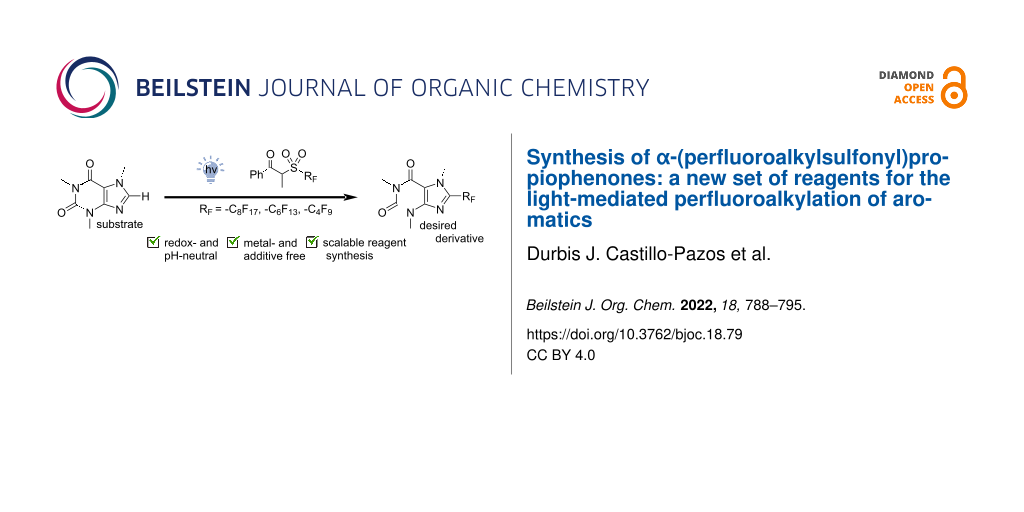
Abstract
In response to the demand for late-stage perfluoroalkylation in synthetic chemistry, we report the synthesis of a series of bench-stable α-(perfluoroalkylsulfonyl)propiophenones. Their application as photocleavable reagents was tested with electron-rich aromatics under metal-free, redox- and pH-neutral conditions to enable late-stage perfluorooctylation, perfluorohexylation, and perfluorobutylation.

Graphical Abstract
Introduction
Perfluorinated compounds are a family of molecules containing a backbone where all C–H bonds have been substituted with fluorine atoms. Within this family of molecules, perfluoroalkyl groups represent an industrially relevant moiety, capable of modifying the physicochemical properties of the scaffold that they are attached to. Such properties and a distinctive reactivity – or inert character – have been harnessed in a plethora of applications in modern life: Teflon in non-stick pans, fire-fighting foams, stain-resistant and weatherproof fabrics, etching of circuit boards, and even imaging agents [1]. Given their importance, multiple synthetic methodologies for the introduction of long perfluorinated chains into aromatic rings have been developed since the first reports of such transformation by George Tiers in 1960, and McLoughlin and Thrower in 1969 [2,3]. Most approaches have made extensive use of organometallic chemistry, radical initiators, photocatalysis, electrochemistry, and more sophisticated platforms such as metal nanoparticles, all of which have been reviewed thoroughly in the literature [4-8].
However, the methods referenced so far display one or more of the following setbacks: involvement of harsh oxidants or reductants, use of expensive metal catalysts, need for superstoichiometric amounts of starting materials, generation of undesired perfluoroalkylated byproducts, and poor chemoselectivity. Modern perfluoralkylation methodologies exceedingly rely on the use of perfluoroalkyl iodides as their principal source of perfluoroalkyl synthons. Indeed, while these molecules are cheap and abundant starting materials their use is fraught with technical complications. This family of molecules is extremely sensitive to bench conditions and requires a carefully controlled refrigeration in addition to low light levels to avoid decomposition. Furthermore, true to the unique solubility of this class of molecules, perfluoroalkyl iodides have a tendency to be weakly soluble in common organic solvents (i.e., ethyl acetate and methanol) rendering their application troublesome [9]. Moreover, the homolysis of the perfluoroalkyl iodide produces iodine radicals that can result in stray halogenation reactions or oxidation. For these reasons, it would be ideal to develop an efficient methodology that allows for the generation of perfluoroalkyl radicals in a mild, redox- and pH-neutral manner, without the assistance of external photocatalysts, heavy metal catalysts, or further additives. Thus, the expansion of our previously reported propiophenone family of reagents was envisioned as suitable alternative to produce a bench stable, organic soluble, and iodine-free perfluroalkylation source.
