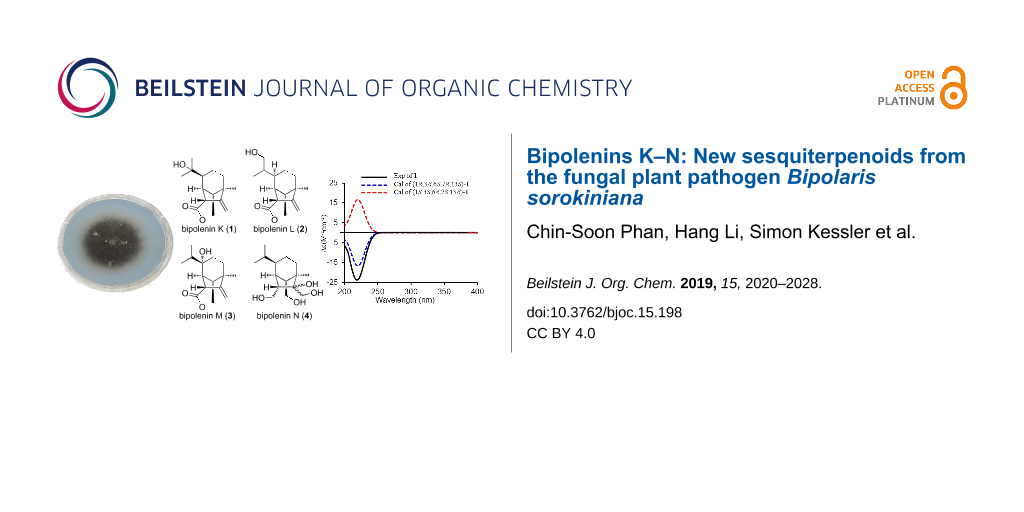
Abstract
Chemical investigation of the barley and wheat fungal pathogen Bipolaris sorokiniana BRIP10943 yielded four new sativene-type sesquiterpenoid natural products, bipolenins K–N (1–4), together with seven related known analogues (5–11), and a sesterterpenoid (12). Their structures were determined by detailed analysis of spectroscopic data, supported by TDDFT calculations and comparison with previously reported analogues. These compounds were evaluated for their phytotoxic activity against wheat seedlings and wheat seed germination. The putative biosynthetic relationships between the isolated sesquiterpenoids were also explored.

Graphical Abstract
Introduction
Fungi belonging to the genus Bipolaris (teleomorph: Cochliobolus) have been reported to produce a diverse array of secondary metabolites, including sesquiterpenes [1-7], sesquiterpene-xanthones [8], diterpenes [9], sesterterpenes [10], cochlioquinones and peptides [11]. Moreover, several of these secondary metabolites are known to play important roles in mediating the virulence of these fungi against plant hosts [12]. Well-known examples include the host-specific toxins victorin and T-toxin and other non-host-specific toxins such as the ophiobolins [11]. Bipolaris sorokiniana (syn. Cochliobolus sativus) has been identified as the causative agent of multiple diseases on wheat and barley and is a major threat to yield improvement and food security in Central Asia [13]. Recent genome sequencing of 35 Australian strains of B. sorokiniana identified a known proteinaceous necrotrophic effector, ToxA, which confers host-specific virulence proteins and is proposed to be acquired through horizontal gene transfer [14]. To date, only three studies have explored phytotoxins from B. sorokiniana [2,7,10]. Therefore, in the framework of furthering our understanding of the roles of B. sorokiniana secondary metabolites in crop disease, we investigated the compounds produced by the ToxA-containing strain BRIP10943 (CS10) [14] and their phytotoxicity. This led to the isolation of four new sativene-type sesquiterpenoid natural products along with seven related known analogues and one sesterterpenoid. Herein, the isolation, structure elucidation and phytotoxic activities of these compounds are presented.
Results and Discussion
B. sorokiniana was cultivated for 22 days in Fries medium supplemented with rolled oats. The resulting broth and mycelia were extracted with methanol and the extracts were partitioned against EtOAc/MeOH/acetic acid (89.9:10:0.1 ratio). The combined organic layer was chromatographed repeatedly with silica gel and RP-HPLC to afford four new sativene-type sesquiterpenoids, bipolenins K–N (1–4), along with eight previously reported compounds (5–12), which were identified as sativene-type sesquiterpenoids prehelminthosporol lactone (5) [1], helminthosporic acid (6) [1], helminthosporol (7) [15], bipolenin A (8) [3], secolongifolene diol (9) [15], dihydroprehelminthosporol (10) [1] and sorokinianin (11) [2], and the cytotoxic sesterterpenoid, terpestacin (12) [16,17] (Figure 1).
![[1860-5397-15-198-1]](/bjoc/content/figures/1860-5397-15-198-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of compounds 1–12 isolated from B. sorokiniana.
Figure 1: Structures of compounds 1–12 isolated from B. sorokiniana.
Bipolenin K (1) was isolated as a colourless oil. Its molecular formula was determined as C15H22O3 from the HRESIMS [M + H]+ ion at m/z 251.1646 (calcd for C15H23O3+, 251.1642), corresponding to five degrees of unsaturation. The IR absorption bands at 3445 and 1729 cm−1 revealed the presence of hydroxy and ester moieties, respectively. The 13C NMR spectrum (Figure S2, Supporting Information File 1) showed 15 distinct carbon signals, while 13C and 1H NMR data (Table 1) indicated an isopropyl unit (δC 28.3, 29.2 and 73.1; δH 1.20 and 1.24), one tertiary methyl (δC 20.1; δH 1.25), a disubstituted olefin (δC 105.7 and 155.2; δH 5.12 and 4.89) and four methines (δC 39.6, 51.1, 51.6 and 54.0; δH 1.73, 1.88, 2.65 and 3.77), which are typical resonances for sativene-type sesquiterpenoids. The NMR data for 1 were very similar to those for prehelminthosporol lactone (5) except for the replacement of a methine group (δC 32.1; δH 1.42) at C-9 in 5 with a hydroxylated quaternary carbon (δC 73.1) in 1. This suggested that 1 was the 9-hydroxy analogue of 5, which was further confirmed by detailed analysis of key 2D NMR correlations (Figure 2). Compound 1 was previously reported in 1970 as a semi-synthetic analogue of 9-hydroxyprehelminthosporol [18], but has not been previously isolated and characterised from a natural source.
