Abstract
The acremines are a family of meroterpenoids isolated from fungi of the genus Acremonium. Here, we present the asymmetric total synthesis of acremine F which hinges on a modestly enantioselective dihydroxylation and a subsequent kinetic resolution via a highly selective asymmetric reduction. Chemoselective oxidation of acremine F gave access to acremines A and B. The dimerization of acremine F to bisacremine E was investigated but could not be achieved, shedding light on the formation of the acremine dimers in nature.

Graphical Abstract
Introduction
Endophytic fungi grow in a symbiotic relationship with their plant hosts [1], which is mediated by secondary metabolites [2]. In 2005, Torta and co-workers reported the isolation of six meroterpenoid natural products, acremines A–F from A20, a strain of Acreonium byssoides, isolated from grapevine leaves that were artificially inoculated with Plasmopora viticola (Figure 1) [3]. This class of natural products is characterized by a highly substituted cyclohexene core, featuring up to three stereogenic carbons, which is linked to a prenyl unit. Nature achieves further diversification by several modes of oxidation and cyclization. While acremine F (5) exhibited no significant bioactivity, acremines A–D showed inhibition of P. viticola sporangia germination.
![[1860-5397-15-219-1]](/bjoc/content/figures/1860-5397-15-219-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected members of the acremine family [3-5].
Figure 1: Selected members of the acremine family [3-5].
In 2015, Wei and co-workers discovered bisacremines E–G, the most complex members of the acremine family, from the soil-derived strain A. persicinum SC0105 [4]. These natural products are presumed to be derived from two acremine F (5) units by a formal [4 + 2] cycloaddition followed by condensation and oxidation.
Given the diversity and structural beauty of this class of natural products, it is not surprising that the acremine family has attracted the attention of the synthetic community [6-8]. Nevertheless, to the best of our knowledge, no asymmetric entry to this class of natural products has been described.
Our retrosynthetic analysis of 5 is depicted in Scheme 1. The prenyl side chain would be introduced by transition metal-catalyzed cross coupling of vinyl iodide 9. Compound 9 in turn could be traced back to silyl enol ether 10. Ent-10 was first reported by Herzon and co-workers [9] and is derived from phenol silyl ether 11 via Birch reduction and dihydroxylation.
![[1860-5397-15-219-i1]](/bjoc/content/inline/1860-5397-15-219-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of acremine F (5).
Scheme 1: Retrosynthetic analysis of acremine F (5).
Results and Discussion
Our synthesis started with meta-cresol (12) which was protected as a TIPS ether and then subjected to Birch reduction conditions to afford cyclohexa-1,4-diene 13 [9]. Enantioselective Sharpless dihydroxylation proceeded in good chemoselectivity but with modest yield and optical purity (25% ee). Unfortunately, all attempts to improve the enantioselectivity of this reaction failed. We discovered, however, that at a later stage the optical purity could be improved (see below). Diol protection gave dioxolane 14, which underwent Saegusa oxidation to afford enone 15. Subsequent α-iodination gave access to α-iodoenone 16, which could be stereoselectively reduced under Corey–Itsuno conditions to yield allylic alcohol 17. The use of a chiral oxazaborolidine catalyst led to kinetic resolution and increased the optical purity of 17 to 95% ee. Deprotection of the diol moiety followed by Stille cross coupling with vinyl stannane 18 finally gave 5 in excellent yield (Scheme 2).
![[1860-5397-15-219-i2]](/bjoc/content/inline/1860-5397-15-219-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Total synthesis of acremine F (5).
Scheme 2: Total synthesis of acremine F (5).
