was not observed (Scheme 2). The stepwise oxidation of the free hydroxy to the carboxy group via intermediary aldehyde was then examined. Swern or pyridinium chlorochromate (PCC) oxidation of 4 also gave a complicated mixture without the desired aldehyde detected. These failed reactions indicated that the 2-isoxazoline moiety could not survive oxidation conditions. Based on this assumption, the corresponding silyl nitronate from 3-nitropropanal or its acetal were not tried for cycloaddition.
![[1860-5397-15-179-i2]](/bjoc/content/inline/1860-5397-15-179-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
We then set to liberate the β-hydroxy ketone synthon by ring opening of the isoxazoline 3 (Scheme 3). Raney-Ni-catalyzed hydrogenolysis in the presence of boronic acid had been widely utilized to disconnect the N–O bond as well as to hydrolyze the resulting imine into a ketone [52]. We applied this method to deprotect the isoxazoline 3. However, the desired β-hydroxy ketone was never obtained. In one instance, the methyl ketone from a retro-aldol reaction of the desired β-hydroxy ketone was observed. In our experience, the hydrogenolysis of a 2-isoxazoline having a 5-ester group was troublesome. Thus, the 5-ester group was reduced with NaBH4 to give 5. The hydroxy group was subsequently protected with benzoyl (Scheme 3), which also worked as a chromophore facilitating HPLC analysis. Afterwards, we tried oxidations once again. After removal of THP from 6, the resulting compound 6' was subjected to oxidation with various reagents (Scheme 4) [53-55]. The expected carboxylic acid or aldehyde was not observed, which further verified the intolerance exemplified in Scheme 2. These results prompted us to try the oxidation in a later stage.
![[1860-5397-15-179-i3]](/bjoc/content/inline/1860-5397-15-179-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparations of 16 and related syn-1,3-diol compounds.
Scheme 3: Preparations of 16 and related syn-1,3-diol compounds.
![[1860-5397-15-179-i4]](/bjoc/content/inline/1860-5397-15-179-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
When 6 was subjected to Raney-Ni-catalyzed hydrogenolysis, the desired β-hydroxy ketone 7 was obtained in 85% yield (Scheme 3). Under the weakly acidic conditions, the THP group survived. Next, a Narasaka–Prasad reduction [4-11] of 7 using Et2BOMe and NaBH4 at −78 °C gave stable ethylboronate 8 in 96% yield. Several ethylboronate compounds have been reported [9-11,56-62]. From 8 to 9, no H2O2 treatment was necessary. Rotary evaporation of 8 with CH3OH at ca. 40 °C easily removed the ethylborane group. Removal of THP in 9 delivered a 1,3,5-trihydroxy compound 10. In another way, 10 could be prepared by treating 8 with PTSA in CH3OH at rt. NMR spectra of 8–10 exhibited only one set of signals corresponding to the syn-dihydroxy products, indicating an extra high diastereoselectivity (syn:anti >99:1) during the reduction. To unambiguously determine the diastereomeric ratio, the anti-1,3-diol corresponding to 10 was prepared from 7 by RuCl3–PPh3-catalyzed hydrogenation [63,64]. However, the two diastereomers had identical proton NMR spectra.
The terminal hydroxy group of 10 was protected with TBS [65-69] and the syn-hydroxy groups subjected to acetonization using PTSA and dimethoxypropane (DMP) to give 12 in 86% total yield [70]. Treatment of 12 with TBAF again liberated the terminal hydroxy group for further oxidation. RuCl3-catalyzed oxidation of 13 with NaIO4 yielded the carboxylic acid 14 in 86% yield [70], which was reacted with Boc2O to get the tert-butyl ester 15 [26,43,71]. The ee of 15 was determined as 74%. The racemic sample of 15 was prepared from racemic diethyl malate following known methods [26,27]. Finally, K2CO3-catalyzed methanolysis gave 16 in 87% yield [26,27]. The absolute stereochemistry of 16 was confirmed by crystal structure analysis [72] and the specific rotation [28] of 17. Centimeter-long prismatic single crystals of 17 were obtained by slow evaporation of a petroleum solution.
Starting from 9, we tested several reactions in order to selectively protect the internal hydroxy groups (Scheme 5). Though not fruitful, these results deserve some comments. The PTSA-catalyzed acetonization of 9 using 2.0 equiv DMP gave the acetonide 18 in a quantitative yield. Treating 18 with a catalytic amount of PTSA in methanol gave 10, with the protecting groups removed except benzoyl. PTSA-catalyzed acetonization of 10 using 2.0 equiv DMP gave a mixture of two acetonides 19 and 13, which are separable by silica gel chromatography (Scheme 5a). In another trial (Scheme 5b), acylation of the two hydroxy groups in 9 yielded 20 in a quantitative yield. PTSA-catalyzed removal of THP in 20 in methanol did occur. However, concomitant monodeacylation as well as further an acyl-transfer reaction also took place, resulting in a mixture. These results indicated THP, isopropylidene or Ac protection to primary or secondary hydroxy groups did not well tolerate PTSA-catalyzed methanolysis.
![[1860-5397-15-179-i5]](/bjoc/content/inline/1860-5397-15-179-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Attempted selective protections of internal 1,3-hydroxy groups: (a) acetonizations of 1,3-diols; (b) removal of co-existing Ac and THP on hydroxy groups.
Scheme 5: Attempted selective protections of internal 1,3-hydroxy groups: (a) acetonizations of 1,3-diols; (b...