In 2017, our group developed a metal-free and redox-neutral protocol for the photoinduced alkylation of aromatics, for which trifluoromethylation was also possible in good to high yields for electron-rich aromatic rings [10]. In this protocol, inspired by Norrish type I reactions and the elimination of β-substituents after ketone photoexcitation [11-13], a series of reagents containing an α-sulfonylpropiophenone moiety readily undergoes homolysis into three parts upon irradiation of light: a propiophenone radical – forming a stabilized and bulky “dummy group” –, a molecule of SO2, and our radical of interest. Once this radical is formed in solution, radical addition to the aromatic substrate undergoes readily, and is subsequently followed by a hydrogen atom transfer (HAT) process aided by the “dummy group” radical. These reagents thus fit the paradigm of a green methodology as their implicit design and photoactivity allows them to react without the use of external metal catalysts. The intrinsic reactivity of these molecules allows this set of reagents to be both redox- and pH-neutral, while also being highly diversifiable. Additionally, all byproducts generated either during its synthesis or use in following reactions have the potential to be recycled, if so desired.
Based on the fact that both, trifluoromethyl radicals and its longer-chain analogues, share a common electrophilic character and a stabilizing stereoelectronic effect [14], we envisioned that the “dummy group” methodology could be translated into the formation of sought after perfluoroalkyl radicals (Scheme 1). In this work, we report the synthesis and application of three new members of the “dummy group” reagent family, based on the α-(perfluoroalkylsulfonyl)propiophenone scaffold for the perfluorobutylation (1a), perfluorohexylation (1b) and perfluorooctylation (1c) of electron-rich aromatics (Scheme 1). With the insights discussed in this paper, the authors hope to provide a new and amenable synthetic tool for the future academic and industrial demand of perfluorinated molecules and materials.
![[1860-5397-18-79-i1]](/bjoc/content/inline/1860-5397-18-79-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Envisioned Minisci perfluoroalkylation facilitated by “dummy group” reagents 1a–c.
Scheme 1: Envisioned Minisci perfluoroalkylation facilitated by “dummy group” reagents 1a–c.
Results and Discussion
To design an efficient and reproducible methodology for the synthesis of α-(perfluoroalkylsulfonyl)propiophenones, we envisioned the bimolecular nucleophilic substitution between an α-halopropiophenone as the “dummy group” scaffold and the corresponding perfluorinated sodium sulfinate salt – also visualized as the installation of the photocleavable moiety onto the perfluoroalkyl chain [15-19]. The precursory sulfinate salts 2a–c were synthesized through the sulfinatodehalogenation reaction, discovered by Huang and co-workers [20,21], and later on adapted by other research groups [22,23]. Pleasingly, the desired C4F9- (2a), C6F13- (2b), and C8F17- (2c) sulfinate salts were obtained from the perfluoroalkyl iodide precursors in good to quantitative yields as previously described. Additionally, we conceived that this methodology should be amenable to the synthesis of one of the limited commercially available secondary perfluoroalkyl groups such as perfluoroisopropyl iodide. However, despite being able to obtain the corresponding sulfinate in limited yield, the decomposition of this compound after several days at 4 °C, and within a few minutes under heating deemed its applicability impractical.
After this first step, we proceeded to test the nucleophilic substitution between our perfluoroalkylsulfinate salts 2 and an α-halopropiophenone. Unfortunately, initial attempts of a nucleophilic attack of sodium perfluorooctylsulfinate (2c) on α-bromopropiophenone (3) were unsuccessful to produce the desired product 1c due to the insufficient nucleophilicity of the sulfinate to substitute a bromide on a secondary carbon atom at 40 °C (Scheme 2).