Table 1: 1H and 13C NMR data for bipolenins K–N (1–4).
| No. | Bipolenin K (1)a | Bipolenin L (2)b | Bipolenin M (3)b | Bipolenin N (4)c | ||||
| δC | δH, mult (J in Hz) | δC | δH, mult (J in Hz) | δC | δH, mult (J in Hz) | δC | δH, mult (J in Hz) | |
| 1 | 54.0 | 3.77, br s | 52.4 | 3.36, br s | 53.1 | 3.31, m | 47.4 | 2.19, dt (10.4, 3.1) |
| 2 | 155.2 | 155.1 | 154.3 | 88.1 | ||||
| 3 | 46.3 | 46.0 | 45.6 | 50.0 | ||||
| 4 | 42.4 | 1.64, m | 42.4 | 1.64, dd (12.8, 4.7) | 39.3 | 1.85, td (14.8, 5.2) | 30.6 | 1.60, m |
| 1.53, m | 1.48, ddd (12.8, 5.6, 2.0) | 1.34, m | ||||||
| 5 | 22.3 | 1.72, m | 26.3 | 1.80, m | 32.5 | 1.69, m | 24.6 | 1.68, m |
| 1.52, m | 1.27, m | 1.34, m | 1.31, m | |||||
| 6 | 51.1 | 1.73, m | 41.9 | 1.70, m | 74.8 | 45.8 | 1.18, m | |
| 7 | 39.6 | 2.65, br s | 40.7 | 2.47, br s | 46.3 | 2.44, br s | 43.5 | 1.53, br s |
| 8 | 20.1 | 1.25, s | 20.3 | 1.25, s | 20.2 | 1.25, s | 19.4 | 0.98, s |
| 9 | 73.1 | 39.7 | 1.44, m | 35.8 | 1.67, septet (6.9) | 30.4 | 1.34, m | |
| 10 | 29.2 | 1.24, s | 15.8 | 1.02, d (6.9) | 16.5 | 0.91, d (6.9) | 21.4 | 0.91, d (6.9) |
| 11 | 28.3 | 1.20, s | 65.8 | 3.58, dd (10.8, 3.8) | 16.5 | 0.91, d (6.9) | 20.3 | 0.84, d (6.9) |
| 3.44, dd (10.8, 6.0) | ||||||||
| 12 | 105.7 | 5.12, s | 106.3 | 5.15, s | 107.4 | 5.19, s | 62.2 | 3.79, d (11.8) |
| 4.89, s | 4.90, s | 4.96, s | 3.65, d (11.8) | |||||
| 13 | 51.6 | 1.88, dd (4.5, 1.8) | 51.0 | 1.92, d (4.5) | 46.0 | 2.38, d (4.4) | 52.2 | 1.50, d (3.5) |
| 14 | 71.3 | 4.46, d (11.7) | 71.6 | 4.48, d (11.7) | 72.1 | 4.53, d (11.7) | 69.8 | 3.90, dd (7.8, 3.5) |
| 4.30, dd (11.7, 4.5) | 4.30, dd (11.7, 4.5) | 4.23, dd (11.7, 4.4) | 3.36, d (7.8) | |||||
| 15 | 174.1 | 174.4 | 174.0 | 62.9 | 3.77, t (10.4) | |||
| 3.47, dd (10.4, 3.1) | ||||||||
aRecorded at 500/125 MHz for 1H/13C in CD3OD; bRecorded at 600/150 MHz for 1H/13C in CD3OD; cRecorded at 600/150 MHz for 1H/13C in CDCl3.
![[1860-5397-15-198-2]](/bjoc/content/figures/1860-5397-15-198-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Key 2D NMR correlations of bipolenins K–N (1–4).
Figure 2: Key 2D NMR correlations of bipolenins K–N (1–4).
The relative configuration of 1 was established based on NOESY correlations (Figure 3) of H-1 to H2-5 and H3-10; and H2-14 to H-7, H3-8 and H-13. Due to the constrained bicyclo[3.2.1]octane ring system, these NOESY correlations indicated that H-1 was β-oriented, while H-6, H-7, H3-8 and H-13 were α-oriented. The absolute configuration of 1 was determined to be 1R,3R,6S,7R,13S by comparison of the experimental electronic circular dichroism (ECD) spectrum with time-dependent density functional theory (TDDFT)-calculated ECD spectra of the two possible enantiomers of 1 (Figure 4).
![[1860-5397-15-198-3]](/bjoc/content/figures/1860-5397-15-198-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Key NOESY correlations of bipolenins K–N (1–4).
Figure 3: Key NOESY correlations of bipolenins K–N (1–4).
![[1860-5397-15-198-4]](/bjoc/content/figures/1860-5397-15-198-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a) Experimental ECD spectrum of 1 (MeOH) compared to TDDFT-calculated spectra (B3LYP-D3/def2-TZVPP) for the two possible enantiomers of 1, which were blue-shifted by 9 nm. (b) Comparison of experimental ECD spectra of 1–3 and 5 (MeOH).
Figure 4: (a) Experimental ECD spectrum of 1 (MeOH) compared to TDDFT-calculated spectra (B3LYP-D3/def2-TZVPP...
Bipolenin L (2) was isolated as a colourless oil. The HRESIMS [M + H]+ ion at m/z 251.1649 corresponded to a molecular formula C15H22O3 (calcd for C15H23O3+, 251.1642), which is isomeric with 1. The 1H and 13C NMR data for 2 (Table 1) were also very similar to those for 5, with the only significant difference being the presence of a hydroxymethylene group (δC 65.8; δH 3.58 and 3.44) in place of the methyl group at C-11. Thus, the structure of 2 was assigned as the 11-hydroxy analogue of 5. The absolute configurations of the chiral centres in 2 were established to be the same as for 1 after investigation of the proton coupling constants (Table 1), NOESY correlations (Figure 3) and ECD spectra (Figure 4). The configuration at C-9 was not determined. Hence, structure 2 was determined as shown in Figure 1.
Bipolenin M (3) was purified as a colourless oil. The molecular formula C15H22O3 was based on a HRESIMS [M + H]+ ion at m/z 251.1647 (calcd for C15H23O3+, 251.1642), and is isomeric with 1 and 2. The 1H and 13C NMR data for 3 (Table 1) were very similar to those for 5, with the only significant difference being the replacement of the methine group (δC 47.1; δH 1.40) at C-6 in 5 with a hydroxylated quaternary carbon (δC 74.8) in 3. This suggested that 3 was the 6-hydroxy analogue of 5, which was further confirmed by detailed analysis of key 2D NMR correlations (Figure 2). The absolute configurations of the stereocentres in 3 were established to be identical to 1 and 2 based on the analysis of proton coupling constants (Table 1), NOESY correlations (Figure 3) and ECD spectra (Figure 4).