Conclusion
In conclusion, we synthesized tert-butyl (3S,5R)-6-hydroxy-3,5-O-isopropylidene-3,5-dihydroxyhexanoate (16), which is enantiomeric to a key intermediate for atorvastatin, from a chiral 2-isoxazoline (3). The β-hydroxy ketone 7 obtained from 3 could be easily converted into several syn-1,3-diol analogs, demonstrating the usefulness of chiral 2-isoxazolines.
Experimental
1: To a dry Schlenk tube were added Cu(OTf)2 (144 mg, 0.4 mmol), chiral bisoxazoline B (139 mg, 0.52 mmol) and anhydrous CH2Cl2 (4 mL) under N2. After stirring at room temperature for 2 h, a clear solution had formed, which was cooled to −50 °C and N-acryloyl-1,3-oxazolidin-2-one (282 mg, 2 mmol) was added. After stirring for 30 min, a solution of the silyl nitronate (3.0 mmol) in anhydrous CH2Cl2 (6 mL) was added. The mixture was stirred for 8 h at −50 °C and monitored by TLC. After the reaction was completed, the product was purified by silica gel chromatography. Yellow oil (923 mg, 95% yield); Rf 0.40 (1:1 hexanes/AcOEt); 1H NMR (400 MHz, CDCl3) δ 5.77–5.74 (m, 1H, CH2CHO), 4.53 (s, 1H, OCHO), 4.44 (t, J = 8.0 Hz, 2H, CH2O), 4.03–3.99 (m, 2H, CH2O), 3.79–3.74 (m, 2H, OCH2CH2), 3.47–3.37 (m, 3H, NCH and NCH2), 2.75–2.66 (m, 1H, CHCH2CH), 2.31–2.27 (m, 1H, CHCH2CH), 2.17–2.12 (m, 1H, CH2CH2), 1.84–1.79 (m, 2H, CH2CH2 and CH2CH2CH2), 1.68–1.49 (m, 6H, CH2CH2CH2), 1.24–1.15 (m, 3H, SiCH), 1.07–1.01 (m, 18H, SiCH(CH3)2); 13C NMR (100 MHz, CDCl3) δ 170.8, 153.1, 98.9, 98.9, 77.4, 77.2, 69.9, 69.8, 65.2, 62.8, 62.5, 62.3, 42.6, 35.6, 35.5, 30.7, 30.6, 29.9, 29.8, 25.5, 19.6, 19.5, 18.1, 18.0, 12.2; IR (cm−1): 3544, 2942, 2867, 2725, 2249, 1780, 1704, 1464, 1386, 1275, 1133, 1035, 883, 806, 677; ESIMS (m/z): [M + Na]+ calcd for C23H42N2O7Si, 509.2659; found, 509.2659.
3: To a solution of 1 (0.86 g, 1.78 mmol) in CHCl3 (15 mL) was added PTSA (31 mg, 0.178 mmol) at 0 °C. The mixture was allowed to warm to room temperature and stirred until complete consumption of the starting material (0.5 h). Vacuum was applied to remove the solvent before Et3N (5 mL) was added. After stirring for 5 min, methanol (30 mL) was added and the mixture was stirred overnight at room temperature. The crude product was purified by column chromatography. Yellow oil (0.41 g, 89% yield); Rf 0.42 (1:1 hexanes/AcOEt); 1H NMR (400 MHz, CDCl3) δ 4.92–4.87 (m, 1H, OCHCO), 4.51–4.50 (m, 1H, OCHO), 3.88–3.82 (m, 1H, CH2O), 3.75–3.71 (m, 1H, CH2O), 3.68 (s, 3H, CH3), 3.56–3.50 (m, 1H, CH2O), 3.42–3.39 (m, 1H, CH2O), 3.24–3.31 (m, 1H, CHCH2CH), 2.62–2.54 (m, 1H, CH2CH2CH), 1.74–1.44 (m, 6H, CH2CH2CH2); 13C NMR (100 MHz, CDCl3) δ 171.0, 156.9, 99.0, 98.9, 64.4, 64.3, 62.5, 62.4, 52.6, 41.6, 30.6, 27.9, 25.4, 19.6, 19.5; IR (cm−1): 3481, 2950, 2873, 2852, 2657, 1756, 1738, 1734, 1628, 1456, 1436, 1367, 1354, 1201, 1134, 1034, 869, 814, 752, 740; ESIMS (m/z): [M + H]+ calcd for C12H19NO5, 258.1341; found, 258.1340.
References
-
Nicolaou, K. C.; Daines, R. A.; Ogawa, Y.; Chakraborty, T. K. J. Am. Chem. Soc. 1988, 110, 4696–4705. doi:10.1021/ja00222a030
Return to citation in text: [1] -
Rychnovsky, S. D.; Griesegraber, G.; Kim, J. J. Am. Chem. Soc. 1994, 116, 2621–2622. doi:10.1021/ja00085a053
and references therein.
