Abstract
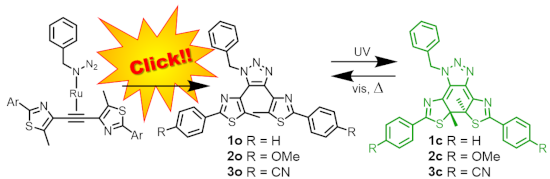
Three new diarylethenes were synthesized from 1,2-bis(5-methyl-2-(4-substituted-phenyl)thiazol-4-yl)ethyne and benzyl azide through Ru(I)-catalyzed Huisgen cyclization reactions. The 4,5-bisthiazolyl-1,2,3-triazoles thus prepared, which belong to the terarylene family, showed thermally reversible photochromism. The absorption maximum wavelengths of the closed forms are longer than other terarylenes reported so far. The thermal back reactions are much faster when the substituents on the terminal phenyl groups are electron-withdrawing cyano groups than when they are electron-donating methoxy groups.
Graphical Abstract
Introduction
Diarylethenes are one of the most widely investigated among the photochromic families [1-3]. They are known to show thermally irreversible photochromism. However, some of them are thermally reversible [4]: (1) when the aromatic stabilization energy of the aromatic rings is large [5]; (2) when the substituent groups on the ring-closing carbon atoms are large [6]; (3) when the substituent groups on the ring-closing carbon atoms are strongly electron-withdrawing [7]; or (4) when the dialkylamino group on the side chain [8] or a carbon atom of the conjugation system in a strained closed form [9] are protonated.
Terarylenes [10,11], one of the closely related families of diarylethenes, are largely thermally reversible [10,12] although some are irreversible when the aromatic stabilization energy of the aromatic rings is small [11]. Their syntheses are usually carried out by the sequential construction of the central aromatic ring at the final stage [10] or the introduction of two aromatic rings to the central aromatic ring [11]. If the construction of three contiguous aromatic-ring arrays can be easily achieved, a new and facile synthesis method for the photochromic family which undergoes 6π-electrocyclization can be realized [13].
Early in the 21st century, Sharpless and co-workers proposed the concept of “click chemistry” [14], which stands for the secure, quick, selective, general and facile reaction between two organic functional groups. In click chemistry, the Huisgen cyclization, which occurs between an organic azide and a terminal alkyne catalyzed by a Cu(I) ion, was regarded as the representative reaction [15,16]. This reaction occurs even in aqueous media, the chemical yield is always high, and the regiochemical structure of the product is always 1,4-disubstituted triazole.
Cu(I)-catalyzed Huisgen cyclization proceeds by the formation of copper acetylide as the intermediate [15,17], resulting in the formation of 1,4-disubstituted triazoles. In contrast, when Ru(I) complexes are employed as the catalyst, the reaction mechanism is different from the case of Cu(I), and the major products are 1,5-disubstituted triazoles. Another more important difference is that Ru(I) catalysts work on the disubstituted alkynes to give 1,4,5-trisubstituted triazoles (Scheme 1) [18,19]. When both substituents of an internal alkyne are aromatic groups, the triazoles thus formed include the hexatriene motif in the structure.
![[1860-5397-15-213-i1]](/bjoc/content/inline/1860-5397-15-213-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction mechanisms of Huisgen cyclization catalyzed by Cu(I) and Ru(I).
Scheme 1: Reaction mechanisms of Huisgen cyclization catalyzed by Cu(I) and Ru(I).
Although a number of photochromic diarylethenes containing triazole groups have been reported [20-31], all of them use the triazole ring as the linker of the second functional molecule with the diarylethene core. To the best of our knowledge, no diarylethenes or terarylenes possessing the triazole ring as one of the components of the hexatriene moiety has been reported to date.
Accordingly, we decided to employ a Ru(I) catalyst to synthesize 4,5-diaryl-1-substituted-1,2,3-triazoles 1o–3o as possible photochromic compounds (Scheme 2).
![[1860-5397-15-213-i2]](/bjoc/content/inline/1860-5397-15-213-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and photochromism of bisthiazolyltriazoles.
Scheme 2: Synthesis and photochromism of bisthiazolyltriazoles.
Results and Discussion
Molecular design and synthesis
As the organic azide we used commercially available benzyl azide. Since 1,2-bis(5-methyl-2-phenylthiazol-4-yl)ethyne was used in our previous research [32-34], we employed bisthiazolylethynes as the foundation for the skeleton of the target compounds. In order to examine the substituent effects of the terminal phenyl groups on the photochromic properties, compounds with methoxy groups or cyano groups at the para-position of the phenyl groups were also synthesized. To avoid the generation of isomers with a substitution pattern by Huisgen cyclization, the same substituents were introduced to both phenyl groups.
