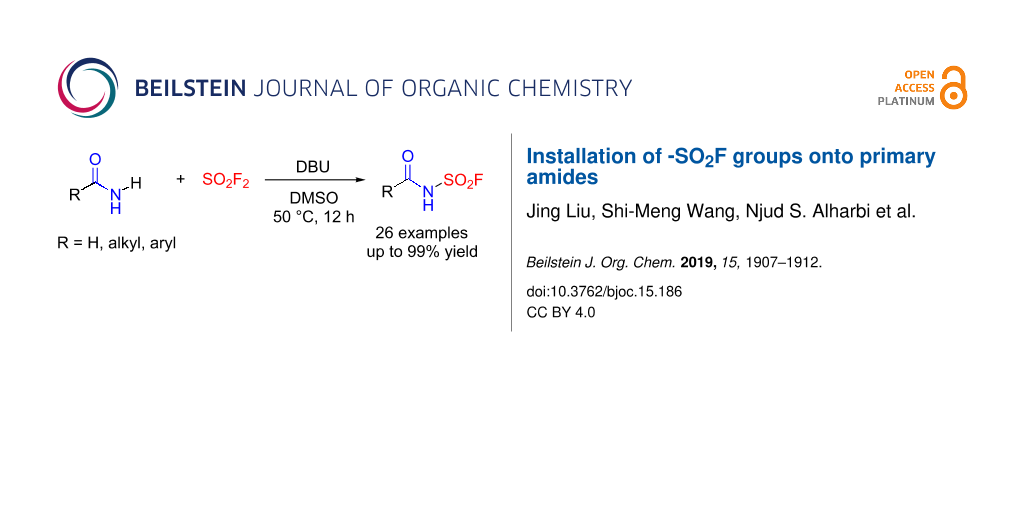
Abstract
A protocol of SO2F2-mediated installation of sulfonyl fluoride onto primary amides has been developed providing a new portal to sulfur(VI) fluoride exchange (SuFEx) click chemistry. The generated molecules contain pharmaceutically important amide and -SO2F moieties for application in the discovery of new therapeutics.

Graphical Abstract
Introduction
Sulfur(VI) fluoride exchange (SuFEx) is a new class of click chemistry developed by Sharpless and co-workers in 2014, for creating molecular connections based on the unique stability–reactivity pattern of the S(VI)–F bond with reliability and efficiency, which has been widely applied in organic synthesis, chemical biology and drug discovery [1-19]. Among all the developed S(VI)–F species, sulfonyl fluoride (RSO2F) was specifically recognized as unique scaffold for covalent protein inhibitors and biological probes with the affinity-driven activation for forming covalent linkages with the amino acid residues of protein binding sites (Figure 1) [20]. The smallest member of this family, methyl sulfonyl fluoride (MSF), is known as a selective and irreversible inhibitor of acetylcholinesterase (AChE) [21,22]. The sulfonyl fluoride inhibitors NSC 127755 was found for specifically modifying tyrosine-31 of DHFR in chicken liver [23]. The nucleotide-derived probe 5’-(para-fluorosulfonylbenzoyl)adenosine (5’-FSBA) was used for labelling the second nucleotide binding site, the adenine nucleotide regulatory site [24]. In addition, aryl fluorosulfates have also been widely applied as sustainable alternative to aryl halides in coupling reactions and as potential covalent probes in protein profiling [14,25-28].
![[1860-5397-15-186-1]](/bjoc/content/figures/1860-5397-15-186-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative sulfonyl fluoride compounds applied in medicinal chemistry and chemical biology.
Figure 1: Representative sulfonyl fluoride compounds applied in medicinal chemistry and chemical biology.
Phenols (or alcohols) and amines as the most common nucleophiles have been found to react with different S(VI) connectors (SO2F2, CH2=CH-SO2F, SOF4 etc.) to provide diversified sulfonyl fluoride derivatives. The reactions of phenols (or alcohols) with SO2F2 [29] or the fluorosulfuryl imidazolium salt were developed for mild and effective formation of the corresponding fluorosulfates to act as biological probes in chemical proteomics studies (Scheme 1, (1)) [1,30]. On the other hand, the reactions of aliphatic or aromatic amines with SO2F2 or the fluorosulfurylimidazolium salt have been achieved for assembly of N-sulfonyl fluorides [1,30], which have served as important active precursors for the development of noncovalent inhibitors (Scheme 1, (1)) [1,30,31]. Amides are the key connections in proteins, amides, and a vast number of synthetic structures, such as polymers, biologically active compounds and pharmaceutical products [32-35]. However, the installation of sulfonyl fluoride (SO2F) onto nitrogen atoms of amides has not been achieved, which, if accomplished, would provide a very important class of sulfonyl fluorides, namely, N-fluorosulfonyl amides, for the development of potential covalent inhibitors [1-24]. The Roesky group described a pioneering protocol for the synthesis of N-fluorosulfonyl amides from fluorosulfonyl isocyanate (Scheme 1, (2)) [36]. The available procedures for the preparation of N-fluorosulfonyl amides are very limited which relied on using either the isocyanate approach, or the amidosulfofluoride (FSO2NH2) (Scheme 1, (2)) [37-39]. Therefore, the development of a new method for the assembly of N-fluorosulfonyl amides from cheap and abundant reagent is highly desirable. Herein, we report the first, to the best of our knowledge, SO2F2-mediated N-fluorosulfonylation [40-42] of amides by using DBU as base for the constructions of a series N-fluorosulfonyl amides (Scheme 1b).