Return to citation in text: [1] -
Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035
Return to citation in text: [1] -
Narasaka, K.; Pai, H. C. Chem. Lett. 1980, 9, 1415–1418. doi:10.1246/cl.1980.1415
Return to citation in text: [1] [2] -
Narasaka, K.; Pai, F.-C. Tetrahedron 1984, 40, 2233–2238. doi:10.1016/0040-4020(84)80006-x
Return to citation in text: [1] [2] -
Kathawala, F. G.; Prager, B.; Prasad, K.; Repič, O.; Shapiro, M. J.; Stabler, R. S.; Widler, L. Helv. Chim. Acta 1986, 69, 803–805. doi:10.1002/hlca.19860690407
Return to citation in text: [1] [2] -
Chen, K.-M.; Hardtmann, G. E.; Prasad, K.; Repič, O.; Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/s0040-4039(00)95673-9
Return to citation in text: [1] [2] -
Chen, K.-M.; Gunderson, K. G.; Hardtmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Chem. Lett. 1987, 16, 1923–1926. doi:10.1246/cl.1987.1923
Return to citation in text: [1] [2] -
Burova, S. A.; McDonald, F. E. J. Am. Chem. Soc. 2002, 124, 8188–8189. doi:10.1021/ja026255p
Return to citation in text: [1] [2] [3] -
Walleser, P.; Brückner, R. Eur. J. Org. Chem. 2010, 4802–4822. doi:10.1002/ejoc.201000280
Return to citation in text: [1] [2] [3] -
Becerril-Jiménez, F.; Ward, D. E. Org. Lett. 2012, 14, 1648–1651. doi:10.1021/ol300432y
Return to citation in text: [1] [2] [3] -
Saito, S.; Hasegawa, T.; Inaba, M.; Nishida, R.; Fujii, T.; Nomizu, S.; Moriwake, T. Chem. Lett. 1984, 13, 1389–1392. doi:10.1246/cl.1984.1389
Return to citation in text: [1] -
Saito, S.; Nagao, Y.; Miyazaki, M.; Inaba, M.; Moriwake, T. Tetrahedron Lett. 1986, 27, 5249–5252. doi:10.1016/s0040-4039(00)85181-3
Return to citation in text: [1] -
Saito, S.; Ishikawa, T.; Kuroda, A.; Koga, K.; Moriwake, T. Tetrahedron 1992, 48, 4067–4086. doi:10.1016/s0040-4020(01)92187-8
Return to citation in text: [1] -
Hamada, Y.; Yokokawa, F.; Kabeya, M.; Hatano, K.; Kurono, Y.; Shioiri, T. Tetrahedron 1996, 52, 8297–8306. doi:10.1016/0040-4020(96)00383-3
Return to citation in text: [1] -
Sauret-Cladière, S.; Jeminet, G. Tetrahedron: Asymmetry 1997, 8, 417–423. doi:10.1016/s0957-4166(96)00517-4
Return to citation in text: [1] -
Nakatani, S.; Ikura, M.; Yamamoto, S.; Nishita, Y.; Itadani, S.; Habashita, H.; Sugiura, T.; Ogawa, K.; Ohno, H.; Takahashi, K.; Nakai, H.; Toda, M. Bioorg. Med. Chem. 2006, 14, 5402–5422. doi:10.1016/j.bmc.2006.03.032
Return to citation in text: [1] -
Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043
Return to citation in text: [1] -
Li, D.; Zhao, Y.; Ye, L.; Chen, C.; Zhang, J. Synthesis 2010, 3325–3331. doi:10.1055/s-0030-1258187
Return to citation in text: [1] -
Kitamura, M.; Ohkuma, T.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1988, 29, 1555–1556. doi:10.1016/s0040-4039(00)80350-0
Return to citation in text: [1] -
Shao, L.; Kawano, H.; Saburi, M.; Uchida, Y. Tetrahedron 1993, 49, 1997–2010. doi:10.1016/s0040-4020(01)86300-6
Return to citation in text: [1] -
Jeulin, S.; Duprat de Paule, S.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Champion, N.; Dellis, P. Angew. Chem., Int. Ed. 2004, 43, 320–325. doi:10.1002/anie.200352453
Return to citation in text: [1] -
Qiu, L.; Wu, J.; Chan, S.; Au-Yeung, T. T.-L.; Ji, J.-X.; Guo, R.; Pai, C.-C.; Zhou, Z.; Li, X.; Fan, Q.-H.; Chan, A. S. C. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5815–5820. doi:10.1073/pnas.0307774101
Return to citation in text: [1] -
Beck, G.; Jendralla, H.; Kesseler, K. Synthesis 1995, 1014–1018. doi:10.1055/s-1995-4028
Return to citation in text: [1] -
Nishiyama, A.; Horikawa, M.; Yasohara, Y.; Ueyama, N.; Inoue, K. Process for preparing optically active 2-[6-(hydroxymethyl)-1,3-dioxan-4-yl]acetic acid derivatives as pharmaceutical intermediates. WO Patent Appl. WO 2001094337 A1, Dec 13, 2001.
Return to citation in text: [1] -
Szpilman, A. M.; Cereghetti, D. M.; Wurtz, N. R.; Manthorpe, J. M.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4335–4338. doi:10.1002/anie.200800589
Return to citation in text: [1] [2] [3] [4] -
Szpilman, A. M.; Cereghetti, D. M.; Manthorpe, J. M.; Wurtz, N. R.; Carreira, E. M. Chem. – Eur. J. 2009, 15, 7117–7128. doi:10.1002/chem.200900231
Return to citation in text: [1] [2] [3] -
Fan, W.; Li, W.; Ma, X.; Tao, X.; Li, X.; Yao, Y.; Xie, X.; Zhang, Z. J. Org. Chem. 2011, 76, 9444–9451. doi:10.1021/jo201822k
Return to citation in text: [1] [2] -
Solladie, G.; Bauder, C.; Rossi, L. J. Org. Chem. 1995, 60, 7774–7777. doi:10.1021/jo00129a018
Return to citation in text: [1] -
Ghosh, A. K.; Lei, H. J. Org. Chem. 2000, 65, 4779–4781. doi:10.1021/jo000528m
Return to citation in text: [1] -
Hubbs, J. L.; Heathcock, C. H. J. Am. Chem. Soc. 2003, 125, 12836–12843. doi:10.1021/ja030316h
Return to citation in text: [1] -
Xu, S. Zhongguo Xinyao Zazhi 2006, 15, 1913.