The Ru(I)-catalyzed Huisgen cyclization reaction proceeded rather smoothly to give 1o, 2o and 3o in moderate chemical yields of 36%, 53% and 20%, respectively. Since the reported chemical yield of the reaction of tolan, the simplest bisarylethyne, and 2-phenylethyl azide was 63% [19], it could be considered reasonable that the reactions of the sterically more congested ethynes and benzyl azide gave triazoles with less chemical yields. Details of the synthesis are described in Supporting Information File 1.
Photochromic reactions
Photochemical cyclizations
Triazoles were dissolved in ethanol (EtOH), acetonitrile (MeCN), ethyl acetate (AcOEt) and toluene, and each solution was irradiated with 313 nm light. The changes in the absorption spectra during UV light irradiation in MeCN are shown in Figure 1 (spectra of the compounds in the other solvents are shown in Figures S1–S9 in Supporting Information File 1). The absorption spectral properties are summarized in Table 1 together with their predicted absorption maxima obtained by TD DFT calculations in vacuum [35]. Although 1o and 2o showed substantial coloration, 3o showed only a slight coloration at room temperature.
![[1860-5397-15-213-1]](/bjoc/content/figures/1860-5397-15-213-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Absorption spectral change of triazoles 1o–3o upon irradiation of 313 nm light in MeCN at 28 °C. Light intensity: 1.8 mW cm−2. (a) 1o. 4.44 × 10−5 mol dm−3. (b) 2o. 4.38 × 10−5 mol dm−3. (c) 3o. 4.73 × 10−5 mol dm−3.
Figure 1: Absorption spectral change of triazoles 1o–3o upon irradiation of 313 nm light in MeCN at 28 °C. Li...
Table 1: Wavelengths of absorption maxima of the closed forms of photochromic triazoles.
| Solvent and calculation method | EtOH | MeCN | AcOEt | toluene | TD DFTa |
|---|---|---|---|---|---|
|
er b
ETN c |
24.55
0.654 |
35.94
0.460 |
6.02
0.228 |
2.38
0.099 |
in vacuum |
| λmax/nm | |||||
| 1c (R = H) | 692 | 695 | 693 | 698 | 704 |
| 2c (R = OMe) | 697 | 696 | 693 | 699 | 699 |
| 3c (R = CN) | 737 | 739 | 737 | 739 | 759 |
aIn vacuum [35]. bRelative dielectric constant. cNormalized ET(30) value.
The solvent effects on the absorption maximum wavelengths of the closed forms were not remarkable. The tendency observed was of the absorption maximum wavelength being a few nanometers longer in less polar toluene than for the other solvents.
When the substituents on the terminal phenyl rings are electron-withdrawing cyano groups, the measured absorption maximum of the closed form was about 40 nm longer than the others in any solvent. The substituent effect observed here is different from the small substituent effect on the closed forms of the representative bisthienylhexafluorocyclopentenes 4c (R = H, 562 nm), 5c (R = OMe, 570 nm) and 6c (R = CN, 570 nm) in hexane (Scheme 3) [36]. In triazoles, while the introduction of electron-donating methoxy groups had little effect on the absorption maximum wavelengths, the introduction of electron-withdrawing cyano groups induced a large bathochromic shift. This observation was well-reproduced by TD DFT calculations (Table 1 and chapter SI-4 in Supporting Information File 1).
![[1860-5397-15-213-i3]](/bjoc/content/inline/1860-5397-15-213-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Wavelengths of absorption maxima of the closed forms of bisthienyletenes in hexane [36].
Scheme 3: Wavelengths of absorption maxima of the closed forms of bisthienyletenes in hexane [36].
When the absorption maximum wavelength of 1c, whose central ethene moiety is triazole, is compared with those of the closely related 7c (cyclopentene) [37], 8c (hexafluorocyclopentene) [38], 9c (thiazole) [39], and 10c (imidazole) [40] in non-polar solvents, 1c has a much longer absorption maximum in toluene (Scheme 4, Table 2). It should also be noted that the absorption maximum wavelength is longer when the central ethene moiety is part of the aromatic ring than when it is an isolated ethene in their open-form structures.
Table 2: Absorption spectral data of triazoles and other related photochromic compounds.
aIn vacuum [35]. bIn toluene. cIn cyclohexane. dIn hexane.
![[1860-5397-15-213-i4]](/bjoc/content/inline/1860-5397-15-213-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Photochromism of closely related compounds.
Scheme 4: Photochromism of closely related compounds.
Thermal back reactions
As expected, all closed forms of triazoles showed thermal back reactions since the photochemical cyclization results in the loss of the aromatic stabilization energy [41] of the three contiguous aromatic rings. The changes in the absorption spectra of thermal decoloration in MeCN at room temperature are shown in Figure 2.