![[1860-5397-15-186-i1]](/bjoc/content/inline/1860-5397-15-186-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis background of N-fluorosulfonyl amides and fluorosulfates.
Scheme 1: Synthesis background of N-fluorosulfonyl amides and fluorosulfates.
Results and Discussions
Initially, benzamide (1a) was selected as model substrate to test the feasibility of this proposed N-fluorosulfonylation reaction in the presence of Cs2CO3 in DMSO under SO2F2 atmosphere (balloon) at 50 °C, and excitingly, the desired product benzoylsulfamoyl fluoride (2a) was obtained in 25% yield (Table 1, entry 1). Encouraged by this preliminary success, several common bases were evaluated, among which, 1,8-diazabicycloundec-7-ene (DBU) catalysed the proposed transformation most effectively to provide the desired product 2a in nearly quantitative yield (Table 1, entries 2–7). Subsequently, different solvents were screened (Table 1, entries 5, 8–12) and DMSO was found to be the best option. Decreasing the temperature from 50 °C to 40 °C or even room temperature, or cutting down the amount of DBU to 4 equivalents resulted in decreased yields (Table 1, entries 13–15).
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-186-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base | Solvent | Temp. (°C) | Yield (2a, %)b |
| 1 | Cs2CO3 | DMSO | 50 | 25 |
| 2 | K2CO3 | DMSO | 50 | 13 |
| 3 | KOH | DMSO | 50 | 19 |
| 4 | NaOH | DMSO | 50 | 15 |
| 5 | DBU | DMSO | 50 | 99 |
| 6 | Et3N | DMSO | 50 | – |
| 7 | DIPEA | DMSO | 50 | – |
| 8 | DBU | NMP | 50 | 81 |
| 9 | DBU | MeCN | 50 | 75 |
| 10 | DBU | toluene | 50 | 87 |
| 11 | DBU | dioxane | 50 | 60 |
| 12 | DBU | THF | 50 | 79 |
| 13 | DBU | DMSO | 40 | 82 |
| 14 | DBU | DMSO | R.T. | 51 |
| 15c | DBU | DMSO | 50 | 69 |
aReaction conditions: benzamide (1a, 1.0 mmol, 1.0 equiv), DBU (5.0 equiv), and DMSO (1.0 mL) stirred with a SO2F2 balloon for 12 h. bIsolated yield. c4 equiv of DBU was used.
With the optimized conditions in hand, we next turned our efforts to investigate the scope of substrates. Under the standard conditions, a variety of substituted amides were examined which were smoothly converted to their corresponding substituted benzoylsulfamoyl fluoride derivatives (Scheme 2) in moderate to excellent isolated yields. Both electron-withdrawing groups, such as halogen atoms (1b–d, 1j, 1m, and 1n), NO2 (1e, 1k) and CF3 (1f), and electron-donating groups, such as Me (1g, 1l, and 1o), tert-butyl (1h) and 2-naphthyl (1i) on the aromatic rings, were well tolerated under the optimized conditions. It was worth noting that not only para- (1b–h) but also meta- (1j–l) and ortho- (1m–o) substituted benzamides afforded the desired products in generally good yields. Arylcarboxylic amides (1p and 1q) bearing bis-substitutions also behaved well under the standard conditions. Heterocyclic aromatic carboxylic amides (1r–u) were well-tolerated and afforded the target products in 56–90% yields. In addition, alkyl carboxylic amides were also smoothly transformed into the corresponding products (2v–z). However, primary amides bearing an amino group or a phenolic hydroxy group were not successfully converted to the corresponding N-fluorosulfonyl amides and only a mixture of undesired products were observed.
![[1860-5397-15-186-i2]](/bjoc/content/inline/1860-5397-15-186-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Screening of the substrate scope of amides. Reaction conditions: a mixture of amides 1 (1.0 mmol), DBU (5.0 mmol, 5.0 equiv), and DMSO (1.0 mL) was added to a reaction flask before SO2F2 was introduced into the stirred reaction mixture by slowly bubbling from a balloon, and the mixture was allowed to stir at 50 °C for 12 h. Isolated yields. a50 °C, 18 h.