Return to citation in text: [1] -
George, S.; Sudalai, A. Tetrahedron Lett. 2007, 48, 8544–8546. doi:10.1016/j.tetlet.2007.09.133
Return to citation in text: [1] -
Bonini, C.; Campaniello, M.; Chiummiento, L.; Videtta, V. Tetrahedron 2008, 64, 8766–8772. doi:10.1016/j.tet.2008.06.094
Return to citation in text: [1] -
Su, Y.; Xu, Y.; Han, J.; Zheng, J.; Qi, J.; Jiang, T.; Pan, X.; She, X. J. Org. Chem. 2009, 74, 2743–2749. doi:10.1021/jo9000146
Return to citation in text: [1] -
Sawant, P.; Maier, M. E. Tetrahedron 2010, 66, 9738–9744. doi:10.1016/j.tet.2010.10.028
Return to citation in text: [1] -
Xiong, F.-J.; Li, J.; Chen, X.-F.; Chen, W.-X.; Chen, F.-E. Tetrahedron: Asymmetry 2014, 25, 1205–1208. doi:10.1016/j.tetasy.2014.07.002
Return to citation in text: [1] -
Brower, P. L.; Butler, D. E.; Deering, C. F.; Le, T. V.; Millar, A.; Nanninga, T. N.; Roth, B. D. Tetrahedron Lett. 1992, 33, 2279–2282. doi:10.1016/s0040-4039(00)74189-x
Return to citation in text: [1] -
Rádl, S. Synth. Commun. 2003, 33, 2275–2283. doi:10.1081/scc-120021507
Return to citation in text: [1] -
Wess, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Beck, G.; Bergmann, A.; Jendralla, H.; Bock, K.; Holzstein, O.; Kleine, H.; Schnierer, M. Tetrahedron Lett. 1990, 31, 2545–2548. doi:10.1016/0040-4039(90)80121-2
Return to citation in text: [1] -
Reddy, M. S. N.; Reddy, B. K.; Reddy, C. K.; Kumar, M. K.; Rajan, S. T.; Reddy, M. S. Orient. J. Chem. 2008, 24, 167–174.
Return to citation in text: [1] -
Gijsen, H. J. M.; Wong, C.-H. J. Am. Chem. Soc. 1994, 116, 8422–8423. doi:10.1021/ja00097a082
Return to citation in text: [1] [2] -
Liu, J.; Hsu, C.-C.; Wong, C.-H. Tetrahedron Lett. 2004, 45, 2439–2441. doi:10.1016/j.tetlet.2004.01.110
Return to citation in text: [1] [2] -
Öhrlein, R.; Baisch, G. Adv. Synth. Catal. 2003, 345, 713–715. doi:10.1002/adsc.200303028
Return to citation in text: [1] -
Greenberg, W. A.; Varvak, A.; Hanson, S. R.; Wong, K.; Huang, H.; Chen, P.; Burk, M. J. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5788–5793. doi:10.1073/pnas.0307563101
Return to citation in text: [1] -
Müller, M. Angew. Chem., Int. Ed. 2005, 44, 362–365. doi:10.1002/anie.200460852
Return to citation in text: [1] -
Guo, Z.; Chen, Y.; Goswami, A.; Hanson, R. L.; Patel, R. N. Tetrahedron: Asymmetry 2006, 17, 1589–1602. doi:10.1016/j.tetasy.2006.05.027
Return to citation in text: [1] -
Sun, F.; Xu, G.; Wu, J.; Yang, L. Tetrahedron: Asymmetry 2007, 18, 2454–2461. doi:10.1016/j.tetasy.2007.09.026
Return to citation in text: [1] -
Dong, L.; Geng, C.; Jiao, P. J. Org. Chem. 2015, 80, 10992–11002. doi:10.1021/acs.joc.5b02035
Return to citation in text: [1] [2] [3] [4] -
Han, X.; Dong, L.; Geng, C.; Jiao, P. Org. Lett. 2015, 17, 3194–3197. doi:10.1021/acs.orglett.5b00826
Return to citation in text: [1] -
Jiang, M.; Feng, L.; Feng, J.; Jiao, P. Org. Lett. 2017, 19, 2210–2213. doi:10.1021/acs.orglett.7b00558
Return to citation in text: [1] -
Curran, D. P. J. Am. Chem. Soc. 1983, 105, 5826–5833. doi:10.1021/ja00356a021
Return to citation in text: [1] -
Song, Z. J.; Zhao, M.; Desmond, R.; Devine, P.; Tschaen, D. M.; Tillyer, R.; Frey, L.; Heid, R.; Xu, F.; Foster, B.; Li, J.; Reamer, R.; Volante, R.; Dolling, U. H.; Reider, P. J.; Okada, S.; Kato, Y.; Mano, E. J. Org. Chem. 1999, 64, 9658–9667. doi:10.1021/jo991292t
Return to citation in text: [1] -
Zhao, M.; Li, J.; Mano, E.; Song, Z.; Tschaen, D. M.; Grabowski, E. J. J.; Reider, P. J. J. Org. Chem. 1999, 64, 2564–2566. doi:10.1021/jo982143y
Return to citation in text: [1] -
Jiang, X.; Zhang, J.; Ma, S. J. Am. Chem. Soc. 2016, 138, 8344–8347. doi:10.1021/jacs.6b03948
Return to citation in text: [1] -
Kim, J.-I.; Lee, S.-K.; Jahng, Y. Arch. Pharmacal Res. 1994, 17, 161–165. doi:10.1007/bf02974252
Return to citation in text: [1] -
Hagiwara, H.; Kon-no, M.; Nakano, T.; Uda, H. J. Chem. Soc., Perkin Trans. 1 1994, 2417. doi:10.1039/p19940002417
Return to citation in text: [1] -
Jefferies, P. R.; Gengo, P. J.; Watson, M. J.; Casida, J. E. J. Med. Chem. 1996, 39, 2339–2346. doi:10.1021/jm950712d
Return to citation in text: [1] -
Karisalmi, K.; Koskinen, A. M. P. Synthesis 2004, 1331–1342. doi:10.1055/s-2004-822389
Return to citation in text: [1] -
Tarur, V. R.; Sathe, D. G.; Rao, N. M.; Bhopalkar, R.; Mahajan, U. S. Process for the preparation of fluvastatin sodium and intermediates. PCT Int. Appl. WO 2007023503 A1, March 1, 2007.