Bipolenin N (4) was acquired as a colourless oil. Its molecular formula was determined to be C15H28O4 from the HRESIMS [M + H − 2H2O]+ ion at m/z 237.1859 (calcd for C15H25O2+, 237.1849). The UV–vis spectra of 1–3 (Figure S37, Supporting Information File 1) and previously reported congener 5 were almost identical, while 4 displayed no significant UV–vis absorptions, suggesting the absence of both the ester and alkene moieties. This was confirmed by the analysis of the 1H and 13C NMR data for 4 (Table 1), which revealed the absence of ester and alkene resonances and the presence of three hydroxylated methylenes at C-12 (δC 62.2; δH 3.79 and 3.65), C-14 (δC 69.8; δH 3.90 and 3.36) and C-15 (δC 62.9; δH 3.77 and 3.47), and one hydroxylated quaternary carbon at C-2 (δC 88.1). This suggested 4 was related to 5, but with reduction of the lactone ring to the dialcohol and dihydroxylation of the Δ2,12 double bond. Detailed analysis of the 2D NMR data for 4 (Figure 2) confirmed the seco-sativene-type scaffold. The relative configurations at C-1, C-3, C-6, C-7 and C-13 were determined to be the same as those of 1–3 and other reported analogues based on NOESY correlations (Figure 3), while the configuration at C-2 was not determined. The ECD spectrum of 4 (Figure S38, Supporting Information File 1) was measured, but no significant Cotton effect was observed. Therefore, the structure of 4 was determined as shown in Figure 1.
Equipped with the compounds, we tested 1–12 for phytotoxic activity against wheat seedlings. The compounds all showed negligible activities at 200 ppm, although 6 and 10 showed signs of necrosis at 500 ppm (Figure S40, Supporting Information File 1). In addition, the activities of 1, 6–10 and 12 against wheat seed germination were also tested, with 7 inhibiting germination at 100 ppm (Figure S41, Supporting Information File 1). This corresponds to a previous report of the inhibitory effects of 7 on lettuce seed germination [19]. This activity could be due to the presence of an aldehyde moiety in 7. Interestingly, an earlier study showed that 7 promoted the elongation of the shoots of rice seedlings [20]. Compound 12 was reported to have a broad spectrum of biological activities, including phytotoxicity on juvenile plant Bromus tectorum [21], syncytium formation inhibitory effects on cells infected with respiratory syncytial virus [22,23], induction of aerial mycelium formation in Fusarium culmorum [24], and as an inhibitor of ubiquinol-cytochrome c reductase binding protein, blocking mitochondrial ROS-mediated vascular endothelial growth factor receptor type 2 signalling pathways in endothelial cells [25]. However, 12 showed no activity against wheat seedlings or wheat seed germination in this study.
The sativene-type sesquiterpenoids contain a bicyclo[3.2.1]octane backbone and are related to seco-sativene and isosativene scaffolds [15] (Figure 5a). They were also proposed to be related to the bicyclo[4.2.1]nonane-containing longifolene and seco-longifolene sesquiterpenoids, as they were often co-isolated [3,4,6,15,26,27]. A closer examination of the biosynthetic relationship between sativene and longifolene scaffolds suggests that the two pathways branched early at the nerolidyl cation (Figure 5b) [28-31]. The biosynthesis of 1–8 and 10–11 are likely to be derived from sativene with a key oxidation at C-15 followed by a Baeyer–Villiger oxidation to break the C-14–C-15 bond (Figure 5c). Based on an isotope labelling study, the γ-butyrolactone moiety on 11 has been proposed to be derived from oxaloacetic acid or similar TCA-cycle intermediates [32]. Compound 9, which contains the seco-longifolene scaffold, is likely to be derived from longifolene via a similar Baeyer–Villiger mechanism proposed above for 1–8 and 10 and 11.
![[1860-5397-15-198-5]](/bjoc/content/figures/1860-5397-15-198-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Relationship of sesquiterpenoids isolated in this study. A) Different groups of sativene/longifolene-type sesquiterpenoid scaffolds; B) The branched pathways to sativene- and longifolene-type sesquiterpenoids. C) Detailed proposed pathways to the sativene-derived sesquiterpenoids from this study.
Figure 5: Relationship of sesquiterpenoids isolated in this study. A) Different groups of sativene/longifolen...
Several sativene-type sesquiterpenoids have been previously reported from fungi, including from Bipolaris sp. [1], B. sorokiniana [2], B. eleusines [3-6,8], Cochliobolus sp. [26], Cochliobolus sativus [18], Helminthosporium sativum [20,33,34], Drechslera sp. [27], Drechslera dematioidea [15], and Veronaea sp. [35], most of which are Dothideomycetes. Significantly, this is the first report pertaining to sativene-type sesquiterpenoids from B. sorokiniana in 25 years, since the first and only literature account was published in 1994 [2]. Furthermore, structure 4 has a seco-sativene type scaffold without an olefin unit at C-1/C-2 or C-2/C-12. In contrast, all the previously known seco-sativene-type sesquiterpenoids possessed a double bond either at C-1/C-2 or C-2/C-12 [1,2,15,18,20,33], except drechslerine C, which contains a decarboxylated seco-sativene-type scaffold [15]. To the best of our knowledge, the previously reported sativene-type sesquiterpenoids 5 (isolated from B. eleusines and Cochliobolus sp., and from semi-synthetic analogue of prehelminthosporol with pyridinium chlorochromate or chromic acid) [1,4,18,26], 8 (isolated from B. eleusines) [3,6], and 10 (isolated from Bipolaris sp. and Cochliobolus sp.) [1,26], were reported in B. sorokiniana for the first time, while, known metabolites 6 and 7 [20], 9 [34], 11 [2], and 12 [16] were previously reported from B. sorokiniana (syn. C. sativus and H. sativum).
The terpene synthase responsible for the biosynthesis of the sativene/longifolene backbone of 1–11 remains unknown. Given that the genome of B. sorokiniana BRIP10943 has been sequenced [14], we surveyed the genome for potential terpene synthases that may be responsible for the biosynthesis of these compounds. Four putative sesquiterpene synthases were found, corresponding to the genes COCSADRAFT_31812, COCSADRAFT_346586, COCSADRAFT_83129 and COCSADRAFT_26102 annotated in the published genome B. sorokiniana ND90Pr in GenBank. However, it is difficult to determine which sesquiterpene synthase is responsible for biosynthesis of the sativene-type sesquiterpene backbone at this stage.