![[1860-5397-15-213-2]](/bjoc/content/figures/1860-5397-15-213-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Absorption spectral change of triazoles 1c–3c during the thermal back reaction after 313-nm light irradiation to 1o–3o in MeCN at 28 °C. Concentration of compounds are the same as in Figure 1. (a) 1c. (b) 2c. (c) 3c.
Figure 2: Absorption spectral change of triazoles 1c–3c during the thermal back reaction after 313-nm light i...
In order to clarify the nature of the thermal back reaction of triazoles, 1o, 2o and 3o were irradiated with 313 nm light in four different solvents, and the decrease in the absorbance of the absorption maximum wavelengths in the visible region was observed at three different temperatures. The first order reaction rate constants of the thermal back reactions at different temperatures were then determined. Arrhenius plots of ln k against 1/T gave pre-exponential factors (A) and Arrhenius activation energy Ea of these thermally reversible photochromic triazoles (Figures S10–S13 in Supporting Information File 1). The kinetic data of the thermal back reactions of 1c–3c in toluene are shown in Table 3 together with the literature data of related compounds 9c [39] and 10c [40] shown in Scheme 4.
Table 3: Kinetic data of thermal back reactions and aromatic stabilization energy of 1c–3c, 9c and 10c.
| A/s−1 | Ea/kJ mol−1 | k (293K)/s−1 | t1/2 (293 K)a | ASEb/kJ mol−1 | |
|---|---|---|---|---|---|
| 9cc | 7.1 × 1011 | 112 | 6.7 × 10−9d | 3.3 years | 72.9 |
| 10cc | 1.3 × 109 | 85 | 8.7 × 10−7d | 9.2 days | 78.6 |
| 1ce | 1.21 × 1013 | 87.8 | 2.80 × 10−3 | 248 s | 102.0 |
| 2ce | 2.79 × 1012 | 87.9 | 6.14 × 10−4 | 1130 s | 102.0 |
| 3ce | 1.26 × 1013 | 81.4 | 3.92 × 10−2 | 17.7 s | 102.0 |
at1/2 (293 K): Half-life at 293 K. bASE: Aromatic stabilization energy of the central aromatic rings when unsubstituted. Data taken from ref. [41]. cIn toluene. Data taken from ref. [39] for 9 and ref. [40] for 10. dCalculated from t1/2 at 293 K.eIn toluene.
As shown in Table 3, the thermal back reaction is fast when the aromatic stabilization energy of the central aromatic ring is large. Although Ea of 10 is smaller than that of 1 and 2, the pre-exponential factors (A) of 1 and 2 are about 104 times and 103 times larger, respectively, than that of 10.
When the thermal kinetic data of 1, 2 and 3 are compared, the thermal back reaction rate of 3 is extremely faster than the others, although the central aromatic ring is common to these compounds.
As shown in Table 4, neither electronic charge distribution on the carbon atoms (natural charge [42]) comprising the thermally breaking C–C bond, its bond length nor its bond order, which were obtained by DFT calculations of these compounds, did not give a clear explanation for the difference in the reaction rate. Possible evidence of the fast back reaction of 3c may be found in the bond lengths and Mulliken bond orders between the atoms constructing the conjugation system around the central cyclohexadiene moiety (Scheme 5).
Table 4: Natural charge, bond length and bond order of the bond-breaking carbon atoms of triazoles 1c – 3c in the thermal back reactions obtained by DFT calculations.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-213-i7.svg?max-width=637&scale=1.0)
|
||||
| Natural chargea | Bond length/Å | Mulliken bond order | ||
|---|---|---|---|---|
| C1 | C2 | C1–C2 | C1–C2 | |
| 1c | −0.198 | −0.204 | 1.547 | 0.933 |
| 2c | −0.195 | −0.204 | 1.546 | 0.933 |
| 3c | −0.198 | −0.204 | 1.546 | 0.933 |
aCalculated by DFT calculations as the number of positive charges [42].
![[1860-5397-15-213-i5]](/bjoc/content/inline/1860-5397-15-213-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Bond length (a) (in Å) and Mulliken bond order (b) of 1c–3c obtained by DFT calculations. Top number of each set: 1c. Second number: 2c. Bottom number: 3c. Red numbers of 3c: indicates stronger double bond character. Green numbers of 3c: indicates stronger single bond character. Blue numbers of 3c: indicates weaker bond character. Italic numbers around the central conjugation systems: no significant difference between the three compounds.
Scheme 5: Bond length (a) (in Å) and Mulliken bond order (b) of 1c–3c obtained by DFT calculations. Top numbe...