Return to citation in text: [1] -
Renata, H.; Zhou, Q.; Baran, P. S. Science 2013, 339, 59–63. doi:10.1126/science.1230631
Return to citation in text: [1] -
Renata, H.; Zhou, Q.; Dünstl, G.; Felding, J.; Merchant, R. R.; Yeh, C.-H.; Baran, P. S. J. Am. Chem. Soc. 2015, 137, 1330–1340. doi:10.1021/ja512022r
Return to citation in text: [1] -
Labeeuw, O.; Roche, C.; Phansavath, P.; Genêt, J.-P. Org. Lett. 2007, 9, 105–108. doi:10.1021/ol062631p
Return to citation in text: [1] -
Roche, C.; Labeeuw, O.; Haddad, M.; Ayad, T.; Genêt, J.-P.; Ratovelomanana-Vidal, V.; Phansavath, P. Eur. J. Org. Chem. 2009, 3977–3986. doi:10.1002/ejoc.200900316
Return to citation in text: [1] -
Besse, P.; Ciblat, S.; Canet, J.-L.; Troin, Y.; Veschambre, H. Tetrahedron: Asymmetry 1999, 10, 2213–2224. doi:10.1016/s0957-4166(99)00234-7
Return to citation in text: [1] -
Koide, K.; Finkelstein, J. M.; Ball, Z.; Verdine, G. L. J. Am. Chem. Soc. 2001, 123, 398–408. doi:10.1021/ja0023377
Return to citation in text: [1] -
Goswami, D.; Koli, M. R.; Chatterjee, S.; Chattopadhyay, S.; Sharma, A. Org. Biomol. Chem. 2017, 15, 3756–3774. doi:10.1039/c7ob00626h
Return to citation in text: [1] -
Ma, X.; Diane, M.; Ralph, G.; Chen, C.; Biscoe, M. R. Angew. Chem., Int. Ed. 2017, 56, 12663–12667. doi:10.1002/anie.201704672
Return to citation in text: [1] -
Ganganna, B.; Srihari, P.; Yadav, J. S. Tetrahedron Lett. 2017, 58, 2685–2689. doi:10.1016/j.tetlet.2017.05.034
Return to citation in text: [1] -
Ghosh, A. K.; Lei, H. J. Org. Chem. 2002, 67, 8783–8788. doi:10.1021/jo020402k
Return to citation in text: [1] [2] -
Das, S.; Goswami, R. K. J. Org. Chem. 2014, 79, 9778–9791. doi:10.1021/jo5019798
Return to citation in text: [1] -
Crystallographic data of 17: CCDC 1826709.
Return to citation in text: [1]
| 1. | Nicolaou, K. C.; Daines, R. A.; Ogawa, Y.; Chakraborty, T. K. J. Am. Chem. Soc. 1988, 110, 4696–4705. doi:10.1021/ja00222a030 |
| 2. |
Rychnovsky, S. D.; Griesegraber, G.; Kim, J. J. Am. Chem. Soc. 1994, 116, 2621–2622. doi:10.1021/ja00085a053
and references therein. |
| 24. | Beck, G.; Jendralla, H.; Kesseler, K. Synthesis 1995, 1014–1018. doi:10.1055/s-1995-4028 |
| 25. | Nishiyama, A.; Horikawa, M.; Yasohara, Y.; Ueyama, N.; Inoue, K. Process for preparing optically active 2-[6-(hydroxymethyl)-1,3-dioxan-4-yl]acetic acid derivatives as pharmaceutical intermediates. WO Patent Appl. WO 2001094337 A1, Dec 13, 2001. |
| 26. | Szpilman, A. M.; Cereghetti, D. M.; Wurtz, N. R.; Manthorpe, J. M.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4335–4338. doi:10.1002/anie.200800589 |
| 27. | Szpilman, A. M.; Cereghetti, D. M.; Manthorpe, J. M.; Wurtz, N. R.; Carreira, E. M. Chem. – Eur. J. 2009, 15, 7117–7128. doi:10.1002/chem.200900231 |
| 28. | Fan, W.; Li, W.; Ma, X.; Tao, X.; Li, X.; Yao, Y.; Xie, X.; Zhang, Z. J. Org. Chem. 2011, 76, 9444–9451. doi:10.1021/jo201822k |
| 29. | Solladie, G.; Bauder, C.; Rossi, L. J. Org. Chem. 1995, 60, 7774–7777. doi:10.1021/jo00129a018 |
| 30. | Ghosh, A. K.; Lei, H. J. Org. Chem. 2000, 65, 4779–4781. doi:10.1021/jo000528m |
| 31. | Hubbs, J. L.; Heathcock, C. H. J. Am. Chem. Soc. 2003, 125, 12836–12843. doi:10.1021/ja030316h |
| 32. | Xu, S. Zhongguo Xinyao Zazhi 2006, 15, 1913. |
| 33. | George, S.; Sudalai, A. Tetrahedron Lett. 2007, 48, 8544–8546. doi:10.1016/j.tetlet.2007.09.133 |
| 34. | Bonini, C.; Campaniello, M.; Chiummiento, L.; Videtta, V. Tetrahedron 2008, 64, 8766–8772. doi:10.1016/j.tet.2008.06.094 |