Scheme 2: Screening of the substrate scope of amides. Reaction conditions: a mixture of amides 1 (1.0 mmol), ...
Interestingly, during the work-up process of drying 2e with Na2SO4, a colourless crystal 4e was observed and its structure was confirmed by XRD analysis (Scheme 3). We speculate that the tautomerism of amides [43] may occur in the reaction process and the tautomer 3e could react with Na2SO4 to generate 4e, which indicated that N–H connected with two electron-withdrawing groups (carbonyl, and SO2F) can behave as an acid to donate a proton for chemical transformations. This property of fluorosulfonyl amides 2 with nucleophilicity may attract significant attention for further applications.
![[1860-5397-15-186-i3]](/bjoc/content/inline/1860-5397-15-186-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Amide resonance model and X-ray single crystal structure of 4e (CCDC 1906002).
Scheme 3: Amide resonance model and X-ray single crystal structure of 4e (CCDC 1906002).
As depicted in Scheme 4, a plausible reaction mechanism is proposed for SO2F2-mediated transformation of amides to N-fluorosulfonyl amides. The reaction was initiated by the deprotonation of amide 1 with the base (DBU) to generate an intermediate A, which subsequently went through a SuFEx process with SO2F2 to deliver the final product 2.
![[1860-5397-15-186-i4]](/bjoc/content/inline/1860-5397-15-186-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The proposed reaction mechanism.
Scheme 4: The proposed reaction mechanism.
Conclusion
In conclusion, we have developed a novel method for N-fluorosulfonylation of amides. This simple, convenient, and mild protocol provides a portal to a class of novel sulfonyl fluorides for SuFEx click chemistry with great potential to be applied in the development of covalent inhibitors. Further studies of this class of molecules in chemical biology and drug discovery are underway in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.7 MB | Download |
| Supporting Information File 2: Crystallographic information file of 4e. | ||
| Format: CIF | Size: 14.6 KB | Download |
| Supporting Information File 3: Checkcif file of 4e. | ||
| Format: PDF | Size: 245.1 KB | Download |
Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grant No. 21772150), the Wuhan applied fundamental research plan of Wuhan Science and Technology Bureau (grant NO. 2017060201010216), the 111 Project (No. B18038) and Wuhan University of Technology for the financial support.
References
-
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Angew. Chem. 2014, 126, 9584–9602. doi:10.1002/ange.201309399
Return to citation in text: [1] [2] [3] [4] [5] -
Wang, H.; Zhou, F.; Ren, G.; Zheng, Q.; Chen, H.; Gao, B.; Klivansky, L.; Liu, Y.; Wu, B.; Xu, Q.; Lu, J.; Sharpless, K. B.; Wu, P. Angew. Chem., Int. Ed. 2017, 56, 11203–11208. doi:10.1002/anie.201701160
Angew. Chem. 2017, 129, 11355–11360. doi:10.1002/ange.201701160
Return to citation in text: [1] [2] -
Gao, B.; Zhang, L.; Zheng, Q.; Zhou, F.; Klivansky, L. M.; Lu, J.; Liu, Y.; Dong, J.; Wu, P.; Sharpless, K. B. Nat. Chem. 2017, 9, 1083–1088. doi:10.1038/nchem.2796
Return to citation in text: [1] [2] -
Liu, Z.; Li, J.; Li, S.; Li, G.; Sharpless, K. B.; Wu, P. J. Am. Chem. Soc. 2018, 140, 2919–2925. doi:10.1021/jacs.7b12788
Return to citation in text: [1] [2] -
Qin, H.-L.; Zheng, Q.; Bare, G. A. L.; Wu, P.; Sharpless, K. B. Angew. Chem., Int. Ed. 2016, 55, 14155–14158. doi:10.1002/anie.201608807
Angew. Chem. 2016, 128, 14361–14364. doi:10.1002/ange.201608807
Return to citation in text: [1] [2] -
Zha, G.-F.; Zheng, Q.; Leng, J.; Wu, P.; Qin, H.-L.; Sharpless, K. B. Angew. Chem., Int. Ed. 2017, 56, 4849–4852. doi:10.1002/anie.201701162
Angew. Chem. 2017, 129, 4927–4930. doi:10.1002/ange.201701162
Return to citation in text: [1] [2] -
Schimler, S. D.; Cismesia, M. A.; Hanley, P. S.; Froese, R. D. J.; Jansma, M. J.; Bland, D. C.; Sanford, M. S. J. Am. Chem. Soc. 2017, 139, 1452–1455. doi:10.1021/jacs.6b12911
Return to citation in text: [1] [2] -