In Scheme 5a, the single bonds of 3c written in red are shorter than in the other two compounds, implying that they have a stronger double bond character. Similarly, the double bonds of 3c written in green are longer, showing the stronger single bond character. This suggests a stronger bond alteration tendency in 3c than in 1c and 2c, and this distinctive character of 3c can be seen more clearly in Scheme 5b.
In addition, since the bond order of the N–Bn bond in blue of 3c is the smallest, it can be reasonably assumed that the lone pair on this nitrogen atom participates in the conjugation in the molecular core more strongly than in 1c and 2c. This distinctive character of 3c originates from the electron-withdrawing cyano groups in the molecule.
Data on the terminal phenyl rings in Scheme 5 do not give any convergent tendency of the substituents due to the electron-donating character of the methoxy groups in 2c and the electron-withdrawing character of the cyano groups in 3c. However, it is safe to say the cyano groups in 3c are pulling the electrons and this effect reaches the nitrogen atom bearing the benzyl group through the conjugation.
Then how does this effect accelerate the thermal back reaction in 3c? We propose the conventional reaction mechanism shown in Scheme 6. When the delocalized lone pair on the nitrogen atom moves back from the resonance structures A and B, it may break the central C–C single bond to give the open form, as shown with the arrows. The resonance structures are stabilized more strongly when R is a cyano group so that the C–C bond scission occurs easily. Thus, it is possible to control the rate of the thermal back reaction by: (1) changing the aromatic ring at the ethene moiety, and (2) changing the substituents on the phenyl rings at the peripheral of the molecule.
![[1860-5397-15-213-i6]](/bjoc/content/inline/1860-5397-15-213-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible reaction mechanism of thermal ring opening of the closed forms.
Scheme 6: Possible reaction mechanism of thermal ring opening of the closed forms.
Finally, we would like to discuss the solvent effects on the thermal back reactions of 1c, 2c, and 3c. As can be seen in Figure S13, the solvents are classified into two categories: (1) polar solvents (EtOH and MeCN) and (2) less polar solvents (AcOEt and toluene). Although the gradient of the lines (Ea) for the same compound in Figure S13a–c are quite similar, the intercepts (A) are clearly different for the two solvent groups of 1c and 2c. As for 3c, although its categorization is not as clear as 1c and 2c, differences can still be observed. In general, the thermal back reactions are faster in the more polar solvents, and faster in MeCN than in EtOH among these two solvents. However, for 2c, it is faster in EtOH than in MeCN. This may have originated from the hydrogen-bond formation between EtOH molecules and the methoxy groups in 2c, which reduces the electrostatic repulsion between the methoxy groups and the negatively charged phenyl groups in the intermediate resonance structures A and B shown in Scheme 6.
In less polar solvents, the thermal back reactions of 1c and 2c are faster in toluene than in AcOEt. However, the reaction of 3c is the opposite. This could be explained by the strong electron-withdrawing power of the cyano groups in 3c, which enhanced the charge-separation character in the intermediate resonance structures A and B. These structures are stabilized more strongly in more polar AcOEt than in toluene, and the thermal back reaction rate in AcOEt was increased.
Conclusion
We have synthesized three novel thermally reversible 4,4'-(1-benzyl-1H-1,2,3-triazole-4,5-diyl)bis(5-methyl-2-(4-substituted-phenyl)thiazole)s 1o–3o by Ru(I)-catalysed Huisgen cyclization, which is a type of “click” reaction. They showed thermally reversible photochromism in various solvents. The absorption maximum wavelengths of 1c (with unsubstituted peripheral phenyl groups) and 2c (with methoxy groups on the phenyl groups) are close to 700 nm, while that of 3c (with cyano groups on the phenyl groups) was about 740 nm. The thermal back reactions of these compounds proved that 3c with the electron-withdrawing cyano groups is the fastest while 2c with electron-donating methoxy groups was the slowest. DFT and TD DFT calculations supported these experimental results.
The Cu(I)-catalyzed Huisgen reaction has been used many times to connect two functional molecules. When the Ru(I)-catalyzed Huisgen reaction is employed to connect two functional molecules, the linker itself possesses the thermally reversible photochromic property. Thus, this work can open the door to the creation of promising new materials with highly integrated functions.
Supporting Information
Experimental details including the synthesis of 1o, 2o and 3o. Changes in the absorption spectra of 1o, 2o and 3o in ethanol, ethyl acetate and toluene. Analysis processes of the thermal back reactions of 1o, 2o and 3o. DFT and TD DFT calculation results of 1c, 2c and 3c and 1H NMR, IR and mass spectra of new compounds.
| Supporting Information File 1: Additional experimental data and spectra. | ||
| Format: PDF | Size: 6.8 MB | Download |
Acknowledgements
This work was supported by MEXT KAKENHI Grant Number JP19050004 in Grant-in-Aid for Scientific Research in Priority Areas “New Frontiers in Photochromism (No. 471)” and JSPS KAKENHI Grant Number JP26107009 in Scientific Research on Innovative Areas “Photosynergetics”. The authors are also grateful to the Instrumental Analysis Center of Yokohama National University for their support in the NMR and MS measurements.