The biosynthetic gene cluster (tpc) for terpestacin (12) has been recently identified from Bipolaris maydis [36]. A didomain sesterterpene synthase (tpcA) with a terpene cyclase domain and polyprenyltransferase domain was demonstrated to be responsible for the production of the sesterterpene backbone of 12. A BLASTp search using tpcA as query against the genome of B. sorokiniana ND90Pr and BRIP10943 identified COCSADRAFT_342920 in ND90Pr (and its homolog in BRIP10943), which shares 96% identity to tpcA. In the vicinity of the sesterterpene synthase gene, we also identified homologs for the two P450 oxygenases (tpcB and tpcC) encoded in the tpc cluster as COCSADRAFT_342924 (92% identity) and COCSADRAFT_146541 (98% identity), respectively, while the tpcD homolog in B. sorokiniana corresponded to COCSADRAFT_94398 (91% identity). This suggests that the homologous gene cluster in B. sorokiniana is likely responsible for production of 12. We are currently investigating the genetic basis for the biosynthesis of the sativene-type terpenoid compounds identified from B. sorokiniana.
Conclusion
Following the first and only reported isolation of a sativene-type sesquiterpenoid, sorokinianin (11), from B. sorokiniana in 1994 [2], we have expanded the number of reported analogues to eleven. These include the new sesquiterpenoid natural products, bipolenins K–N (1–4), as well as the previously reported sesquiterpenoids prehelminthosporol lactone (5), helminthosporic acid (6), helminthosporol (7), bipolenin A (8), secolongifolene diol (9), dihydroprehelminthosporol (10) and sorokinianin (11), together with a sesterterpenoid, terpestacin (12). We demonstrated that 6 and 10 have weak necrotic activity against wheat leaves, while 7 inhibited wheat seed germination at 100 ppm. These compounds served as markers for identifying putative sesquiterpene synthase genes in the genome of B. sorokiniana BRIP10943, allowing the molecular genetic basis for their biosynthesis and their roles in mediating the virulence of B. sorokiniana against wheat to be explored.
Experimental
General experimental procedures
Optical rotations were measured on an A. Krüss Optronic P8000 polarimeter. The IR spectra were collected on a Perkin Elmer Spectrum One FTIR spectrometer. The HR-ESIMS spectra were recorded on a Waters LCT Premier XE mass spectrometer. The ESIMS spectra were recorded on an Agilent 1260 LC system equipped with a DAD detector and coupled to an Agilent 6130 Quadrupole MS with an ESI source. The NMR spectra were recorded on Bruker Avance III HD 500 or AV600 spectrometers. The ECD spectra were recorded on a Jasco J-810 spectropolarimeter with MeOH as solvent. Flash cartridge (Reveleris, HP-silica, 12 g, 20 µm), Kinetex C18 (Phenomenex, 2.6 µm, 2.1 × 100 mm), and semi-preparative C18 (Grace, 5 µm, 10 × 250 mm) were used. All solvents used for extraction were analytical grade, and solvents for HPLC were HPLC grade.
Biological material
The fungal strain B. sorokiniana BRIP10943 was obtained from Queensland Plant Pathology Herbarium (BRIP). It was isolated from a wheat field at Hermitage, QLD, Australia. The fungus was maintained on potato dextrose agar (PDA).
Extraction and isolation
B. sorokiniana was cultured on 16 plates of V8PDA at 25 °C for 14 days, then inoculated in 4 L shake-flask culture (25 °C, 180 rpm for 22 days) in Fries medium supplemented with oat. The Fries medium was filtered and extracted by partition with EtOAc/MeOH/acetic acid at 89.9:10:0.1 ratio. The cells were extracted with MeOH and partition with EtOAc/MeOH/acetic acid at 89.7:10:0.3 ratio. Both organic partitioned layers were combined to obtain a light-yellow crude extract (205 mg), which was fractionated on a Reveleris flash chromatography (Grace) using gradient mode of H2O/MeOH equipped with the flash cartridge, UV and evaporative light scattering detector. The resulting fractions were further purified by RP-HPLC on gradient mode of H2O/MeCN equipped with the C18 column, and DAD detector to yield 1 (1.5 mg), 2 (0.4 mg), 3 (0.4 mg), 4 (0.4 mg), 5 (0.5 mg), 6 (1.0 mg), 7 (4.0 mg), 8 (1.8 mg), 9 (2.8 mg), 10 (3.0 mg), 11 (0.4 mg), and 12 (1.3 mg). The Kinetex C18 on RP-HPLC (Phenomenex, 2.6 µm, 2.1 × 100 mm, 0.75 mL/min, DAD detection 200–800 nm, gradient: 0–10 min 5–95% MeCN with 0.1% formic acid, 10–15 min 95% MeCN with 0.1% formic acid) eluted 1 (tR 5.10 min), 2 (tR 4.79 min), 3 (tR 5.28 min), 4 (tR 5.96 min), 5 (tR 7.64 min), 6 (tR 6.00 min), 7 (tR 6.45 min), 8 (tR 4.85 min), 9 (tR 5.69 min), 10 (tR 6.37 min), 11 (tR 6.48 min) and 12 (tR 6.88 min).
Bipolenin K (1): Colourless oil; [α]D20 −59 (c 0.15, MeOH); IR (KBr) λmax 3445, 2926 and 1729 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z: [M + H]+ calcd for C15H23O3+, 251.1642; found, 251.1646, and [M + H − H2O]+ calcd for C15H21O2+, 233.1536; found, 233.1531.
Bipolenin L (2): Colourless oil; [α]D20 −63 (c 0.04, MeOH); IR (KBr) λmax 3419, 2920 and 1730 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z: [M + H]+ calcd for C15H23O3+, 251.1642; found, 251.1649, and [M + H − H2O]+ calcd for C15H21O2+, 233.1536; found, 233.1535.
Bipolenin M (3): Colourless oil; [α]D20 −57 (c 0.04, MeOH); IR (KBr) λmax 3418, 2927 and 1720 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z: [M + H]+ calcd for C15H23O3+, 251.1642; found, 251.1647, and [M + H − H2O]+ calcd for C15H21O2+, 233.1536; found, 233.1545.