Epifanov, M.; Foth, P. J.; Gu, F.; Barrillon, C.; Kanani, S. S.; Higman, C. S.; Hein, J. E.; Sammis, G. M. J. Am. Chem. Soc. 2018, 140, 16464–16468. doi:10.1021/jacs.8b11309
Return to citation in text: [1] [2] -
Liang, Q.; Xing, P.; Huang, Z.; Dong, J.; Sharpless, K. B.; Li, X.; Jiang, B. Org. Lett. 2015, 17, 1942–1945. doi:10.1021/acs.orglett.5b00654
Return to citation in text: [1] [2] -
Zhang, E.; Tang, J.; Li, S.; Wu, P.; Moses, J. E.; Sharpless, K. B. Chem. – Eur. J. 2016, 22, 5692–5697. doi:10.1002/chem.201600167
Return to citation in text: [1] [2] -
Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891
Return to citation in text: [1] [2] -
Wang, X.-Y.; Leng, J.; Wang, S.-M.; Asiri, A. M.; Marwani, H. M.; Qin, H.-L. Tetrahedron Lett. 2017, 58, 2340–2343. doi:10.1016/j.tetlet.2017.04.070
Return to citation in text: [1] [2] -
Fang, W.-Y.; Huang, Y.-M.; Leng, J.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 751–756. doi:10.1002/ajoc.201800037
Return to citation in text: [1] [2] -
Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K. P.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 662–682. doi:10.1002/ajoc.201700591
Return to citation in text: [1] [2] [3] -
Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a
Return to citation in text: [1] [2] -
Zha, G.-F.; Fang, W.-Y.; Li, Y.-G.; Leng, J.; Chen, X.; Qin, H.-L. J. Am. Chem. Soc. 2018, 140, 17666–17673. doi:10.1021/jacs.8b10069
Return to citation in text: [1] [2] -
Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Eur. J. Org. Chem. 2019, 1801–1807. doi:10.1002/ejoc.201801888
Return to citation in text: [1] [2] -
Zhang, X.; Rakesh, K. P.; Qin, H.-L. Chem. Commun. 2019, 55, 2845–2848. doi:10.1039/c8cc09693g
Return to citation in text: [1] [2] -
Wang, S.-M.; Zhao, C.; Zhang, X.; Qin, H.-L. Org. Biomol. Chem. 2019, 17, 4087–4101. doi:10.1039/c9ob00699k
Return to citation in text: [1] [2] -
Narayanan, A.; Jones, L. H. Chem. Sci. 2015, 6, 2650–2659. doi:10.1039/c5sc00408j
Return to citation in text: [1] [2] -
Moss, D. E.; Berlanga, P.; Hagan, M. M.; Sandoval, H.; Ishida, C. Alzheimer Dis. Assoc. Disord. 1999, 13, 20–25. doi:10.1097/00002093-199903000-00003
Return to citation in text: [1] [2] -
Kitz, R.; Wilson, I. B. J. Biol. Chem. 1962, 237, 3245–3249.
Return to citation in text: [1] [2] -
Kumar, A. A.; Mangum, J. H.; Blankenship, D. T.; Freisheim, J. H. J. Biol. Chem. 1981, 256, 8970–8976.
Return to citation in text: [1] [2] -
Esch, F. S.; Allison, W. S. J. Biol. Chem. 1978, 253, 6100–6106.
Return to citation in text: [1] [2] -
Hanley, P. S.; Clark, T. P.; Krasovskiy, A. L.; Ober, M. S.; O’Brien, J. P.; Staton, T. S. ACS Catal. 2016, 6, 3515–3519. doi:10.1021/acscatal.6b00865
Return to citation in text: [1] -
Mortenson, D. E.; Brighty, G. J.; Plate, L.; Bare, G.; Chen, W.; Li, S.; Wang, H.; Cravatt, B. F.; Forli, S.; Powers, E. T.; Sharpless, K. B.; Wilson, I. A.; Kelly, J. W. J. Am. Chem. Soc. 2018, 140, 200–210. doi:10.1021/jacs.7b08366
Return to citation in text: [1] -
Chen, W.; Dong, J.; Plate, L.; Mortenson, D. E.; Brighty, G. J.; Li, S.; Liu, Y.; Galmozzi, A.; Lee, P. S.; Hulce, J. J.; Cravatt, B. F.; Saez, E.; Powers, E. T.; Wilson, I. A.; Sharpless, K. B.; Kelly, J. W. J. Am. Chem. Soc. 2016, 138, 7353–7364. doi:10.1021/jacs.6b02960
Return to citation in text: [1] -
Gilles, P.; Veryser, C.; Vangrunderbeeck, S.; Ceusters, S.; Van Meervelt, L.; De Borggraeve, W. M. J. Org. Chem. 2019, 84, 1070–1078. doi:10.1021/acs.joc.8b02785
Return to citation in text: [1] -
Sulbaek Andersen, M. P.; Blake, D. R.; Rowland, F. S.; Hurley, M. D.; Wallington, T. J. Environ. Sci. Technol. 2009, 43, 1067–1070. doi:10.1021/es802439f
Return to citation in text: [1] -
Guo, T.; Meng, G.; Zhan, X.; Yang, Q.; Ma, T.; Xu, L.; Sharpless, K. B.; Dong, J. Angew. Chem., Int. Ed. 2018, 57, 2605–2610. doi:10.1002/anie.201712429
Angew. Chem. 2018, 130, 2635–2640. doi:10.1002/ange.201712429
Return to citation in text: [1] [2] [3] -
Spillane, W.; Malaubier, J.-B. Chem. Rev. 2014, 114, 2507–2586. doi:10.1021/cr400230c
Return to citation in text: [1] -
Greenberg, A.; Breneman, C. M.; Liebman, J. F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; Wiley-Interscience: Hoboken, NJ, 2000.