| 35. | Su, Y.; Xu, Y.; Han, J.; Zheng, J.; Qi, J.; Jiang, T.; Pan, X.; She, X. J. Org. Chem. 2009, 74, 2743–2749. doi:10.1021/jo9000146 |
| 36. | Sawant, P.; Maier, M. E. Tetrahedron 2010, 66, 9738–9744. doi:10.1016/j.tet.2010.10.028 |
| 37. | Xiong, F.-J.; Li, J.; Chen, X.-F.; Chen, W.-X.; Chen, F.-E. Tetrahedron: Asymmetry 2014, 25, 1205–1208. doi:10.1016/j.tetasy.2014.07.002 |
| 4. | Narasaka, K.; Pai, H. C. Chem. Lett. 1980, 9, 1415–1418. doi:10.1246/cl.1980.1415 |
| 5. | Narasaka, K.; Pai, F.-C. Tetrahedron 1984, 40, 2233–2238. doi:10.1016/0040-4020(84)80006-x |
| 6. | Kathawala, F. G.; Prager, B.; Prasad, K.; Repič, O.; Shapiro, M. J.; Stabler, R. S.; Widler, L. Helv. Chim. Acta 1986, 69, 803–805. doi:10.1002/hlca.19860690407 |
| 7. | Chen, K.-M.; Hardtmann, G. E.; Prasad, K.; Repič, O.; Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/s0040-4039(00)95673-9 |
| 8. | Chen, K.-M.; Gunderson, K. G.; Hardtmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Chem. Lett. 1987, 16, 1923–1926. doi:10.1246/cl.1987.1923 |
| 9. | Burova, S. A.; McDonald, F. E. J. Am. Chem. Soc. 2002, 124, 8188–8189. doi:10.1021/ja026255p |
| 10. | Walleser, P.; Brückner, R. Eur. J. Org. Chem. 2010, 4802–4822. doi:10.1002/ejoc.201000280 |
| 11. | Becerril-Jiménez, F.; Ward, D. E. Org. Lett. 2012, 14, 1648–1651. doi:10.1021/ol300432y |
| 12. | Saito, S.; Hasegawa, T.; Inaba, M.; Nishida, R.; Fujii, T.; Nomizu, S.; Moriwake, T. Chem. Lett. 1984, 13, 1389–1392. doi:10.1246/cl.1984.1389 |
| 13. | Saito, S.; Nagao, Y.; Miyazaki, M.; Inaba, M.; Moriwake, T. Tetrahedron Lett. 1986, 27, 5249–5252. doi:10.1016/s0040-4039(00)85181-3 |
| 14. | Saito, S.; Ishikawa, T.; Kuroda, A.; Koga, K.; Moriwake, T. Tetrahedron 1992, 48, 4067–4086. doi:10.1016/s0040-4020(01)92187-8 |
| 15. | Hamada, Y.; Yokokawa, F.; Kabeya, M.; Hatano, K.; Kurono, Y.; Shioiri, T. Tetrahedron 1996, 52, 8297–8306. doi:10.1016/0040-4020(96)00383-3 |
| 16. | Sauret-Cladière, S.; Jeminet, G. Tetrahedron: Asymmetry 1997, 8, 417–423. doi:10.1016/s0957-4166(96)00517-4 |
| 17. | Nakatani, S.; Ikura, M.; Yamamoto, S.; Nishita, Y.; Itadani, S.; Habashita, H.; Sugiura, T.; Ogawa, K.; Ohno, H.; Takahashi, K.; Nakai, H.; Toda, M. Bioorg. Med. Chem. 2006, 14, 5402–5422. doi:10.1016/j.bmc.2006.03.032 |
| 18. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 19. | Li, D.; Zhao, Y.; Ye, L.; Chen, C.; Zhang, J. Synthesis 2010, 3325–3331. doi:10.1055/s-0030-1258187 |
| 20. | Kitamura, M.; Ohkuma, T.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1988, 29, 1555–1556. doi:10.1016/s0040-4039(00)80350-0 |
| 21. | Shao, L.; Kawano, H.; Saburi, M.; Uchida, Y. Tetrahedron 1993, 49, 1997–2010. doi:10.1016/s0040-4020(01)86300-6 |
| 22. | Jeulin, S.; Duprat de Paule, S.; Ratovelomanana-Vidal, V.; Genêt, J.-P.; Champion, N.; Dellis, P. Angew. Chem., Int. Ed. 2004, 43, 320–325. doi:10.1002/anie.200352453 |
| 23. | Qiu, L.; Wu, J.; Chan, S.; Au-Yeung, T. T.-L.; Ji, J.-X.; Guo, R.; Pai, C.-C.; Zhou, Z.; Li, X.; Fan, Q.-H.; Chan, A. S. C. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5815–5820. doi:10.1073/pnas.0307774101 |
| 9. | Burova, S. A.; McDonald, F. E. J. Am. Chem. Soc. 2002, 124, 8188–8189. doi:10.1021/ja026255p |
| 10. | Walleser, P.; Brückner, R. Eur. J. Org. Chem. 2010, 4802–4822. doi:10.1002/ejoc.201000280 |
| 11. | Becerril-Jiménez, F.; Ward, D. E. Org. Lett. 2012, 14, 1648–1651. doi:10.1021/ol300432y |
| 56. | Kim, J.-I.; Lee, S.-K.; Jahng, Y. Arch. Pharmacal Res. 1994, 17, 161–165. doi:10.1007/bf02974252 |
| 57. | Hagiwara, H.; Kon-no, M.; Nakano, T.; Uda, H. J. Chem. Soc., Perkin Trans. 1 1994, 2417. doi:10.1039/p19940002417 |