![[1860-5397-18-79-i2]](/bjoc/content/inline/1860-5397-18-79-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Control experiments for the nucleophilic substitution of perfluoroalkylsulfinates 2 and halogenated electrophilic partners.
Scheme 2: Control experiments for the nucleophilic substitution of perfluoroalkylsulfinates 2 and halogenated...
Furthermore, increasing the temperature to 70 °C was not found to generate the product and instead resulted in slight decomposition of the starting materials. Attempting to trap the sulfinate nucleophiles with primary benzyl bromide (4) with catalytic sodium iodide under thermal conditions afforded product 5 in only 30% yield and underscored the sluggish reactivity of these sulfinate derivatives towards undergoing nucleophilic substitution (Scheme 2). To solve this problem, we turned to the use of α-iodopropiophenone (6), generated from its bromide counterpart through a simple Finkelstein reaction [24]. After performing a control experiment between α-iodopropiophenone (6) and sodium triflinate (2d) that afforded a quantitative yield of 1d, we proceeded to optimize the conditions for the nucleophilic substitution on this substrate by sodium perfluorohexylsulfinate (2b) to synthesize α-(perfluorohexyl)propiophenone (1b, Table 1). Temperature displayed a pivotal role in this synthesis: while room temperature proved insufficient to promote the substitution, the use of heat beyond 70 °C was detrimental for the reaction due to decomposition of the product. Once established that 40 °C was enough to promote the reaction, while limiting decomposition, we proceeded to screen the possible molar ratios between both components in the reaction. Given the higher economic value of the perfluorinated salts 2, we decided to vary the amounts of α-iodopropiophenone 6 to increase the molar ratio. Ranging from a 2.5:1 until a 10:1 molar ratio, yields increased significantly from 20% to 68%; however, a 5:1 ratio offered us a similar yield with a much shorter workup time when the reaction concentration was doubled.
Table 1: Optimization for the nucleophilic substitution between α-iodopropiophenone (6) and sodium perfluorohexylsulfinate (2b).
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-79-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry |
Molar ratio 6/2b
X:Y |
Volume DMA (mL) | Temperature (°C) | Time (h) | NMR yield 1b (%)a |
| 1 | 1:1.5 | 1 | 70 | 16 | n.d. |
| 2 | 1:1.5 | 1 | 40 | 16 | 18 |
| 3 | 1:1.5 | 1 | 20 | 16 | traces |
| 4 | 2.5:1 | 0.5 | 40 | 18 | 20 |
| 5 | 4:1 | 0.5 | 40 | 18 | 33 |
| 6 | 10:1 | 0.5 | 40 | 18 | 68 |
| 7 | 40:1 | 0.5 | 40 | 18 | 48 |
| 8 | 3:1 | 0.5 | 50 | 18 | 12 |
| 9 | 4:1 | 0.5 | 50 | 18 | 19 |
| 10 | 5:1 | 1 | 40 | 18 | 24 |
| 11 | 5:1 | 0.5 | 40 | 18 | 38 |
| 12 | 5:1 | 0.25 | 40 | 18 | 51 |
| 13 | 5:1 | 0.125 | 40 | 18 | 53 |
aUsing dimethylsulfone as a standard.
Knowing that nucleophilicity is a key factor in this reaction, we also employed crown ethers, 15-crown-5 and 18-crown-6, to test whether a “naked” sulfinate ion would help us achieve a better yield. Unfortunately, the addition of such ethers shut down all reactivity, most likely due to side reactions with the sulfinate salt. Moreover, it is worth mentioning that, while other synthetic approaches were explored to obtain these reagents, the SN2 strategy described in this work was the most efficient. Such synthetic alternatives included: first, a sulfur(VI) fluoride exchange (SuFEx) between perfluoroalkylsulfonyl fluorides and the corresponding silyl enol ether generated in situ from propiophenone, and second, the deprotection of propiophenone α-thioesters in the presence of perfluoroalkyliodides and subsequent oxidation of the formed perfluorothioether into the sulfone. However, none of these proposed pathways gave yields high enough for the reaction to be scalable (i.e., a maximum of 15% by 1H NMR).