Return to citation in text: [1] -
Wieland, T.; Bodanszky, M. The World of Peptides: A Brief History of Peptide Chemistry; Springer-Verlag: New York, 1991.
Return to citation in text: [1] -
de Figueiredo, R. M.; Suppo, J.-S.; Campagne, J.-M. Chem. Rev. 2016, 116, 12029–12122. doi:10.1021/acs.chemrev.6b00237
Return to citation in text: [1] -
Crespo, L.; Sanclimens, G.; Pons, M.; Giralt, E.; Royo, M.; Albericio, F. Chem. Rev. 2005, 105, 1663–1682. doi:10.1021/cr030449l
Return to citation in text: [1] -
Roesky, H. W.; Giere, H.-H. Chem. Ber. 1969, 102, 3707–3712. doi:10.1002/cber.19691021112
Return to citation in text: [1] -
Clauβ, K.; Friedrich, H.-J.; Jensen, H. Justus Liebigs Ann. Chem. 1974, 561–592. doi:10.1002/jlac.197419740404
Return to citation in text: [1] -
Pietsch, H.; Clauss, K.; Jensen, H.; Schmidt, E. Verfahren Zur Herstellung Von Acetoacetamid-N-sulfofluorid. Ger. Pat. Appl. DE2453063A1, May 13, 1976.
Return to citation in text: [1] -
Linkies, A.; Reuschling, D. Process for preparing crystalline salts of acetoacetamide-N-sulfofluoride. U.S. Patent US4618455, Oct 21, 1986.
Return to citation in text: [1] -
Appel, R.; Rittersbacher, H. Chem. Ber. 1964, 97, 849–851. doi:10.1002/cber.19640970330
Return to citation in text: [1] -
Appel, R.; Montenarh, M. Chem. Ber. 1976, 109, 2437–2441. doi:10.1002/cber.19761090710
Return to citation in text: [1] -
Beran, M.; Příhoda, J.; Taraba, J. Polyhedron 2010, 29, 991–994. doi:10.1016/j.poly.2009.11.024
Return to citation in text: [1] -
Liu, C.; Shi, S.; Liu, Y.; Liu, R.; Lalancette, R.; Szostak, R.; Szostak, M. Org. Lett. 2018, 20, 7771–7774. doi:10.1021/acs.orglett.8b03175
Return to citation in text: [1]
| 1. |
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Angew. Chem. 2014, 126, 9584–9602. doi:10.1002/ange.201309399 |
| 2. |
Wang, H.; Zhou, F.; Ren, G.; Zheng, Q.; Chen, H.; Gao, B.; Klivansky, L.; Liu, Y.; Wu, B.; Xu, Q.; Lu, J.; Sharpless, K. B.; Wu, P. Angew. Chem., Int. Ed. 2017, 56, 11203–11208. doi:10.1002/anie.201701160
Angew. Chem. 2017, 129, 11355–11360. doi:10.1002/ange.201701160 |
| 3. | Gao, B.; Zhang, L.; Zheng, Q.; Zhou, F.; Klivansky, L. M.; Lu, J.; Liu, Y.; Dong, J.; Wu, P.; Sharpless, K. B. Nat. Chem. 2017, 9, 1083–1088. doi:10.1038/nchem.2796 |
| 4. | Liu, Z.; Li, J.; Li, S.; Li, G.; Sharpless, K. B.; Wu, P. J. Am. Chem. Soc. 2018, 140, 2919–2925. doi:10.1021/jacs.7b12788 |
| 5. |
Qin, H.-L.; Zheng, Q.; Bare, G. A. L.; Wu, P.; Sharpless, K. B. Angew. Chem., Int. Ed. 2016, 55, 14155–14158. doi:10.1002/anie.201608807
Angew. Chem. 2016, 128, 14361–14364. doi:10.1002/ange.201608807 |
| 6. |
Zha, G.-F.; Zheng, Q.; Leng, J.; Wu, P.; Qin, H.-L.; Sharpless, K. B. Angew. Chem., Int. Ed. 2017, 56, 4849–4852. doi:10.1002/anie.201701162
Angew. Chem. 2017, 129, 4927–4930. doi:10.1002/ange.201701162 |
| 7. | Schimler, S. D.; Cismesia, M. A.; Hanley, P. S.; Froese, R. D. J.; Jansma, M. J.; Bland, D. C.; Sanford, M. S. J. Am. Chem. Soc. 2017, 139, 1452–1455. doi:10.1021/jacs.6b12911 |
| 8. | Epifanov, M.; Foth, P. J.; Gu, F.; Barrillon, C.; Kanani, S. S.; Higman, C. S.; Hein, J. E.; Sammis, G. M. J. Am. Chem. Soc. 2018, 140, 16464–16468. doi:10.1021/jacs.8b11309 |