Bipolenin N (4): Colourless oil; [α]D20 +38 (c 0.04, MeOH); IR (KBr) λmax 3334, 2927 and 1045 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z: [M + H − 2H2O]+ calcd for C15H25O2+, 237.1849; found, 237.1859.
Phytotoxicity assays
The phytotoxicity assays on wheat leaves and seeds were carried out as previously reported [37]. Briefly, the leaves of 17-days-old wheat seedlings in 10 cm planting pots were grown at 20 °C under a 16 h/8 h light/dark cycle regime. Compounds were dissolved in 0.2% MeOH/H2O and 30 µL of dissolved solution was infiltrated on the adaxial face of leaves at concentrations of 100, 200 and 500 ppm (serial dilution) using a 1 mL syringe. The leaves were examined for the presence of necrosis or chlorosis after 24 h and 48 h. The control consisted of 30 µL of 0.2% MeOH/H2O without dissolved compound. Two wheat seeds (sterilised by 10% EtOH) were placed on top of the agar (1.5% agar in 1 mL of tap water) containing 100 ppm of compound. The control was agar containing 30 µL of MeOH. The seeds were monitored for the progress of germination on day 5 and day 7.
Calculation of ECD spectra
Structures were initially subjected to a LowModeMD conformational search using the Molecular Operating Environment 2019.0101 package. The lowest energy geometry for each molecule was further optimised by DFT at the B3LYP-D3/def2-TZVPP level of theory using Turbomole 7.1 [38] and ECD spectra were calculated in Turbomole using TDDFT (B3LYP-D3/def2-TZVPP).
Supporting Information
| Supporting Information File 1: NMR, IR and MS spectra of compounds 1–4. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgements
This study was funded, in part, by the Australian Research Council (FT130100142 and FT160100233) and the Cooperative Research Centres Projects scheme (CRCPFIVE000119). The research was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. Authors acknowledge the facilities of Microscopy Australia at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.
References
-
Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Ai, H.-L.; Yang, M.-S.; Zi, S.-H.; Guo, H.-C. J. Asian Nat. Prod. Res. 2015, 17, 982–987. doi:10.1080/10286020.2015.1041929
Return to citation in text: [1] [2] [3] [4] [5] -
Yang, M.-S.; Cai, X.-Y.; He, Y.-Y.; Lu, M.-Y.; Liu, S.; Wang, W.-X.; Li, Z.-H.; Ai, H.-L.; Feng, T. Nat. Prod. Bioprospect. 2017, 7, 147–150. doi:10.1007/s13659-016-0116-4
Return to citation in text: [1] [2] [3] [4] -
Li, Z.-H.; Ai, H.-L.; Yang, M.-S.; He, J.; Feng, T. Phytochem. Lett. 2018, 27, 87–89. doi:10.1016/j.phytol.2018.07.007
Return to citation in text: [1] [2] -
He, J.; Li, Z.-H.; Ai, H.-L.; Feng, T.; Liu, J.-K. Nat. Prod. Res. 2018. doi:10.1080/14786419.2018.1486313
Return to citation in text: [1] [2] [3] [4] -
Jahani, M.; Aggarwal, R.; Gupta, S.; Sharma, S.; Dureja, P. Cereal Res. Commun. 2014, 42, 252–261. doi:10.1556/crc.2013.0053
Return to citation in text: [1] [2] -
He, J.; Yang, M.-S.; Wang, W.-X.; Li, Z.-H.; Elkhateeb, W. A. M.; Wen, T.-C.; Ai, H.-L.; Feng, T. RSC Adv. 2019, 9, 128–131. doi:10.1039/c8ra09861a
Return to citation in text: [1] [2] -
Wang, Q.-x.; Qi, Q.-y.; Wang, K.; Li, L.; Bao, L.; Han, J.-j.; Liu, M.-m.; Zhang, L.-x.; Cai, L.; Liu, H.-w. Org. Lett. 2013, 15, 3982–3985. doi:10.1021/ol401736z
Return to citation in text: [1] -
Nihashi, Y.; Lim, C. H.; Tanaka, C.; Miyagawa, H.; Ueno, T. Biosci., Biotechnol., Biochem. 2002, 66, 685–688. doi:10.1271/bbb.66.685
Return to citation in text: [1] [2] -
Muria-Gonzalez, M. J.; Chooi, Y.-H.; Breen, S.; Solomon, P. S. Mol. Plant Pathol. 2015, 16, 92–107. doi:10.1111/mpp.12162
Return to citation in text: [1] [2] -
Chooi, Y.-H.; Solomon, P. S. Front. Microbiol. 2014, 5, No. 640. doi:10.3389/fmicb.2014.00640
Return to citation in text: [1] -
Kumar, J.; Schäfer, P.; Hückelhoven, R.; Langen, G.; Baltruschat, H.; Stein, E.; Nagarajan, S.; Kogel, K.-H. Mol. Plant Pathol. 2002, 3, 185–195. doi:10.1046/j.1364-3703.2002.00120.x
Return to citation in text: [1] -
McDonald, M. C.; Ahren, D.; Simpfendorfer, S.; Milgate, A.; Solomon, P. S. Mol. Plant Pathol. 2018, 19, 432–439. doi:10.1111/mpp.12535
Return to citation in text: [1] [2] [3] -
Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Lim, C. H.; Miyagawa, H.; Ueno, T.; Takenaka, H.; Sung, N. D. Agric. Chem. Biotechnol. (Engl. Ed.) 1996, 39, 241–244.