Finally, due to the concentration of α-iodopropiophenone (6) employed, we detected the formation of a byproduct in the last stages of the optimization, namely the condensation of the desired product with α-propiophenone in the form of an enol ether. Once this byproduct was fully characterized by NMR, and the structure was confirmed by SCXRD, we conceived a hydrolysis protocol to break apart the formed enol ether (fully described in section 2.4 of Supporting Information File 1). After brief optimization, we succeeded at recovering the portion of perfluoroalkylating reagent that participated in such condensation (around 30%), giving us the final yields of perfluoroalkylating reagents 1a–c displayed in Scheme 3. Afterward, to show the practicality of application of these reagents in industry, we proceeded to scale up their synthesis in gram-scale. Satisfactorily, the developed synthesis and workup allowed us to produce the desired products in batches of up to six grams, with no decomposition observed over the course of 6 months.
![[1860-5397-18-79-i3]](/bjoc/content/inline/1860-5397-18-79-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Left: isolated yields of synthesized perfluoroalkylating reagents: perfluorobutyl (1a), perfluorohexyl (1b), and perfluorooctyl (1c) analogues (after conversion of byproduct); middle: gram amounts of perfluorooctyl product 1c; right: UV–vis absorption of reagents 1b and 1c.
Scheme 3: Left: isolated yields of synthesized perfluoroalkylating reagents: perfluorobutyl (1a), perfluorohe...
For the last section of this work, we proceeded to test the capacity of our reagents to generate the desired perfluoroalkyl radicals under light irradiation for the diversification of arenes. To verify the generation of perfluoroalkyl radicals from compounds 1, we conducted an experiment with perfluorohexyl analogue 1b and 1,1-diphenylethylene (7) as a radical trapping agent (Scheme 4). Gratifyingly we observed the formation of 2-(perfluorohexyl)-1,1-diphenylethylene (8), and propiophenone through GC–MS analysis. Additionally, the presence of free SO2 gas was confirmed by the reaction of acidic potassium dichromate solution on paper (green coloration of the exposed surface). See Supporting Information File 1 for details.
![[1860-5397-18-79-i4]](/bjoc/content/inline/1860-5397-18-79-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Radical trapping experiment with 1,1-diphenylethylene (7) and 1b confirming the initially proposed mechanism.
Scheme 4: Radical trapping experiment with 1,1-diphenylethylene (7) and 1b confirming the initially proposed ...
Using 1,3,5-trimethoxybenzene (TMB, 9) as a model substrate, we optimized the perfluoroalkylation reaction under irradiation of a 300 W Xenon arc lamp (Table 2). Based on the UV–vis absorption of our reagents 1, we used long-pass filters at either 280 nm or 295 nm to avoid side reactions caused by shorter wavelengths. During this optimization, the use of 2 to 3 equivalents of the reagents 1 resulted in better yields, along with more concentrated reaction mixtures, reaching almost quantitative yields (by NMR) for the perfluorohexylation of TMB (10b) and 83% NMR yield for its perfluorooctylation (10c), both in less than 6 hours (Scheme 5). Unsubstituted arenes such as naphthalene were well tolerated in this methodology and produced 72% isolated yield of the perfluorohexylated product 11b. The radical addition to unsubstituted benzene was also found to be possible affording perfluorooctylated product 12c in 68% isolated yield, but as tends to be the case for inactivated substrates, excess quantities of benzene (50% v/v) were required. Compounds containing esters such as methyl 3,4,5-trimethoxybenzoate and naproxen methyl ester were also tolerated and the desired products 13b and 14b were isolated in yields of 64% and 20%, respectively. Arenes containing halogens were attempted; however, in accordance to previous reported literature, the compounds were found to decompose under the ultraviolet radiation necessary for the homolysis of the reagent [25]. Lastly, some heteroaromatic substrates such as N-phenylpyrrole and 2-phenylindole were found to produce large quantities of the desired perfluorohexyl and perfluorooctyl analogues as observed by both 1H NMR and GC–MS analysis. However, these molecules generated large concentrations of fluorinated byproducts which rendered separation of the products impossible. Furthermore, we tested this methodology on caffeine (Scheme 5), leading to a lower yield of products 15b and c, due to its less electron-rich nature [26]. However, this yield was concordant with other radical innate functionalizations reported in the literature, showing the potential of these reagents as late-stage functionalization agents [26,27]. For a trend in reactivity, a more comprehensive scope of arenes and heteroarenes has been explored with the innate trifluoromethylation methodology previously reported by our group [10].