| 58. | Jefferies, P. R.; Gengo, P. J.; Watson, M. J.; Casida, J. E. J. Med. Chem. 1996, 39, 2339–2346. doi:10.1021/jm950712d |
| 59. | Karisalmi, K.; Koskinen, A. M. P. Synthesis 2004, 1331–1342. doi:10.1055/s-2004-822389 |
| 60. | Tarur, V. R.; Sathe, D. G.; Rao, N. M.; Bhopalkar, R.; Mahajan, U. S. Process for the preparation of fluvastatin sodium and intermediates. PCT Int. Appl. WO 2007023503 A1, March 1, 2007. |
| 61. | Renata, H.; Zhou, Q.; Baran, P. S. Science 2013, 339, 59–63. doi:10.1126/science.1230631 |
| 62. | Renata, H.; Zhou, Q.; Dünstl, G.; Felding, J.; Merchant, R. R.; Yeh, C.-H.; Baran, P. S. J. Am. Chem. Soc. 2015, 137, 1330–1340. doi:10.1021/ja512022r |
| 4. | Narasaka, K.; Pai, H. C. Chem. Lett. 1980, 9, 1415–1418. doi:10.1246/cl.1980.1415 |
| 5. | Narasaka, K.; Pai, F.-C. Tetrahedron 1984, 40, 2233–2238. doi:10.1016/0040-4020(84)80006-x |
| 6. | Kathawala, F. G.; Prager, B.; Prasad, K.; Repič, O.; Shapiro, M. J.; Stabler, R. S.; Widler, L. Helv. Chim. Acta 1986, 69, 803–805. doi:10.1002/hlca.19860690407 |
| 7. | Chen, K.-M.; Hardtmann, G. E.; Prasad, K.; Repič, O.; Shapiro, M. J. Tetrahedron Lett. 1987, 28, 155–158. doi:10.1016/s0040-4039(00)95673-9 |
| 8. | Chen, K.-M.; Gunderson, K. G.; Hardtmann, G. E.; Prasad, K.; Repic, O.; Shapiro, M. J. Chem. Lett. 1987, 16, 1923–1926. doi:10.1246/cl.1987.1923 |
| 9. | Burova, S. A.; McDonald, F. E. J. Am. Chem. Soc. 2002, 124, 8188–8189. doi:10.1021/ja026255p |
| 10. | Walleser, P.; Brückner, R. Eur. J. Org. Chem. 2010, 4802–4822. doi:10.1002/ejoc.201000280 |
| 11. | Becerril-Jiménez, F.; Ward, D. E. Org. Lett. 2012, 14, 1648–1651. doi:10.1021/ol300432y |
| 52. | Curran, D. P. J. Am. Chem. Soc. 1983, 105, 5826–5833. doi:10.1021/ja00356a021 |
| 3. | Evans, D. A.; Chapman, K. T.; Carreira, E. M. J. Am. Chem. Soc. 1988, 110, 3560–3578. doi:10.1021/ja00219a035 |
| 53. | Song, Z. J.; Zhao, M.; Desmond, R.; Devine, P.; Tschaen, D. M.; Tillyer, R.; Frey, L.; Heid, R.; Xu, F.; Foster, B.; Li, J.; Reamer, R.; Volante, R.; Dolling, U. H.; Reider, P. J.; Okada, S.; Kato, Y.; Mano, E. J. Org. Chem. 1999, 64, 9658–9667. doi:10.1021/jo991292t |
| 54. | Zhao, M.; Li, J.; Mano, E.; Song, Z.; Tschaen, D. M.; Grabowski, E. J. J.; Reider, P. J. J. Org. Chem. 1999, 64, 2564–2566. doi:10.1021/jo982143y |
| 55. | Jiang, X.; Zhang, J.; Ma, S. J. Am. Chem. Soc. 2016, 138, 8344–8347. doi:10.1021/jacs.6b03948 |
| 49. | Dong, L.; Geng, C.; Jiao, P. J. Org. Chem. 2015, 80, 10992–11002. doi:10.1021/acs.joc.5b02035 |
| 50. | Han, X.; Dong, L.; Geng, C.; Jiao, P. Org. Lett. 2015, 17, 3194–3197. doi:10.1021/acs.orglett.5b00826 |
| 51. | Jiang, M.; Feng, L.; Feng, J.; Jiao, P. Org. Lett. 2017, 19, 2210–2213. doi:10.1021/acs.orglett.7b00558 |
| 49. | Dong, L.; Geng, C.; Jiao, P. J. Org. Chem. 2015, 80, 10992–11002. doi:10.1021/acs.joc.5b02035 |
| 42. | Gijsen, H. J. M.; Wong, C.-H. J. Am. Chem. Soc. 1994, 116, 8422–8423. doi:10.1021/ja00097a082 |
| 49. | Dong, L.; Geng, C.; Jiao, P. J. Org. Chem. 2015, 80, 10992–11002. doi:10.1021/acs.joc.5b02035 |
| 42. | Gijsen, H. J. M.; Wong, C.-H. J. Am. Chem. Soc. 1994, 116, 8422–8423. doi:10.1021/ja00097a082 |
| 43. | Liu, J.; Hsu, C.-C.; Wong, C.-H. Tetrahedron Lett. 2004, 45, 2439–2441. doi:10.1016/j.tetlet.2004.01.110 |
| 44. | Öhrlein, R.; Baisch, G. Adv. Synth. Catal. 2003, 345, 713–715. doi:10.1002/adsc.200303028 |
| 45. | Greenberg, W. A.; Varvak, A.; Hanson, S. R.; Wong, K.; Huang, H.; Chen, P.; Burk, M. J. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5788–5793. doi:10.1073/pnas.0307563101 |