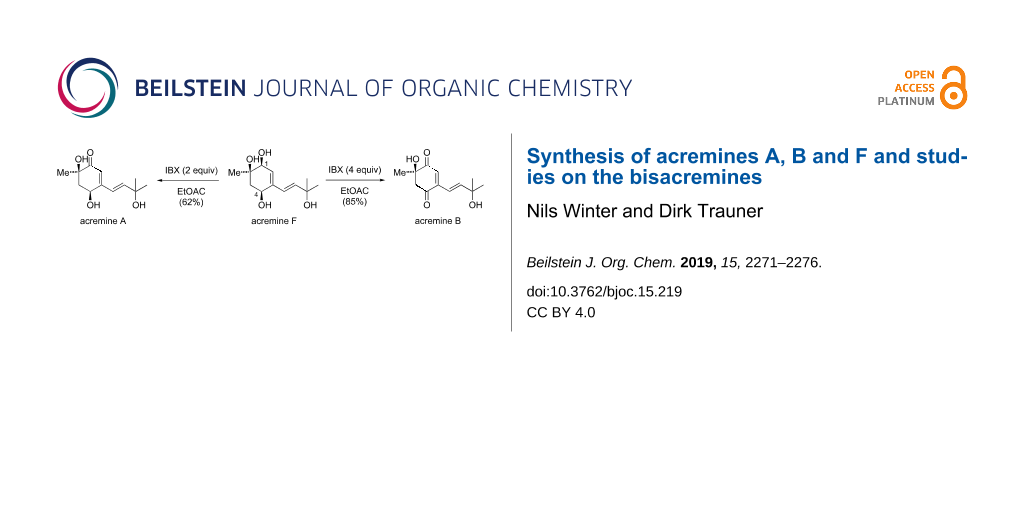
Since acremine F (5) can be expected to be the biogenetic precursor of acremines A (1) and B (2), we wanted to access these antifungal derivatives through selective oxidations. Indeed, treatment of 5 with IBX preferentially oxidized the C1-allylic alcohol, giving 1 in respectable yield. Prolonged treatment (9 h) of 5 with a large excess of IBX oxidized both secondary allylic alcohols and afforded 2 in good overall yield (Scheme 3).
![[1860-5397-15-219-i3]](/bjoc/content/inline/1860-5397-15-219-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of acremines A and B through selective oxidation of acremine F.
Scheme 3: Synthesis of acremines A and B through selective oxidation of acremine F.
Bisacremine E (7) was proposed to be formed in nature via [4 + 2] cycloaddition involving two acremine F (5) units [4]. Although dimerization of 5 through a Diels–Alder cycloaddition is not electronically favorable, we speculated that this reaction might proceed through ionic intermediates (Scheme 4).
![[1860-5397-15-219-i4]](/bjoc/content/inline/1860-5397-15-219-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed biomimetic dimerization of 5.
Scheme 4: Proposed biomimetic dimerization of 5.
To probe this hypothesis, we tried to generate allylic cation 19 by treatment with different acids or under thermal conditions (Table 1). Unfortunately, all conditions led to either decomposition of the starting material or elimination of the tertiary allylic alcohol to the unstable triene 6. As we were able to observe elimination as well as acetate incorporation into the molecule the desired cation was clearly formed under a variety of conditions. Nevertheless, none of these could affect the desired cyclization. Attempts to enhance the stability of the molecule, and therefore prevent decomposition, by protection of the non-participating alcohols and attempts to generate the allylic cation from a cyclic ketal [10-12], aiming for a Gassman-type reaction mechanism, were also unfruitful.