Table 2: Optimization for the perfluoroalkylation of aromatics under UV light.
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-79-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Equiv reagenta | Vol. MeCN (mL) | Time (h) | Filter (nm) | NMR yield 10 (%)b |
| 1 | 1 | 0.75 | 6 | >295 | 20 |
| 2 | 2 | 0.75 | 6 | >280 | 25 |
| 3 | 3 | 0.75 | 6 | >295 | 35 |
| 4 | 1 | 0.75 | 12 | >295 | 20 |
| 5 | 1 | 0.75 | 6 | no filter | 20 |
| 6 | 1 | 0.75 | 24 | CFLc | traces |
| 7 | 1 | 0.75 | 18 | >295 | 20 |
| 8 | 1 | 0.50 | 6 | >295 | 47 |
| 9 | 1 | 0.25 | 6 | >280 | 47 |
| 10 | 2 | 0.25 | 6 | >280 | 97 |
| 11 | 3 | 0.25 | 6 | >295 | 97 |
| 12 | 2 | 0.25 | 4 | >295 | 90 |
| 13 | 1 | 0.75 | 6 | >295 | 36 |
| 14 | 2 | 0.5 | 6 | >295 | 83 |
aEntries 1–12 were carried out with the perfluorohexyl analogue 1b, entries 13 and 14 with the perfluorooctyl analogue 1c; busing dimethylsulfone as a standard; ccompact fluorescent lamp, 23 W.
![[1860-5397-18-79-i5]](/bjoc/content/inline/1860-5397-18-79-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Demonstrative scope for the perfluoroalkylation of aromatics. Isolated yields are shown in parentheses.
Scheme 5: Demonstrative scope for the perfluoroalkylation of aromatics. Isolated yields are shown in parenthe...
Conclusion
In summary, we have successfully developed a robust synthetic methodology for α-(perfluoroalkylsulfonyl)propiophenones, envisioned as new members of photocleavable perfluoroalkylating reagents. In this work, we have demonstrated their scalability and applicability in the metal-, catalyst- and additive-free, redox- and pH-neutral perfluoroalkylation of electron-rich aromatics, as well as in the late-stage functionalization of small molecules such as caffeine, which is of great interest in the current literature [28]. In future work, we will explore the reach and applicability of these reagents for the functionalization of compounds of interest in academia and industry, namely, for the synthesis of molecules with novel properties in the fields of material and bioorganic chemistry.
Supporting Information
CCDC 2163755 contains the supplementary crystallographic data of byproduct B (perfluorooctyl analogue). These data can be obtained free of charge through the Cambridge Crystallographic Data Center (http://www.ccdc.cam.ac.uk/data_request/cif).
| Supporting Information File 1: Detailed experimental procedures and compound characterization data. | ||
| Format: PDF | Size: 4.8 MB | Download |
Acknowledgements
We gratefully acknowledge the McGill Chemistry Characterization Facility (MC2) for their contribution to this work, especially Robin Stein and Tara Sprules on the NMR data, Hatem Titi on SCXRD interpretation, Nadim Saadé and Alexander Wahba on HRMS, and Petr Fiurasek and Ehsan Hamzehpoor on UV–vis spectroscopy.
Funding
The authors acknowledge the Canada Research Chair (Tier I) foundation, the E.B. Eddy endowment fund, the CFI, NSERC, and FRQNT for support of our research. DJCP acknowledges the financial support granted by the Vanier Canada Graduate Scholarships, CONACYT Mexico, and the Richard H. Tomlinson Fellowship.