| 9. | Liang, Q.; Xing, P.; Huang, Z.; Dong, J.; Sharpless, K. B.; Li, X.; Jiang, B. Org. Lett. 2015, 17, 1942–1945. doi:10.1021/acs.orglett.5b00654 |
| 10. | Zhang, E.; Tang, J.; Li, S.; Wu, P.; Moses, J. E.; Sharpless, K. B. Chem. – Eur. J. 2016, 22, 5692–5697. doi:10.1002/chem.201600167 |
| 11. | Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891 |
| 12. | Wang, X.-Y.; Leng, J.; Wang, S.-M.; Asiri, A. M.; Marwani, H. M.; Qin, H.-L. Tetrahedron Lett. 2017, 58, 2340–2343. doi:10.1016/j.tetlet.2017.04.070 |
| 13. | Fang, W.-Y.; Huang, Y.-M.; Leng, J.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 751–756. doi:10.1002/ajoc.201800037 |
| 14. | Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K. P.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 662–682. doi:10.1002/ajoc.201700591 |
| 15. | Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a |
| 16. | Zha, G.-F.; Fang, W.-Y.; Li, Y.-G.; Leng, J.; Chen, X.; Qin, H.-L. J. Am. Chem. Soc. 2018, 140, 17666–17673. doi:10.1021/jacs.8b10069 |
| 17. | Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Eur. J. Org. Chem. 2019, 1801–1807. doi:10.1002/ejoc.201801888 |
| 18. | Zhang, X.; Rakesh, K. P.; Qin, H.-L. Chem. Commun. 2019, 55, 2845–2848. doi:10.1039/c8cc09693g |
| 19. | Wang, S.-M.; Zhao, C.; Zhang, X.; Qin, H.-L. Org. Biomol. Chem. 2019, 17, 4087–4101. doi:10.1039/c9ob00699k |
| 40. | Appel, R.; Rittersbacher, H. Chem. Ber. 1964, 97, 849–851. doi:10.1002/cber.19640970330 |
| 41. | Appel, R.; Montenarh, M. Chem. Ber. 1976, 109, 2437–2441. doi:10.1002/cber.19761090710 |
| 42. | Beran, M.; Příhoda, J.; Taraba, J. Polyhedron 2010, 29, 991–994. doi:10.1016/j.poly.2009.11.024 |
| 23. | Kumar, A. A.; Mangum, J. H.; Blankenship, D. T.; Freisheim, J. H. J. Biol. Chem. 1981, 256, 8970–8976. |
| 43. | Liu, C.; Shi, S.; Liu, Y.; Liu, R.; Lalancette, R.; Szostak, R.; Szostak, M. Org. Lett. 2018, 20, 7771–7774. doi:10.1021/acs.orglett.8b03175 |
| 21. | Moss, D. E.; Berlanga, P.; Hagan, M. M.; Sandoval, H.; Ishida, C. Alzheimer Dis. Assoc. Disord. 1999, 13, 20–25. doi:10.1097/00002093-199903000-00003 |
| 22. | Kitz, R.; Wilson, I. B. J. Biol. Chem. 1962, 237, 3245–3249. |
| 36. | Roesky, H. W.; Giere, H.-H. Chem. Ber. 1969, 102, 3707–3712. doi:10.1002/cber.19691021112 |
| 20. | Narayanan, A.; Jones, L. H. Chem. Sci. 2015, 6, 2650–2659. doi:10.1039/c5sc00408j |
| 37. | Clauβ, K.; Friedrich, H.-J.; Jensen, H. Justus Liebigs Ann. Chem. 1974, 561–592. doi:10.1002/jlac.197419740404 |
| 38. | Pietsch, H.; Clauss, K.; Jensen, H.; Schmidt, E. Verfahren Zur Herstellung Von Acetoacetamid-N-sulfofluorid. Ger. Pat. Appl. DE2453063A1, May 13, 1976. |
| 39. | Linkies, A.; Reuschling, D. Process for preparing crystalline salts of acetoacetamide-N-sulfofluoride. U.S. Patent US4618455, Oct 21, 1986. |
| 1. |
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Angew. Chem. 2014, 126, 9584–9602. doi:10.1002/ange.201309399 |
| 30. |
Guo, T.; Meng, G.; Zhan, X.; Yang, Q.; Ma, T.; Xu, L.; Sharpless, K. B.; Dong, J. Angew. Chem., Int. Ed. 2018, 57, 2605–2610. doi:10.1002/anie.201712429