Table 1: Representative screening conditions for the biomimetic cascade.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-219-i7.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent | additive | temperature | observation |
| 1 | H2O | none | 85 °C | 6 |
| 2 | H2O | none | 100 °C | decomposition |
| 3 | PhMe | none | 150 °C | 6 |
| 4 | oDCB | none | 160 °C | decomposition |
| 5 | Et2O | 4 M LiClO4 | rt | 6 |
| 6 | neat | none | 45 °C | 6 |
| 7 | neat | none | 110 °C | decomposition |
| 8 | MeCN | 12 kbar | rt | starting material |
| 9 | MeCN | AcOH, 12 kbar | 60 °C | decomposition |
| 10 | MeCN | CSA | −40 °C to rt | 6 |
| 11 | neat | CSA | rt | decomposition |
| 12 | MeCN | AcOH | 85 °C | 6 + acetate trapping |
| 13 | MeCN | HCOOH | 90 °C | 6 |
| 14 | H2O | H2SO4 | 85 °C | decomposition |
| 15 | DMF | Mes-Acr-Ph, lighta | rt | decomposition |
| 16 | PhMe | Ni(cod)2, PPh3 | 80 °C | starting material |
| 17 | MeCN | DCB, lightb | rt | 22 |
| 18 | acetone | lightb | rt | 22 |
aBlue LED, bmedium pressure Hg lamp.
While radical cations are known to undergo Diels–Alder reactions with electron-rich dienophiles [13-17], treatment of 5 with Fukuzumi’s catalyst [18] under illumination with blue light only led to decomposition of the starting material (Table 1, entry 15). Notably, a photoredox catalyst with a lower oxidation potential could not affect any reaction. Next, we tried to enhance the possibility for productive reactivity by tethering two units of 5 and therefore executed the reaction in an intramolecular way (Scheme 5). Unfortunately, treatment of 23 with various redox catalysts led either to decomposition or recovery of the starting material.
![[1860-5397-15-219-i5]](/bjoc/content/inline/1860-5397-15-219-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Attempted intramolecular cyclization of 23.
Scheme 5: Attempted intramolecular cyclization of 23.
Next, we tried to trigger the cyclization through a [2 + 2] cycloaddition followed by vinyl cyclobutane rearrangement [19,20]. We reasoned that the initially formed divinyl cyclobutane [21] should undergo an allylic rearrangement to furnish the decalin system [22,23]. Condensation should then close the tetrahydrofuran ring of the natural product. Upon irradiation of 5 using a Hg lamp, however, the only productive pathway which could be observed was isomerization of the disubstituted double bond (Table 1, entries 17 and 18). Again, we attempted to promote the reaction by tethering two acremine units through a dioxysilane (Scheme 6) [24-26]. Unfortunately, irradiation of 25 with and without the presence of different photosensitizers only led to decomposition.
![[1860-5397-15-219-i6]](/bjoc/content/inline/1860-5397-15-219-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Attempted photochemical cyclization of 25.
Scheme 6: Attempted photochemical cyclization of 25.
Conclusion
In conclusion, we report the first asymmetric synthesis of acremine F (5) relying on the combination of a modestly enantioselective oxidation and a highly enantioselective reduction with kinetic resolution to access the acremine framework. The route proved to be scalable and delivered 300 mg of the natural product. Acremine F could further be converted into acremines A (1) and B (2) by a selective oxidation providing a versatile entry into this class of natural products. Furthermore, we investigated the proposed biomimetic dimerization of 5 to bisacremine E. Since these extensive studies were unsuccessful and no dimers could be observed under a variety of biomimetic conditions, it appears that this transformation requires enzymatic catalysis in nature.
Supporting Information
Experimental procedures, spectroscopic data and copies of NMR spectra (PDF) as well as crystallographic data of compounds 16 and 17. CIF files for 16 (CCDC 1854563) and 17 (CCDC 1854564) are available free from charge on https://www.ccdc.cam.ac.uk/structures/).