References
-
Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Chem. Rev. 2014, 114, 12174–12277. doi:10.1021/cr500249p
Return to citation in text: [1] -
Tian, H.; Yang, S. Chem. Soc. Rev. 2004, 33, 85–97. doi:10.1039/b302356g
Return to citation in text: [1] -
Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d
Return to citation in text: [1] -
Kitagawa, D.; Sasaki, K.; Kobatake, S. Bull. Chem. Soc. Jpn. 2011, 84, 141–147. doi:10.1246/bcsj.20100274
Return to citation in text: [1] -
Nakamura, S.; Irie, M. J. Org. Chem. 1988, 53, 6136–6138. doi:10.1021/jo00261a035
Return to citation in text: [1] -
Kobatake, S.; Uchida, K.; Tsuchida, E.; Irie, M. Chem. Lett. 2000, 29, 1340–1341. doi:10.1246/cl.2000.1340
Return to citation in text: [1] -
Gilat, S. L.; Kawai, S. H.; Lehn, J.-M. Chem. – Eur. J. 1995, 1, 275–284. doi:10.1002/chem.19950010504
Return to citation in text: [1] -
Kobatake, S.; Terakawa, Y. Chem. Commun. 2007, 1698–1700. doi:10.1039/b700177k
Return to citation in text: [1] -
Jurissek, C.; Berger, F.; Eisenreich, F.; Kathan, M.; Hecht, S. Angew. Chem., Int. Ed. 2019, 58, 1945–1949. doi:10.1002/anie.201812284
Return to citation in text: [1] -
Krayushkin, M. M.; Ivanov, S. N.; Martynkin, A. Y.; Lichitsky, B. V.; Dudinov, A. A.; Uzhinov, B. M. Russ. Chem. Bull. 2001, 50, 116–121. doi:10.1023/a:1009541605405
Return to citation in text: [1] [2] [3] -
Kawai, T.; Iseda, T.; Irie, M. Chem. Commun. 2004, 72–73. doi:10.1039/b311334e
Return to citation in text: [1] [2] [3] -
Kawai, S.; Nakashima, T.; Atsumi, K.; Sakai, T.; Harigai, M.; Imamoto, Y.; Kamikubo, H.; Kataoka, M.; Kawai, T. Chem. Mater. 2007, 19, 3479–3483. doi:10.1021/cm0630340
Return to citation in text: [1] -
Kochi, J.; Ubukata, T.; Yokoyama, Y. J. Org. Chem. 2018, 83, 10695–10700. doi:10.1021/acs.joc.8b01341
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.3.co;2-x
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4
Return to citation in text: [1] [2] -
Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128–1137. doi:10.1016/s1359-6446(03)02933-7
Return to citation in text: [1] -
Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457–460. doi:10.1126/science.1229506
Return to citation in text: [1] -
Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s
Return to citation in text: [1] -
Boren, B. C.; Narayan, S.; Rasmussen, L. K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V. V. J. Am. Chem. Soc. 2008, 130, 8923–8930. doi:10.1021/ja0749993
Return to citation in text: [1] [2] -
Finden, J.; Kunz, T. K.; Branda, N. R.; Wolf, M. O. Adv. Mater. (Weinheim, Ger.) 2008, 20, 1998–2002. doi:10.1002/adma.200702455
Return to citation in text: [1] -
Ouhenia-Ouadahi, K.; Métivier, R.; Maisonneuve, S.; Jacquart, A.; Xie, J.; Léaustic, A.; Yu, P.; Nakatani, K. Photochem. Photobiol. Sci. 2012, 11, 1705–1714. doi:10.1039/c2pp25129a
Return to citation in text: [1] -
Ding, H.; Liu, G.; Pu, S.; Zheng, C. Dyes Pigm. 2014, 103, 82–88. doi:10.1016/j.dyepig.2013.11.022
Return to citation in text: [1] -
Pu, S.; Ding, H.; Liu, G.; Zheng, C.; Xu, H. J. Phys. Chem. C 2014, 118, 7010–7017. doi:10.1021/jp5001495
Return to citation in text: [1] -
Maisonneuve, S.; Métivier, R.; Yu, P.; Nakatani, K.; Xie, J. Beilstein J. Org. Chem. 2014, 10, 1471–1481. doi:10.3762/bjoc.10.151