References
-
Glüge, J.; Scheringer, M.; Cousins, I. T.; DeWitt, J. C.; Goldenman, G.; Herzke, D.; Lohmann, R.; Ng, C. A.; Trier, X.; Wang, Z. Environ. Sci.: Processes Impacts 2020, 22, 2345–2373. doi:10.1039/d0em00291g
Return to citation in text: [1] -
Tiers, G. V. D. J. Am. Chem. Soc. 1960, 82, 5513. doi:10.1021/ja01505a059
Return to citation in text: [1] -
McLoughlin, V. C. R.; Thrower, J. Tetrahedron 1969, 25, 5921–5940. doi:10.1016/s0040-4020(01)83100-8
Return to citation in text: [1] -
Castillo-Pazos, D. J.; Lasso, J. D.; Li, C.-J. Org. Biomol. Chem. 2021, 19, 7116–7128. doi:10.1039/d1ob01122g
Return to citation in text: [1] -
Barata-Vallejo, S.; Bonesi, S. M.; Postigo, A. RSC Adv. 2015, 5, 62498–62518. doi:10.1039/c5ra11337g
Return to citation in text: [1] -
Postigo, A. Eur. J. Org. Chem. 2018, 6391–6404. doi:10.1002/ejoc.201801079
Return to citation in text: [1] -
Barata-Vallejo, S.; Cooke, M. V.; Postigo, A. ACS Catal. 2018, 8, 7287–7307. doi:10.1021/acscatal.8b02066
Return to citation in text: [1] -
Yerien, D. E.; Lantaño, B.; Barata‐Vallejo, S.; Postigo, A. ChemCatChem 2021, 13, 4497–4506. doi:10.1002/cctc.202100997
Return to citation in text: [1] -
These properties are usually reflected in the material safety data sheet of any of these building blocks. For example: Perfluorooctyl iodide, MSDS No. P286505; Toronto Research Chemicals: Toronto, ON, 2020. https://www.trc-canada.com/prod-img/MSDS/P286505MSDS.pdf (accessed May 24, 2022).
Return to citation in text: [1] -
Liu, P.; Liu, W.; Li, C.-J. J. Am. Chem. Soc. 2017, 139, 14315–14321. doi:10.1021/jacs.7b08685
Return to citation in text: [1] [2] -
Zimmerman, H. E.; Hancock, K. G.; Licke, G. C. J. Am. Chem. Soc. 1968, 90, 4892–4911. doi:10.1021/ja01020a024
Return to citation in text: [1] -
Sheehan, J. C.; Wilson, R. M.; Oxford, A. W. J. Am. Chem. Soc. 1971, 93, 7222–7228. doi:10.1021/ja00755a017
Return to citation in text: [1] -
Sheehan, J. C.; Wilson, R. M. J. Am. Chem. Soc. 1964, 86, 5277–5281. doi:10.1021/ja01077a046
Return to citation in text: [1] -
Song, H. Research progress on trifluoromethyl-based radical reaction process. In IOP Conference Series: Earth and Environmental Science, Volume 100, 1st International Global on Renewable Energy and Development (IGRED 2017), Singapore, Dec 22–25, 2017; IOP Publishing, 2017; pp 012061 ff. doi:10.1088/1755-1315/100/1/012061
Return to citation in text: [1] -
Fujiwara, Y.; Dixon, J. A.; O’Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herlé, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95–99. doi:10.1038/nature11680
Return to citation in text: [1] -
O’Hara, F.; Blackmond, D. G.; Baran, P. S. J. Am. Chem. Soc. 2013, 135, 12122–12134. doi:10.1021/ja406223k
Return to citation in text: [1] -
Fujiwara, Y.; Baran, P. S. Radical-Based Late Stage C–H Functionalization of Heteroaromatics in Drug Discovery. In New Horizons of Process Chemistry: Scalable Reactions and Technologies; Tomioka, K.; Shioiri, T.; Sajiki, H., Eds.; Springer: Singapore, 2017; pp 103–120. doi:10.1007/978-981-10-3421-3_8
Return to citation in text: [1] -
Aziz, J.; Hamze, A. Org. Biomol. Chem. 2020, 18, 9136–9159. doi:10.1039/d0ob01718c
Return to citation in text: [1] -
Liang, S.; Hofman, K.; Friedrich, M.; Manolikakes, G. Eur. J. Org. Chem. 2020, 4664–4676. doi:10.1002/ejoc.202000403
Return to citation in text: [1] -
Huang, X.-T.; Long, Z.-Y.; Chen, Q.-Y. J. Fluorine Chem. 2001, 111, 107–113. doi:10.1016/s0022-1139(01)00442-0
Return to citation in text: [1] -
Huang, W.; Huang, B.; Wang, W. Acta Chim. Sin. (Chin. Ed.) 1985, 43, 663–668.