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 6.0 MB | Download |
References
-
Wilson, D. Oikos 1995, 73, 274–276. doi:10.2307/3545919
Return to citation in text: [1] -
Arnone, A.; Nasini, G.; Panzeri, W.; de Pava, O. V.; Malpezzi, L. J. Nat. Prod. 2008, 71, 146–149. doi:10.1021/np070413e
Return to citation in text: [1] -
Assante, G.; Dallavalle, S.; Malpezzi, L.; Nasini, G.; Burruano, S.; Torta, L. Tetrahedron 2005, 61, 7686–7692. doi:10.1016/j.tet.2005.05.094
Return to citation in text: [1] [2] -
Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Org. Lett. 2015, 17, 4922–4925. doi:10.1021/acs.orglett.5b02536
Return to citation in text: [1] [2] [3] -
Wu, P.; Yao, L.; Xu, L.; Xue, J.; Wei, X. J. Nat. Prod. 2015, 78, 2161–2166. doi:10.1021/np501037x
Return to citation in text: [1] -
Mehta, G.; Sunil Kumar, Y. C.; Khan, T. B. Tetrahedron Lett. 2010, 51, 5112–5115. doi:10.1016/j.tetlet.2010.07.110
Return to citation in text: [1] -
Arkoudis, E.; Lykakis, I. N.; Gryparis, C.; Stratakis, M. Org. Lett. 2009, 11, 2988–2991. doi:10.1021/ol901004e
Return to citation in text: [1] -
Mehta, G.; Khan, T. B.; Sunil Kumar, Y. C. Tetrahedron Lett. 2010, 51, 5116–5119. doi:10.1016/j.tetlet.2010.07.109
Return to citation in text: [1] -
Woo, C. M.; Lu, L.; Gholap, S. L.; Smith, D. R.; Herzon, S. B. J. Am. Chem. Soc. 2010, 132, 2540–2541. doi:10.1021/ja910769j
Return to citation in text: [1] [2] -
Gassman, P. G.; Singleton, D. A.; Wilwerding, J. J.; Chavan, S. P. J. Am. Chem. Soc. 1987, 109, 2182–2184. doi:10.1021/ja00241a047
Return to citation in text: [1] -
Gassman, P. G.; Lottes, A. C. Tetrahedron Lett. 1992, 33, 157–160. doi:10.1016/0040-4039(92)88038-7
Return to citation in text: [1] -
Sammakia, T.; Berliner, M. A. J. Org. Chem. 1994, 59, 6890–6891. doi:10.1021/jo00102a006
Return to citation in text: [1] -
Pabon, R. A.; Bellville, D. J.; Bauld, N. L. J. Am. Chem. Soc. 1983, 105, 5158–5159. doi:10.1021/ja00353a065
Return to citation in text: [1] -
Mlcoch, J.; Steckhan, E. Angew. Chem., Int. Ed. Engl. 1985, 24, 412–414. doi:10.1002/anie.198504121
Return to citation in text: [1] -
Nicewicz, D. A.; Nguyen, T. M. ACS Catal. 2014, 4, 355–360. doi:10.1021/cs400956a
Return to citation in text: [1] -
Bellville, D. J.; Bauld, N. L.; Pabon, R.; Gardner, S. A. J. Am. Chem. Soc. 1983, 105, 3584–3588. doi:10.1021/ja00349a038
Return to citation in text: [1] -
Drew, S. L.; Lawrence, A. L.; Sherburn, M. S. Angew. Chem., Int. Ed. 2013, 52, 4221–4224. doi:10.1002/anie.201210084
Return to citation in text: [1] -
Fukuzumi, S.; Kotani, H.; Ohkubo, K.; Ogo, S.; Tkachenko, N. V.; Lemmetyinen, H. J. Am. Chem. Soc. 2004, 126, 1600–1601. doi:10.1021/ja038656q
Return to citation in text: [1] -
Grée, R.; Laabassi, M.; Mosset, P.; Carrié, R. Tetrahedron Lett. 1985, 26, 2317–2318. doi:10.1016/s0040-4039(00)95085-8
Return to citation in text: [1] -