Return to citation in text: [1] -
Ma, L.; Liu, G.; Pu, S.; Ding, H.; Li, G. Tetrahedron 2016, 72, 985–991. doi:10.1016/j.tet.2015.12.068
Return to citation in text: [1] -
Duan, F.; Liu, G.; Fan, C.; Pu, S. Tetrahedron Lett. 2016, 57, 1963–1966. doi:10.1016/j.tetlet.2016.03.079
Return to citation in text: [1] -
Xue, D.; Zheng, C.; Qu, S.; Liao, G.; Fan, C.; Liu, G.; Pu, S. Luminescence 2017, 32, 652–660. doi:10.1002/bio.3234
Return to citation in text: [1] -
Singh, R.; Wu, H.-Y.; Kumar Dwivedi, A.; Singh, A.; Lin, C.-M.; Raghunath, P.; Lin, M.-C.; Wu, T.-K.; Wei, K.-H.; Lin, H.-C. J. Mater. Chem. C 2017, 5, 9952–9962. doi:10.1039/c7tc03071a
Return to citation in text: [1] -
Wang, N.; Wang, R.; Tu, Y.; Pu, S.; Liu, G. Spectrochim. Acta, Part A 2018, 196, 303–310. doi:10.1016/j.saa.2018.02.018
Return to citation in text: [1] -
Guo, S.; Liu, G.; Fan, C.; Pu, S. RSC Adv. 2018, 8, 22786–22798. doi:10.1039/c8ra03443e
Return to citation in text: [1] -
Kang, H.; Fan, C.; Xu, H.; Liu, G.; Pu, S. Tetrahedron 2018, 74, 4390–4399. doi:10.1016/j.tet.2018.07.002
Return to citation in text: [1] -
Morinaka, K.; Ubukata, T.; Yokoyama, Y. Org. Lett. 2009, 11, 3890–3893. doi:10.1021/ol901497t
Return to citation in text: [1] -
Ogawa, H.; Takagi, K.; Ubukata, T.; Okamoto, A.; Yonezawa, N.; Delbaere, S.; Yokoyama, Y. Chem. Commun. 2012, 48, 11838–11840. doi:10.1039/c2cc35793c
Return to citation in text: [1] -
Nakagawa, T.; Miyasaka, Y.; Yokoyama, Y. Chem. Commun. 2018, 54, 3207–3210. doi:10.1039/c8cc00566d
Return to citation in text: [1] -
DFT and TD DFT calculations were carried out with Spartan’18 (Wavefunction) at the B3LYP/6-31G* level.
Return to citation in text: [1] [2] [3] -
Irie, M.; Sakemura, K.; Okinaka, M.; Uchida, K. J. Org. Chem. 1995, 60, 8305–8309. doi:10.1021/jo00130a035
Return to citation in text: [1] [2] -
Herder, M.; Schmidt, B. M.; Grubert, L.; Pätzel, M.; Schwarz, J.; Hecht, S. J. Am. Chem. Soc. 2015, 137, 2738–2747. doi:10.1021/ja513027s
Return to citation in text: [1] [2] -
Uchida, K.; Ishikawa, T.; Takeshita, M.; Irie, M. Tetrahedron 1998, 54, 6627–6638. doi:10.1016/s0040-4020(98)00330-5
Return to citation in text: [1] [2] -
Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074
Return to citation in text: [1] [2] [3] [4] -
Nakashima, T.; Goto, M.; Kawai, S.; Kawai, T. J. Am. Chem. Soc. 2008, 130, 14570–14575. doi:10.1021/ja802986y
Return to citation in text: [1] [2] [3] [4] -
Cyrañski, M. K.; Krygowski, T. M.; Katritzky, A. R.; Schleyer, P. v. R. J. Org. Chem. 2002, 67, 1333–1338. doi:10.1021/jo016255s
Return to citation in text: [1] [2] -
Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899–926. doi:10.1021/cr00088a005
Return to citation in text: [1] [2]
| 40. | Nakashima, T.; Goto, M.; Kawai, S.; Kawai, T. J. Am. Chem. Soc. 2008, 130, 14570–14575. doi:10.1021/ja802986y |
| 37. | Herder, M.; Schmidt, B. M.; Grubert, L.; Pätzel, M.; Schwarz, J.; Hecht, S. J. Am. Chem. Soc. 2015, 137, 2738–2747. doi:10.1021/ja513027s |
| 38. | Uchida, K.; Ishikawa, T.; Takeshita, M.; Irie, M. Tetrahedron 1998, 54, 6627–6638. doi:10.1016/s0040-4020(98)00330-5 |
| 1. | Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Chem. Rev. 2014, 114, 12174–12277. doi:10.1021/cr500249p |
| 2. | Tian, H.; Yang, S. Chem. Soc. Rev. 2004, 33, 85–97. doi:10.1039/b302356g |