Return to citation in text: [1] -
Cao, H.-P.; Chen, Q.-Y. J. Fluorine Chem. 2007, 128, 1187–1190. doi:10.1016/j.jfluchem.2007.04.018
Return to citation in text: [1] -
Dmowski, W.; Piasecka-Maciejewska, K. J. Fluorine Chem. 2005, 126, 877–882. doi:10.1016/j.jfluchem.2005.02.014
Return to citation in text: [1] -
Finkelstein, H. Ber. Dtsch. Chem. Ges. 1910, 43, 1528–1532. doi:10.1002/cber.19100430257
Return to citation in text: [1] -
Li, L.; Liu, W.; Zeng, H.; Mu, X.; Cosa, G.; Mi, Z.; Li, C.-J. J. Am. Chem. Soc. 2015, 137, 8328–8331. doi:10.1021/jacs.5b03220
Return to citation in text: [1] -
Beatty, J. W.; Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. Nat. Commun. 2015, 6, 7919. doi:10.1038/ncomms8919
Return to citation in text: [1] [2] -
Yin, D.; Su, D.; Jin, J. Cell Rep. Phys. Sci. 2020, 1, 100141. doi:10.1016/j.xcrp.2020.100141
Return to citation in text: [1] -
Lasso, J. D.; Castillo-Pazos, D. J.; Li, C.-J. Chem. Soc. Rev. 2021, 50, 10955–10982. doi:10.1039/d1cs00380a
Return to citation in text: [1]
| 1. | Glüge, J.; Scheringer, M.; Cousins, I. T.; DeWitt, J. C.; Goldenman, G.; Herzke, D.; Lohmann, R.; Ng, C. A.; Trier, X.; Wang, Z. Environ. Sci.: Processes Impacts 2020, 22, 2345–2373. doi:10.1039/d0em00291g |
| 10. | Liu, P.; Liu, W.; Li, C.-J. J. Am. Chem. Soc. 2017, 139, 14315–14321. doi:10.1021/jacs.7b08685 |
| 10. | Liu, P.; Liu, W.; Li, C.-J. J. Am. Chem. Soc. 2017, 139, 14315–14321. doi:10.1021/jacs.7b08685 |
| 9. | These properties are usually reflected in the material safety data sheet of any of these building blocks. For example: Perfluorooctyl iodide, MSDS No. P286505; Toronto Research Chemicals: Toronto, ON, 2020. https://www.trc-canada.com/prod-img/MSDS/P286505MSDS.pdf (accessed May 24, 2022). |
| 28. | Lasso, J. D.; Castillo-Pazos, D. J.; Li, C.-J. Chem. Soc. Rev. 2021, 50, 10955–10982. doi:10.1039/d1cs00380a |
| 4. | Castillo-Pazos, D. J.; Lasso, J. D.; Li, C.-J. Org. Biomol. Chem. 2021, 19, 7116–7128. doi:10.1039/d1ob01122g |
| 5. | Barata-Vallejo, S.; Bonesi, S. M.; Postigo, A. RSC Adv. 2015, 5, 62498–62518. doi:10.1039/c5ra11337g |
| 6. | Postigo, A. Eur. J. Org. Chem. 2018, 6391–6404. doi:10.1002/ejoc.201801079 |
| 7. | Barata-Vallejo, S.; Cooke, M. V.; Postigo, A. ACS Catal. 2018, 8, 7287–7307. doi:10.1021/acscatal.8b02066 |
| 8. | Yerien, D. E.; Lantaño, B.; Barata‐Vallejo, S.; Postigo, A. ChemCatChem 2021, 13, 4497–4506. doi:10.1002/cctc.202100997 |
| 26. | Beatty, J. W.; Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. Nat. Commun. 2015, 6, 7919. doi:10.1038/ncomms8919 |
| 2. | Tiers, G. V. D. J. Am. Chem. Soc. 1960, 82, 5513. doi:10.1021/ja01505a059 |