Return to citation in text: [1] [2] -
Chan, J.; Jamison, T. F. J. Am. Chem. Soc. 2004, 126, 10682–10691. doi:10.1021/ja0470968
Return to citation in text: [1] -
Aldridge, D. C.; Turner, W. B. J. Chem. Soc. C 1970, 686–688. doi:10.1039/j39700000686
Return to citation in text: [1] [2] [3] [4] -
Qader, M. M.; Kumar, N. S.; Jayasinghe, L.; Araya, H.; Fujimoto, Y. Mycology 2017, 8, 17–20. doi:10.1080/21501203.2016.1269844
Return to citation in text: [1] -
Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Agric. Biol. Chem. 1965, 29, 216–221. doi:10.1080/00021369.1965.10858370
Return to citation in text: [1] [2] [3] [4] -
Masi, M.; Meyer, S.; Górecki, M.; Pescitelli, G.; Clement, S.; Cimmino, A.; Evidente, A. Molecules 2018, 23, 1734. doi:10.3390/molecules23071734
Return to citation in text: [1] -
Oka, M.; Iimura, S.; Narita, Y.; Furumai, T.; Konishi, M.; Oki, T.; Gao, Q.; Kakisawa, H. J. Org. Chem. 1993, 58, 1875–1881. doi:10.1021/jo00059a045
Return to citation in text: [1] -
Oka, M.; Iimura, S.; Tenmyo, O.; Sawada, Y.; Sugawara, M.; Okhusa, N.; Yamamoto, H.; Kawano, K.; Hu, S. L.; Fukagawa, Y.; Oki, T. J. Antibiot. 1993, 46, 367–373. doi:10.7164/antibiotics.46.367
Return to citation in text: [1] -
Schlegel, B.; Schmidtke, M.; Dörfelt, H.; Kleinwächter, P.; Gräfe, U. J. Basic Microbiol. 2001, 41, 179–183. doi:10.1002/1521-4028(200107)41:3/4<179::aid-jobm179>3.0.co;2-h
Return to citation in text: [1] -
Jung, H. J.; Kim, Y.; Chang, J.; Kang, S. W.; Kim, J. H.; Kwon, H. J. J. Mol. Med. (Heidelberg, Ger.) 2013, 91, 1117–1128. doi:10.1007/s00109-013-1049-6
Return to citation in text: [1] -
Zhang, G.-F.; Guo, Z.-K.; Wang, W.; Cui, J.-T.; Tan, R.-X.; Ge, H.-M. J. Asian Nat. Prod. Res. 2011, 13, 761–764. doi:10.1080/10286020.2011.585608
Return to citation in text: [1] [2] [3] [4] -
Abdel-Lateff, A.; Okino, T.; Alarif, W. M.; Al-Lihaibi, S. S. J. Saudi Chem. Soc. 2013, 17, 161–165. doi:10.1016/j.jscs.2011.03.002
Return to citation in text: [1] [2] -
Lodewyk, M. W.; Gutta, P.; Tantillo, D. J. J. Org. Chem. 2008, 73, 6570–6579. doi:10.1021/jo800868r
Return to citation in text: [1] -
Little, D. B.; Croteau, R. B. Arch. Biochem. Biophys. 2002, 402, 120–135. doi:10.1016/s0003-9861(02)00068-1
Return to citation in text: [1] -
Steele, C. L.; Crock, J.; Bohlmann, J.; Croteau, R. J. Biol. Chem. 1998, 273, 2078–2089. doi:10.1074/jbc.273.4.2078
Return to citation in text: [1] -
Van Vranken, D.; Weiss, G. A. Introduction to Bioorganic Chemistry and Chemical Biology, 1st ed.; Garland Science: New York, U.S.A., 2013.
Return to citation in text: [1] -
Nakajima, H.; Toratsu, Y.; Fujii, Y.; Ichinoe, M.; Hamasaki, T. Tetrahedron Lett. 1998, 39, 1013–1016. doi:10.1016/s0040-4039(97)10803-6
Return to citation in text: [1] -
de Mayo, P.; Williams, R. E. J. Am. Chem. Soc. 1965, 87, 3275. doi:10.1021/ja01092a066
Return to citation in text: [1] [2] -
Dorn, F.; Arigoni, D. Experientia 1974, 30, 851–852. doi:10.1007/bf01938319
Return to citation in text: [1] [2] -
Zhou, L.; Zheng, X.; Wan, C.-P.; Yu, Z.-F.; Zhang, K.-Q.; Li, G.-H. Chem. Nat. Compd. 2015, 51, 270–272. doi:10.1007/s10600-015-1259-y
Return to citation in text: [1] -
Narita, K.; Minami, A.; Ozaki, T.; Liu, C.; Kodama, M.; Oikawa, H. J. Org. Chem. 2018, 83, 7042–7048. doi:10.1021/acs.joc.7b03220
Return to citation in text: [1] -
Li, H.; Hu, J.; Wei, H.; Solomon, P. S.; Vuong, D.; Lacey, E.; Stubbs, K. A.; Piggott, A. M.; Chooi, Y.-H. Org. Lett. 2018, 20, 6148–6152. doi:10.1021/acs.orglett.8b02617
Return to citation in text: [1] -
TURBOMOLE V7.1; a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
Return to citation in text: [1]
| 3. | Ai, H.-L.; Yang, M.-S.; Zi, S.-H.; Guo, H.-C. J. Asian Nat. Prod. Res. 2015, 17, 982–987. doi:10.1080/10286020.2015.1041929 |
| 4. | Yang, M.-S.; Cai, X.-Y.; He, Y.-Y.; Lu, M.-Y.; Liu, S.; Wang, W.-X.; Li, Z.-H.; Ai, H.-L.; Feng, T. Nat. Prod. Bioprospect. 2017, 7, 147–150. doi:10.1007/s13659-016-0116-4 |
| 6. | He, J.; Li, Z.-H.; Ai, H.-L.; Feng, T.; Liu, J.-K. Nat. Prod. Res. 2018. doi:10.1080/14786419.2018.1486313 |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 26. | Zhang, G.-F.; Guo, Z.-K.; Wang, W.; Cui, J.-T.; Tan, R.-X.; Ge, H.-M. J. Asian Nat. Prod. Res. 2011, 13, 761–764. doi:10.1080/10286020.2011.585608 |
| 27. | Abdel-Lateff, A.; Okino, T.; Alarif, W. M.; Al-Lihaibi, S. S. J. Saudi Chem. Soc. 2013, 17, 161–165. doi:10.1016/j.jscs.2011.03.002 |
| 28. | Lodewyk, M. W.; Gutta, P.; Tantillo, D. J. J. Org. Chem. 2008, 73, 6570–6579. doi:10.1021/jo800868r |