| 3. | Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d |
| 7. | Gilat, S. L.; Kawai, S. H.; Lehn, J.-M. Chem. – Eur. J. 1995, 1, 275–284. doi:10.1002/chem.19950010504 |
| 15. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 16. | Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128–1137. doi:10.1016/s1359-6446(03)02933-7 |
| 41. | Cyrañski, M. K.; Krygowski, T. M.; Katritzky, A. R.; Schleyer, P. v. R. J. Org. Chem. 2002, 67, 1333–1338. doi:10.1021/jo016255s |
| 6. | Kobatake, S.; Uchida, K.; Tsuchida, E.; Irie, M. Chem. Lett. 2000, 29, 1340–1341. doi:10.1246/cl.2000.1340 |
| 15. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::aid-anie2596>3.0.co;2-4 |
| 17. | Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457–460. doi:10.1126/science.1229506 |
| 39. | Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074 |
| 5. | Nakamura, S.; Irie, M. J. Org. Chem. 1988, 53, 6136–6138. doi:10.1021/jo00261a035 |
| 13. | Kochi, J.; Ubukata, T.; Yokoyama, Y. J. Org. Chem. 2018, 83, 10695–10700. doi:10.1021/acs.joc.8b01341 |
| 39. | Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074 |
| 4. | Kitagawa, D.; Sasaki, K.; Kobatake, S. Bull. Chem. Soc. Jpn. 2011, 84, 141–147. doi:10.1246/bcsj.20100274 |
| 14. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::aid-anie2004>3.3.co;2-x |
| 40. | Nakashima, T.; Goto, M.; Kawai, S.; Kawai, T. J. Am. Chem. Soc. 2008, 130, 14570–14575. doi:10.1021/ja802986y |
| 10. | Krayushkin, M. M.; Ivanov, S. N.; Martynkin, A. Y.; Lichitsky, B. V.; Dudinov, A. A.; Uzhinov, B. M. Russ. Chem. Bull. 2001, 50, 116–121. doi:10.1023/a:1009541605405 |
| 12. | Kawai, S.; Nakashima, T.; Atsumi, K.; Sakai, T.; Harigai, M.; Imamoto, Y.; Kamikubo, H.; Kataoka, M.; Kawai, T. Chem. Mater. 2007, 19, 3479–3483. doi:10.1021/cm0630340 |
| 10. | Krayushkin, M. M.; Ivanov, S. N.; Martynkin, A. Y.; Lichitsky, B. V.; Dudinov, A. A.; Uzhinov, B. M. Russ. Chem. Bull. 2001, 50, 116–121. doi:10.1023/a:1009541605405 |
| 35. | DFT and TD DFT calculations were carried out with Spartan’18 (Wavefunction) at the B3LYP/6-31G* level. |
| 10. | Krayushkin, M. M.; Ivanov, S. N.; Martynkin, A. Y.; Lichitsky, B. V.; Dudinov, A. A.; Uzhinov, B. M. Russ. Chem. Bull. 2001, 50, 116–121. doi:10.1023/a:1009541605405 |
| 11. | Kawai, T.; Iseda, T.; Irie, M. Chem. Commun. 2004, 72–73. doi:10.1039/b311334e |
| 11. | Kawai, T.; Iseda, T.; Irie, M. Chem. Commun. 2004, 72–73. doi:10.1039/b311334e |
| 41. | Cyrañski, M. K.; Krygowski, T. M.; Katritzky, A. R.; Schleyer, P. v. R. J. Org. Chem. 2002, 67, 1333–1338. doi:10.1021/jo016255s |
| 9. | Jurissek, C.; Berger, F.; Eisenreich, F.; Kathan, M.; Hecht, S. Angew. Chem., Int. Ed. 2019, 58, 1945–1949. doi:10.1002/anie.201812284 |
| 39. | Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074 |
| 11. | Kawai, T.; Iseda, T.; Irie, M. Chem. Commun. 2004, 72–73. doi:10.1039/b311334e |
| 40. | Nakashima, T.; Goto, M.; Kawai, S.; Kawai, T. J. Am. Chem. Soc. 2008, 130, 14570–14575. doi:10.1021/ja802986y |
| 32. | Morinaka, K.; Ubukata, T.; Yokoyama, Y. Org. Lett. 2009, 11, 3890–3893. doi:10.1021/ol901497t |
| 33. | Ogawa, H.; Takagi, K.; Ubukata, T.; Okamoto, A.; Yonezawa, N.; Delbaere, S.; Yokoyama, Y. Chem. Commun. 2012, 48, 11838–11840. doi:10.1039/c2cc35793c |