Angew. Chem. 2018, 130, 2635–2640. doi:10.1002/ange.201712429 |
| 32. | Greenberg, A.; Breneman, C. M.; Liebman, J. F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; Wiley-Interscience: Hoboken, NJ, 2000. |
| 33. | Wieland, T.; Bodanszky, M. The World of Peptides: A Brief History of Peptide Chemistry; Springer-Verlag: New York, 1991. |
| 34. | de Figueiredo, R. M.; Suppo, J.-S.; Campagne, J.-M. Chem. Rev. 2016, 116, 12029–12122. doi:10.1021/acs.chemrev.6b00237 |
| 35. | Crespo, L.; Sanclimens, G.; Pons, M.; Giralt, E.; Royo, M.; Albericio, F. Chem. Rev. 2005, 105, 1663–1682. doi:10.1021/cr030449l |
| 1. |
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Angew. Chem. 2014, 126, 9584–9602. doi:10.1002/ange.201309399 |
| 30. |
Guo, T.; Meng, G.; Zhan, X.; Yang, Q.; Ma, T.; Xu, L.; Sharpless, K. B.; Dong, J. Angew. Chem., Int. Ed. 2018, 57, 2605–2610. doi:10.1002/anie.201712429
Angew. Chem. 2018, 130, 2635–2640. doi:10.1002/ange.201712429 |
| 1. |
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Angew. Chem. 2014, 126, 9584–9602. doi:10.1002/ange.201309399 |
| 2. |
Wang, H.; Zhou, F.; Ren, G.; Zheng, Q.; Chen, H.; Gao, B.; Klivansky, L.; Liu, Y.; Wu, B.; Xu, Q.; Lu, J.; Sharpless, K. B.; Wu, P. Angew. Chem., Int. Ed. 2017, 56, 11203–11208. doi:10.1002/anie.201701160
Angew. Chem. 2017, 129, 11355–11360. doi:10.1002/ange.201701160 |
| 3. | Gao, B.; Zhang, L.; Zheng, Q.; Zhou, F.; Klivansky, L. M.; Lu, J.; Liu, Y.; Dong, J.; Wu, P.; Sharpless, K. B. Nat. Chem. 2017, 9, 1083–1088. doi:10.1038/nchem.2796 |
| 4. | Liu, Z.; Li, J.; Li, S.; Li, G.; Sharpless, K. B.; Wu, P. J. Am. Chem. Soc. 2018, 140, 2919–2925. doi:10.1021/jacs.7b12788 |
| 5. |
Qin, H.-L.; Zheng, Q.; Bare, G. A. L.; Wu, P.; Sharpless, K. B. Angew. Chem., Int. Ed. 2016, 55, 14155–14158. doi:10.1002/anie.201608807
Angew. Chem. 2016, 128, 14361–14364. doi:10.1002/ange.201608807 |
| 6. |
Zha, G.-F.; Zheng, Q.; Leng, J.; Wu, P.; Qin, H.-L.; Sharpless, K. B. Angew. Chem., Int. Ed. 2017, 56, 4849–4852. doi:10.1002/anie.201701162
Angew. Chem. 2017, 129, 4927–4930. doi:10.1002/ange.201701162 |
| 7. | Schimler, S. D.; Cismesia, M. A.; Hanley, P. S.; Froese, R. D. J.; Jansma, M. J.; Bland, D. C.; Sanford, M. S. J. Am. Chem. Soc. 2017, 139, 1452–1455. doi:10.1021/jacs.6b12911 |
| 8. | Epifanov, M.; Foth, P. J.; Gu, F.; Barrillon, C.; Kanani, S. S.; Higman, C. S.; Hein, J. E.; Sammis, G. M. J. Am. Chem. Soc. 2018, 140, 16464–16468. doi:10.1021/jacs.8b11309 |
| 9. | Liang, Q.; Xing, P.; Huang, Z.; Dong, J.; Sharpless, K. B.; Li, X.; Jiang, B. Org. Lett. 2015, 17, 1942–1945. doi:10.1021/acs.orglett.5b00654 |
| 10. | Zhang, E.; Tang, J.; Li, S.; Wu, P.; Moses, J. E.; Sharpless, K. B. Chem. – Eur. J. 2016, 22, 5692–5697. doi:10.1002/chem.201600167 |
| 11. | Fang, W.-Y.; Leng, J.; Qin, H.-L. Chem. – Asian J. 2017, 12, 2323–2331. doi:10.1002/asia.201700891 |
| 12. | Wang, X.-Y.; Leng, J.; Wang, S.-M.; Asiri, A. M.; Marwani, H. M.; Qin, H.-L. Tetrahedron Lett. 2017, 58, 2340–2343. doi:10.1016/j.tetlet.2017.04.070 |