| 29. | Little, D. B.; Croteau, R. B. Arch. Biochem. Biophys. 2002, 402, 120–135. doi:10.1016/s0003-9861(02)00068-1 |
| 30. | Steele, C. L.; Crock, J.; Bohlmann, J.; Croteau, R. J. Biol. Chem. 1998, 273, 2078–2089. doi:10.1074/jbc.273.4.2078 |
| 31. | Van Vranken, D.; Weiss, G. A. Introduction to Bioorganic Chemistry and Chemical Biology, 1st ed.; Garland Science: New York, U.S.A., 2013. |
| 32. | Nakajima, H.; Toratsu, Y.; Fujii, Y.; Ichinoe, M.; Hamasaki, T. Tetrahedron Lett. 1998, 39, 1013–1016. doi:10.1016/s0040-4039(97)10803-6 |
| 27. | Abdel-Lateff, A.; Okino, T.; Alarif, W. M.; Al-Lihaibi, S. S. J. Saudi Chem. Soc. 2013, 17, 161–165. doi:10.1016/j.jscs.2011.03.002 |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 18. | Aldridge, D. C.; Turner, W. B. J. Chem. Soc. C 1970, 686–688. doi:10.1039/j39700000686 |
| 20. | Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Agric. Biol. Chem. 1965, 29, 216–221. doi:10.1080/00021369.1965.10858370 |
| 33. | de Mayo, P.; Williams, R. E. J. Am. Chem. Soc. 1965, 87, 3275. doi:10.1021/ja01092a066 |
| 34. | Dorn, F.; Arigoni, D. Experientia 1974, 30, 851–852. doi:10.1007/bf01938319 |
| 3. | Ai, H.-L.; Yang, M.-S.; Zi, S.-H.; Guo, H.-C. J. Asian Nat. Prod. Res. 2015, 17, 982–987. doi:10.1080/10286020.2015.1041929 |
| 4. | Yang, M.-S.; Cai, X.-Y.; He, Y.-Y.; Lu, M.-Y.; Liu, S.; Wang, W.-X.; Li, Z.-H.; Ai, H.-L.; Feng, T. Nat. Prod. Bioprospect. 2017, 7, 147–150. doi:10.1007/s13659-016-0116-4 |
| 5. | Li, Z.-H.; Ai, H.-L.; Yang, M.-S.; He, J.; Feng, T. Phytochem. Lett. 2018, 27, 87–89. doi:10.1016/j.phytol.2018.07.007 |
| 6. | He, J.; Li, Z.-H.; Ai, H.-L.; Feng, T.; Liu, J.-K. Nat. Prod. Res. 2018. doi:10.1080/14786419.2018.1486313 |
| 8. | He, J.; Yang, M.-S.; Wang, W.-X.; Li, Z.-H.; Elkhateeb, W. A. M.; Wen, T.-C.; Ai, H.-L.; Feng, T. RSC Adv. 2019, 9, 128–131. doi:10.1039/c8ra09861a |
| 26. | Zhang, G.-F.; Guo, Z.-K.; Wang, W.; Cui, J.-T.; Tan, R.-X.; Ge, H.-M. J. Asian Nat. Prod. Res. 2011, 13, 761–764. doi:10.1080/10286020.2011.585608 |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 35. | Zhou, L.; Zheng, X.; Wan, C.-P.; Yu, Z.-F.; Zhang, K.-Q.; Li, G.-H. Chem. Nat. Compd. 2015, 51, 270–272. doi:10.1007/s10600-015-1259-y |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 18. | Aldridge, D. C.; Turner, W. B. J. Chem. Soc. C 1970, 686–688. doi:10.1039/j39700000686 |
| 20. | Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Agric. Biol. Chem. 1965, 29, 216–221. doi:10.1080/00021369.1965.10858370 |
| 33. | de Mayo, P.; Williams, R. E. J. Am. Chem. Soc. 1965, 87, 3275. doi:10.1021/ja01092a066 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 16. | Lim, C. H.; Miyagawa, H.; Ueno, T.; Takenaka, H.; Sung, N. D. Agric. Chem. Biotechnol. (Engl. Ed.) 1996, 39, 241–244. |
| 20. | Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Agric. Biol. Chem. 1965, 29, 216–221. doi:10.1080/00021369.1965.10858370 |
| 3. | Ai, H.-L.; Yang, M.-S.; Zi, S.-H.; Guo, H.-C. J. Asian Nat. Prod. Res. 2015, 17, 982–987. doi:10.1080/10286020.2015.1041929 |
| 6. | He, J.; Li, Z.-H.; Ai, H.-L.; Feng, T.; Liu, J.-K. Nat. Prod. Res. 2018. doi:10.1080/14786419.2018.1486313 |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 26. | Zhang, G.-F.; Guo, Z.-K.; Wang, W.; Cui, J.-T.; Tan, R.-X.; Ge, H.-M. J. Asian Nat. Prod. Res. 2011, 13, 761–764. doi:10.1080/10286020.2011.585608 |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 4. | Yang, M.-S.; Cai, X.-Y.; He, Y.-Y.; Lu, M.-Y.; Liu, S.; Wang, W.-X.; Li, Z.-H.; Ai, H.-L.; Feng, T. Nat. Prod. Bioprospect. 2017, 7, 147–150. doi:10.1007/s13659-016-0116-4 |
| 18. | Aldridge, D. C.; Turner, W. B. J. Chem. Soc. C 1970, 686–688. doi:10.1039/j39700000686 |
| 26. | Zhang, G.-F.; Guo, Z.-K.; Wang, W.; Cui, J.-T.; Tan, R.-X.; Ge, H.-M. J. Asian Nat. Prod. Res. 2011, 13, 761–764. doi:10.1080/10286020.2011.585608 |
| 36. | Narita, K.; Minami, A.; Ozaki, T.; Liu, C.; Kodama, M.; Oikawa, H. J. Org. Chem. 2018, 83, 7042–7048. doi:10.1021/acs.joc.7b03220 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 14. | McDonald, M. C.; Ahren, D.; Simpfendorfer, S.; Milgate, A.; Solomon, P. S. Mol. Plant Pathol. 2018, 19, 432–439. doi:10.1111/mpp.12535 |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 3. | Ai, H.-L.; Yang, M.-S.; Zi, S.-H.; Guo, H.-C. J. Asian Nat. Prod. Res. 2015, 17, 982–987. doi:10.1080/10286020.2015.1041929 |
| 4. | Yang, M.-S.; Cai, X.-Y.; He, Y.-Y.; Lu, M.-Y.; Liu, S.; Wang, W.-X.; Li, Z.-H.; Ai, H.-L.; Feng, T. Nat. Prod. Bioprospect. 2017, 7, 147–150. doi:10.1007/s13659-016-0116-4 |