| 34. | Nakagawa, T.; Miyasaka, Y.; Yokoyama, Y. Chem. Commun. 2018, 54, 3207–3210. doi:10.1039/c8cc00566d |
| 18. | Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s |
| 19. | Boren, B. C.; Narayan, S.; Rasmussen, L. K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V. V. J. Am. Chem. Soc. 2008, 130, 8923–8930. doi:10.1021/ja0749993 |
| 40. | Nakashima, T.; Goto, M.; Kawai, S.; Kawai, T. J. Am. Chem. Soc. 2008, 130, 14570–14575. doi:10.1021/ja802986y |
| 20. | Finden, J.; Kunz, T. K.; Branda, N. R.; Wolf, M. O. Adv. Mater. (Weinheim, Ger.) 2008, 20, 1998–2002. doi:10.1002/adma.200702455 |
| 21. | Ouhenia-Ouadahi, K.; Métivier, R.; Maisonneuve, S.; Jacquart, A.; Xie, J.; Léaustic, A.; Yu, P.; Nakatani, K. Photochem. Photobiol. Sci. 2012, 11, 1705–1714. doi:10.1039/c2pp25129a |
| 22. | Ding, H.; Liu, G.; Pu, S.; Zheng, C. Dyes Pigm. 2014, 103, 82–88. doi:10.1016/j.dyepig.2013.11.022 |
| 23. | Pu, S.; Ding, H.; Liu, G.; Zheng, C.; Xu, H. J. Phys. Chem. C 2014, 118, 7010–7017. doi:10.1021/jp5001495 |
| 24. | Maisonneuve, S.; Métivier, R.; Yu, P.; Nakatani, K.; Xie, J. Beilstein J. Org. Chem. 2014, 10, 1471–1481. doi:10.3762/bjoc.10.151 |
| 25. | Ma, L.; Liu, G.; Pu, S.; Ding, H.; Li, G. Tetrahedron 2016, 72, 985–991. doi:10.1016/j.tet.2015.12.068 |
| 26. | Duan, F.; Liu, G.; Fan, C.; Pu, S. Tetrahedron Lett. 2016, 57, 1963–1966. doi:10.1016/j.tetlet.2016.03.079 |
| 27. | Xue, D.; Zheng, C.; Qu, S.; Liao, G.; Fan, C.; Liu, G.; Pu, S. Luminescence 2017, 32, 652–660. doi:10.1002/bio.3234 |
| 28. | Singh, R.; Wu, H.-Y.; Kumar Dwivedi, A.; Singh, A.; Lin, C.-M.; Raghunath, P.; Lin, M.-C.; Wu, T.-K.; Wei, K.-H.; Lin, H.-C. J. Mater. Chem. C 2017, 5, 9952–9962. doi:10.1039/c7tc03071a |
| 29. | Wang, N.; Wang, R.; Tu, Y.; Pu, S.; Liu, G. Spectrochim. Acta, Part A 2018, 196, 303–310. doi:10.1016/j.saa.2018.02.018 |
| 30. | Guo, S.; Liu, G.; Fan, C.; Pu, S. RSC Adv. 2018, 8, 22786–22798. doi:10.1039/c8ra03443e |
| 31. | Kang, H.; Fan, C.; Xu, H.; Liu, G.; Pu, S. Tetrahedron 2018, 74, 4390–4399. doi:10.1016/j.tet.2018.07.002 |
| 42. | Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899–926. doi:10.1021/cr00088a005 |
| 42. | Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899–926. doi:10.1021/cr00088a005 |
| 38. | Uchida, K.; Ishikawa, T.; Takeshita, M.; Irie, M. Tetrahedron 1998, 54, 6627–6638. doi:10.1016/s0040-4020(98)00330-5 |
| 39. | Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074 |
| 36. | Irie, M.; Sakemura, K.; Okinaka, M.; Uchida, K. J. Org. Chem. 1995, 60, 8305–8309. doi:10.1021/jo00130a035 |
| 37. | Herder, M.; Schmidt, B. M.; Grubert, L.; Pätzel, M.; Schwarz, J.; Hecht, S. J. Am. Chem. Soc. 2015, 137, 2738–2747. doi:10.1021/ja513027s |
| 35. | DFT and TD DFT calculations were carried out with Spartan’18 (Wavefunction) at the B3LYP/6-31G* level. |
| 36. | Irie, M.; Sakemura, K.; Okinaka, M.; Uchida, K. J. Org. Chem. 1995, 60, 8305–8309. doi:10.1021/jo00130a035 |
| 19. | Boren, B. C.; Narayan, S.; Rasmussen, L. K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V. V. J. Am. Chem. Soc. 2008, 130, 8923–8930. doi:10.1021/ja0749993 |
| 35. | DFT and TD DFT calculations were carried out with Spartan’18 (Wavefunction) at the B3LYP/6-31G* level. |
© 2019 Zhang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)