| 46. | Müller, M. Angew. Chem., Int. Ed. 2005, 44, 362–365. doi:10.1002/anie.200460852 |
| 47. | Guo, Z.; Chen, Y.; Goswami, A.; Hanson, R. L.; Patel, R. N. Tetrahedron: Asymmetry 2006, 17, 1589–1602. doi:10.1016/j.tetasy.2006.05.027 |
| 48. | Sun, F.; Xu, G.; Wu, J.; Yang, L. Tetrahedron: Asymmetry 2007, 18, 2454–2461. doi:10.1016/j.tetasy.2007.09.026 |
| 38. | Brower, P. L.; Butler, D. E.; Deering, C. F.; Le, T. V.; Millar, A.; Nanninga, T. N.; Roth, B. D. Tetrahedron Lett. 1992, 33, 2279–2282. doi:10.1016/s0040-4039(00)74189-x |
| 39. | Rádl, S. Synth. Commun. 2003, 33, 2275–2283. doi:10.1081/scc-120021507 |
| 40. | Wess, G.; Kesseler, K.; Baader, E.; Bartmann, W.; Beck, G.; Bergmann, A.; Jendralla, H.; Bock, K.; Holzstein, O.; Kleine, H.; Schnierer, M. Tetrahedron Lett. 1990, 31, 2545–2548. doi:10.1016/0040-4039(90)80121-2 |
| 41. | Reddy, M. S. N.; Reddy, B. K.; Reddy, C. K.; Kumar, M. K.; Rajan, S. T.; Reddy, M. S. Orient. J. Chem. 2008, 24, 167–174. |
| 49. | Dong, L.; Geng, C.; Jiao, P. J. Org. Chem. 2015, 80, 10992–11002. doi:10.1021/acs.joc.5b02035 |
| 70. | Ghosh, A. K.; Lei, H. J. Org. Chem. 2002, 67, 8783–8788. doi:10.1021/jo020402k |
| 63. | Labeeuw, O.; Roche, C.; Phansavath, P.; Genêt, J.-P. Org. Lett. 2007, 9, 105–108. doi:10.1021/ol062631p |
| 64. | Roche, C.; Labeeuw, O.; Haddad, M.; Ayad, T.; Genêt, J.-P.; Ratovelomanana-Vidal, V.; Phansavath, P. Eur. J. Org. Chem. 2009, 3977–3986. doi:10.1002/ejoc.200900316 |
| 65. | Besse, P.; Ciblat, S.; Canet, J.-L.; Troin, Y.; Veschambre, H. Tetrahedron: Asymmetry 1999, 10, 2213–2224. doi:10.1016/s0957-4166(99)00234-7 |
| 66. | Koide, K.; Finkelstein, J. M.; Ball, Z.; Verdine, G. L. J. Am. Chem. Soc. 2001, 123, 398–408. doi:10.1021/ja0023377 |
| 67. | Goswami, D.; Koli, M. R.; Chatterjee, S.; Chattopadhyay, S.; Sharma, A. Org. Biomol. Chem. 2017, 15, 3756–3774. doi:10.1039/c7ob00626h |
| 68. | Ma, X.; Diane, M.; Ralph, G.; Chen, C.; Biscoe, M. R. Angew. Chem., Int. Ed. 2017, 56, 12663–12667. doi:10.1002/anie.201704672 |
| 69. | Ganganna, B.; Srihari, P.; Yadav, J. S. Tetrahedron Lett. 2017, 58, 2685–2689. doi:10.1016/j.tetlet.2017.05.034 |
| 28. | Fan, W.; Li, W.; Ma, X.; Tao, X.; Li, X.; Yao, Y.; Xie, X.; Zhang, Z. J. Org. Chem. 2011, 76, 9444–9451. doi:10.1021/jo201822k |
| 26. | Szpilman, A. M.; Cereghetti, D. M.; Wurtz, N. R.; Manthorpe, J. M.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4335–4338. doi:10.1002/anie.200800589 |
| 27. | Szpilman, A. M.; Cereghetti, D. M.; Manthorpe, J. M.; Wurtz, N. R.; Carreira, E. M. Chem. – Eur. J. 2009, 15, 7117–7128. doi:10.1002/chem.200900231 |
| 26. | Szpilman, A. M.; Cereghetti, D. M.; Wurtz, N. R.; Manthorpe, J. M.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4335–4338. doi:10.1002/anie.200800589 |
| 27. | Szpilman, A. M.; Cereghetti, D. M.; Manthorpe, J. M.; Wurtz, N. R.; Carreira, E. M. Chem. – Eur. J. 2009, 15, 7117–7128. doi:10.1002/chem.200900231 |
| 70. | Ghosh, A. K.; Lei, H. J. Org. Chem. 2002, 67, 8783–8788. doi:10.1021/jo020402k |
| 26. | Szpilman, A. M.; Cereghetti, D. M.; Wurtz, N. R.; Manthorpe, J. M.; Carreira, E. M. Angew. Chem., Int. Ed. 2008, 47, 4335–4338. doi:10.1002/anie.200800589 |
| 43. | Liu, J.; Hsu, C.-C.; Wong, C.-H. Tetrahedron Lett. 2004, 45, 2439–2441. doi:10.1016/j.tetlet.2004.01.110 |
| 71. | Das, S.; Goswami, R. K. J. Org. Chem. 2014, 79, 9778–9791. doi:10.1021/jo5019798 |
© 2019 Feng et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)