| 5. | Li, Z.-H.; Ai, H.-L.; Yang, M.-S.; He, J.; Feng, T. Phytochem. Lett. 2018, 27, 87–89. doi:10.1016/j.phytol.2018.07.007 |
| 6. | He, J.; Li, Z.-H.; Ai, H.-L.; Feng, T.; Liu, J.-K. Nat. Prod. Res. 2018. doi:10.1080/14786419.2018.1486313 |
| 7. | Jahani, M.; Aggarwal, R.; Gupta, S.; Sharma, S.; Dureja, P. Cereal Res. Commun. 2014, 42, 252–261. doi:10.1556/crc.2013.0053 |
| 11. | Muria-Gonzalez, M. J.; Chooi, Y.-H.; Breen, S.; Solomon, P. S. Mol. Plant Pathol. 2015, 16, 92–107. doi:10.1111/mpp.12162 |
| 3. | Ai, H.-L.; Yang, M.-S.; Zi, S.-H.; Guo, H.-C. J. Asian Nat. Prod. Res. 2015, 17, 982–987. doi:10.1080/10286020.2015.1041929 |
| 10. | Nihashi, Y.; Lim, C. H.; Tanaka, C.; Miyagawa, H.; Ueno, T. Biosci., Biotechnol., Biochem. 2002, 66, 685–688. doi:10.1271/bbb.66.685 |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 9. | Wang, Q.-x.; Qi, Q.-y.; Wang, K.; Li, L.; Bao, L.; Han, J.-j.; Liu, M.-m.; Zhang, L.-x.; Cai, L.; Liu, H.-w. Org. Lett. 2013, 15, 3982–3985. doi:10.1021/ol401736z |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 8. | He, J.; Yang, M.-S.; Wang, W.-X.; Li, Z.-H.; Elkhateeb, W. A. M.; Wen, T.-C.; Ai, H.-L.; Feng, T. RSC Adv. 2019, 9, 128–131. doi:10.1039/c8ra09861a |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 14. | McDonald, M. C.; Ahren, D.; Simpfendorfer, S.; Milgate, A.; Solomon, P. S. Mol. Plant Pathol. 2018, 19, 432–439. doi:10.1111/mpp.12535 |
| 14. | McDonald, M. C.; Ahren, D.; Simpfendorfer, S.; Milgate, A.; Solomon, P. S. Mol. Plant Pathol. 2018, 19, 432–439. doi:10.1111/mpp.12535 |
| 13. | Kumar, J.; Schäfer, P.; Hückelhoven, R.; Langen, G.; Baltruschat, H.; Stein, E.; Nagarajan, S.; Kogel, K.-H. Mol. Plant Pathol. 2002, 3, 185–195. doi:10.1046/j.1364-3703.2002.00120.x |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 11. | Muria-Gonzalez, M. J.; Chooi, Y.-H.; Breen, S.; Solomon, P. S. Mol. Plant Pathol. 2015, 16, 92–107. doi:10.1111/mpp.12162 |
| 37. | Li, H.; Hu, J.; Wei, H.; Solomon, P. S.; Vuong, D.; Lacey, E.; Stubbs, K. A.; Piggott, A. M.; Chooi, Y.-H. Org. Lett. 2018, 20, 6148–6152. doi:10.1021/acs.orglett.8b02617 |
| 12. | Chooi, Y.-H.; Solomon, P. S. Front. Microbiol. 2014, 5, No. 640. doi:10.3389/fmicb.2014.00640 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 7. | Jahani, M.; Aggarwal, R.; Gupta, S.; Sharma, S.; Dureja, P. Cereal Res. Commun. 2014, 42, 252–261. doi:10.1556/crc.2013.0053 |
| 10. | Nihashi, Y.; Lim, C. H.; Tanaka, C.; Miyagawa, H.; Ueno, T. Biosci., Biotechnol., Biochem. 2002, 66, 685–688. doi:10.1271/bbb.66.685 |
| 38. | TURBOMOLE V7.1; a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com. |
| 16. | Lim, C. H.; Miyagawa, H.; Ueno, T.; Takenaka, H.; Sung, N. D. Agric. Chem. Biotechnol. (Engl. Ed.) 1996, 39, 241–244. |
| 17. | Chan, J.; Jamison, T. F. J. Am. Chem. Soc. 2004, 126, 10682–10691. doi:10.1021/ja0470968 |
| 1. | Pena-Rodriguez, L. M.; Armingeon, N. A.; Chilton, W. S. J. Nat. Prod. 1988, 51, 821–828. doi:10.1021/np50059a001 |
| 2. | Nakajima, H.; Isomi, K.; Hamasaki, T.; Ichinoe, M. Tetrahedron Lett. 1994, 35, 9597–9600. doi:10.1016/0040-4039(94)88520-6 |
| 25. | Jung, H. J.; Kim, Y.; Chang, J.; Kang, S. W.; Kim, J. H.; Kwon, H. J. J. Mol. Med. (Heidelberg, Ger.) 2013, 91, 1117–1128. doi:10.1007/s00109-013-1049-6 |
| 15. | Osterhage, C.; König, G. M.; Höller, U.; Wright, A. D. J. Nat. Prod. 2002, 65, 306–313. doi:10.1021/np010092l |
| 22. | Oka, M.; Iimura, S.; Narita, Y.; Furumai, T.; Konishi, M.; Oki, T.; Gao, Q.; Kakisawa, H. J. Org. Chem. 1993, 58, 1875–1881. doi:10.1021/jo00059a045 |
| 23. | Oka, M.; Iimura, S.; Tenmyo, O.; Sawada, Y.; Sugawara, M.; Okhusa, N.; Yamamoto, H.; Kawano, K.; Hu, S. L.; Fukagawa, Y.; Oki, T. J. Antibiot. 1993, 46, 367–373. doi:10.7164/antibiotics.46.367 |
| 24. | Schlegel, B.; Schmidtke, M.; Dörfelt, H.; Kleinwächter, P.; Gräfe, U. J. Basic Microbiol. 2001, 41, 179–183. doi:10.1002/1521-4028(200107)41:3/4<179::aid-jobm179>3.0.co;2-h |
| 20. | Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Agric. Biol. Chem. 1965, 29, 216–221. doi:10.1080/00021369.1965.10858370 |
| 21. | Masi, M.; Meyer, S.; Górecki, M.; Pescitelli, G.; Clement, S.; Cimmino, A.; Evidente, A. Molecules 2018, 23, 1734. doi:10.3390/molecules23071734 |
| 18. | Aldridge, D. C.; Turner, W. B. J. Chem. Soc. C 1970, 686–688. doi:10.1039/j39700000686 |
| 19. | Qader, M. M.; Kumar, N. S.; Jayasinghe, L.; Araya, H.; Fujimoto, Y. Mycology 2017, 8, 17–20. doi:10.1080/21501203.2016.1269844 |
© 2019 Phan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)