| 13. | Fang, W.-Y.; Huang, Y.-M.; Leng, J.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 751–756. doi:10.1002/ajoc.201800037 |
| 14. | Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K. P.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 662–682. doi:10.1002/ajoc.201700591 |
| 15. | Zhao, C.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Org. Chem. Front. 2018, 5, 1835–1839. doi:10.1039/c8qo00295a |
| 16. | Zha, G.-F.; Fang, W.-Y.; Li, Y.-G.; Leng, J.; Chen, X.; Qin, H.-L. J. Am. Chem. Soc. 2018, 140, 17666–17673. doi:10.1021/jacs.8b10069 |
| 17. | Zhao, C.; Zha, G.-F.; Fang, W.-Y.; Rakesh, K. P.; Qin, H.-L. Eur. J. Org. Chem. 2019, 1801–1807. doi:10.1002/ejoc.201801888 |
| 18. | Zhang, X.; Rakesh, K. P.; Qin, H.-L. Chem. Commun. 2019, 55, 2845–2848. doi:10.1039/c8cc09693g |
| 19. | Wang, S.-M.; Zhao, C.; Zhang, X.; Qin, H.-L. Org. Biomol. Chem. 2019, 17, 4087–4101. doi:10.1039/c9ob00699k |
| 20. | Narayanan, A.; Jones, L. H. Chem. Sci. 2015, 6, 2650–2659. doi:10.1039/c5sc00408j |
| 21. | Moss, D. E.; Berlanga, P.; Hagan, M. M.; Sandoval, H.; Ishida, C. Alzheimer Dis. Assoc. Disord. 1999, 13, 20–25. doi:10.1097/00002093-199903000-00003 |
| 22. | Kitz, R.; Wilson, I. B. J. Biol. Chem. 1962, 237, 3245–3249. |
| 23. | Kumar, A. A.; Mangum, J. H.; Blankenship, D. T.; Freisheim, J. H. J. Biol. Chem. 1981, 256, 8970–8976. |
| 24. | Esch, F. S.; Allison, W. S. J. Biol. Chem. 1978, 253, 6100–6106. |
| 29. | Sulbaek Andersen, M. P.; Blake, D. R.; Rowland, F. S.; Hurley, M. D.; Wallington, T. J. Environ. Sci. Technol. 2009, 43, 1067–1070. doi:10.1021/es802439f |
| 14. | Revathi, L.; Ravindar, L.; Leng, J.; Rakesh, K. P.; Qin, H.-L. Asian J. Org. Chem. 2018, 7, 662–682. doi:10.1002/ajoc.201700591 |
| 25. | Hanley, P. S.; Clark, T. P.; Krasovskiy, A. L.; Ober, M. S.; O’Brien, J. P.; Staton, T. S. ACS Catal. 2016, 6, 3515–3519. doi:10.1021/acscatal.6b00865 |
| 26. | Mortenson, D. E.; Brighty, G. J.; Plate, L.; Bare, G.; Chen, W.; Li, S.; Wang, H.; Cravatt, B. F.; Forli, S.; Powers, E. T.; Sharpless, K. B.; Wilson, I. A.; Kelly, J. W. J. Am. Chem. Soc. 2018, 140, 200–210. doi:10.1021/jacs.7b08366 |
| 27. | Chen, W.; Dong, J.; Plate, L.; Mortenson, D. E.; Brighty, G. J.; Li, S.; Liu, Y.; Galmozzi, A.; Lee, P. S.; Hulce, J. J.; Cravatt, B. F.; Saez, E.; Powers, E. T.; Wilson, I. A.; Sharpless, K. B.; Kelly, J. W. J. Am. Chem. Soc. 2016, 138, 7353–7364. doi:10.1021/jacs.6b02960 |
| 28. | Gilles, P.; Veryser, C.; Vangrunderbeeck, S.; Ceusters, S.; Van Meervelt, L.; De Borggraeve, W. M. J. Org. Chem. 2019, 84, 1070–1078. doi:10.1021/acs.joc.8b02785 |
| 1. |
Dong, J.; Krasnova, L.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. doi:10.1002/anie.201309399
Angew. Chem. 2014, 126, 9584–9602. doi:10.1002/ange.201309399 |
| 30. |
Guo, T.; Meng, G.; Zhan, X.; Yang, Q.; Ma, T.; Xu, L.; Sharpless, K. B.; Dong, J. Angew. Chem., Int. Ed. 2018, 57, 2605–2610. doi:10.1002/anie.201712429
Angew. Chem. 2018, 130, 2635–2640. doi:10.1002/ange.201712429 |
| 31. | Spillane, W.; Malaubier, J.-B. Chem. Rev. 2014, 114, 2507–2586. doi:10.1021/cr400230c |
© 2019 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)