| 3. | McLoughlin, V. C. R.; Thrower, J. Tetrahedron 1969, 25, 5921–5940. doi:10.1016/s0040-4020(01)83100-8 |
| 26. | Beatty, J. W.; Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. Nat. Commun. 2015, 6, 7919. doi:10.1038/ncomms8919 |
| 27. | Yin, D.; Su, D.; Jin, J. Cell Rep. Phys. Sci. 2020, 1, 100141. doi:10.1016/j.xcrp.2020.100141 |
| 20. | Huang, X.-T.; Long, Z.-Y.; Chen, Q.-Y. J. Fluorine Chem. 2001, 111, 107–113. doi:10.1016/s0022-1139(01)00442-0 |
| 21. | Huang, W.; Huang, B.; Wang, W. Acta Chim. Sin. (Chin. Ed.) 1985, 43, 663–668. |
| 24. | Finkelstein, H. Ber. Dtsch. Chem. Ges. 1910, 43, 1528–1532. doi:10.1002/cber.19100430257 |
| 15. | Fujiwara, Y.; Dixon, J. A.; O’Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herlé, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95–99. doi:10.1038/nature11680 |
| 16. | O’Hara, F.; Blackmond, D. G.; Baran, P. S. J. Am. Chem. Soc. 2013, 135, 12122–12134. doi:10.1021/ja406223k |
| 17. | Fujiwara, Y.; Baran, P. S. Radical-Based Late Stage C–H Functionalization of Heteroaromatics in Drug Discovery. In New Horizons of Process Chemistry: Scalable Reactions and Technologies; Tomioka, K.; Shioiri, T.; Sajiki, H., Eds.; Springer: Singapore, 2017; pp 103–120. doi:10.1007/978-981-10-3421-3_8 |
| 18. | Aziz, J.; Hamze, A. Org. Biomol. Chem. 2020, 18, 9136–9159. doi:10.1039/d0ob01718c |
| 19. | Liang, S.; Hofman, K.; Friedrich, M.; Manolikakes, G. Eur. J. Org. Chem. 2020, 4664–4676. doi:10.1002/ejoc.202000403 |
| 25. | Li, L.; Liu, W.; Zeng, H.; Mu, X.; Cosa, G.; Mi, Z.; Li, C.-J. J. Am. Chem. Soc. 2015, 137, 8328–8331. doi:10.1021/jacs.5b03220 |
| 14. | Song, H. Research progress on trifluoromethyl-based radical reaction process. In IOP Conference Series: Earth and Environmental Science, Volume 100, 1st International Global on Renewable Energy and Development (IGRED 2017), Singapore, Dec 22–25, 2017; IOP Publishing, 2017; pp 012061 ff. doi:10.1088/1755-1315/100/1/012061 |
| 11. | Zimmerman, H. E.; Hancock, K. G.; Licke, G. C. J. Am. Chem. Soc. 1968, 90, 4892–4911. doi:10.1021/ja01020a024 |
| 12. | Sheehan, J. C.; Wilson, R. M.; Oxford, A. W. J. Am. Chem. Soc. 1971, 93, 7222–7228. doi:10.1021/ja00755a017 |
| 13. | Sheehan, J. C.; Wilson, R. M. J. Am. Chem. Soc. 1964, 86, 5277–5281. doi:10.1021/ja01077a046 |
| 22. | Cao, H.-P.; Chen, Q.-Y. J. Fluorine Chem. 2007, 128, 1187–1190. doi:10.1016/j.jfluchem.2007.04.018 |
| 23. | Dmowski, W.; Piasecka-Maciejewska, K. J. Fluorine Chem. 2005, 126, 877–882. doi:10.1016/j.jfluchem.2005.02.014 |
© 2022 Castillo-Pazos et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.