Wender, P. A.; Correia, C. R. D. J. Am. Chem. Soc. 1987, 109, 2523–2525. doi:10.1021/ja00242a053
Return to citation in text: [1] -
Hammond, G. S.; Turro, N. J.; Liu, R. S. H. J. Org. Chem. 1963, 28, 3297–3303. doi:10.1021/jo01047a005
Return to citation in text: [1] -
Trecker, D. J.; Henry, J. P. J. Am. Chem. Soc. 1964, 86, 902–905. doi:10.1021/ja01059a032
Return to citation in text: [1] -
Hammond, G. S.; DeBoer, C. D. J. Am. Chem. Soc. 1964, 86, 899–902. doi:10.1021/ja01059a031
Return to citation in text: [1] -
Fleming, S. A.; Parent, A. A.; Parent, E. E.; Pincock, J. A.; Renault, L. J. Org. Chem. 2007, 72, 9464–9470. doi:10.1021/jo7014664
Return to citation in text: [1] -
Ward, S. C.; Fleming, S. A. J. Org. Chem. 1994, 59, 6476–6479. doi:10.1021/jo00100a063
Return to citation in text: [1] -
Fleming, S. A.; Ward, S. C. Tetrahedron Lett. 1992, 33, 1013–1016. doi:10.1016/s0040-4039(00)91847-1
Return to citation in text: [1]
| 4. | Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Org. Lett. 2015, 17, 4922–4925. doi:10.1021/acs.orglett.5b02536 |
| 22. | Trecker, D. J.; Henry, J. P. J. Am. Chem. Soc. 1964, 86, 902–905. doi:10.1021/ja01059a032 |
| 23. | Hammond, G. S.; DeBoer, C. D. J. Am. Chem. Soc. 1964, 86, 899–902. doi:10.1021/ja01059a031 |
| 3. | Assante, G.; Dallavalle, S.; Malpezzi, L.; Nasini, G.; Burruano, S.; Torta, L. Tetrahedron 2005, 61, 7686–7692. doi:10.1016/j.tet.2005.05.094 |
| 4. | Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Org. Lett. 2015, 17, 4922–4925. doi:10.1021/acs.orglett.5b02536 |
| 5. | Wu, P.; Yao, L.; Xu, L.; Xue, J.; Wei, X. J. Nat. Prod. 2015, 78, 2161–2166. doi:10.1021/np501037x |
| 24. | Fleming, S. A.; Parent, A. A.; Parent, E. E.; Pincock, J. A.; Renault, L. J. Org. Chem. 2007, 72, 9464–9470. doi:10.1021/jo7014664 |
| 25. | Ward, S. C.; Fleming, S. A. J. Org. Chem. 1994, 59, 6476–6479. doi:10.1021/jo00100a063 |
| 26. | Fleming, S. A.; Ward, S. C. Tetrahedron Lett. 1992, 33, 1013–1016. doi:10.1016/s0040-4039(00)91847-1 |
| 3. | Assante, G.; Dallavalle, S.; Malpezzi, L.; Nasini, G.; Burruano, S.; Torta, L. Tetrahedron 2005, 61, 7686–7692. doi:10.1016/j.tet.2005.05.094 |
| 19. | Grée, R.; Laabassi, M.; Mosset, P.; Carrié, R. Tetrahedron Lett. 1985, 26, 2317–2318. doi:10.1016/s0040-4039(00)95085-8 |
| 20. | Wender, P. A.; Correia, C. R. D. J. Am. Chem. Soc. 1987, 109, 2523–2525. doi:10.1021/ja00242a053 |
| 2. | Arnone, A.; Nasini, G.; Panzeri, W.; de Pava, O. V.; Malpezzi, L. J. Nat. Prod. 2008, 71, 146–149. doi:10.1021/np070413e |
| 21. | Hammond, G. S.; Turro, N. J.; Liu, R. S. H. J. Org. Chem. 1963, 28, 3297–3303. doi:10.1021/jo01047a005 |
| 4. | Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Org. Lett. 2015, 17, 4922–4925. doi:10.1021/acs.orglett.5b02536 |
| 13. | Pabon, R. A.; Bellville, D. J.; Bauld, N. L. J. Am. Chem. Soc. 1983, 105, 5158–5159. doi:10.1021/ja00353a065 |
| 14. | Mlcoch, J.; Steckhan, E. Angew. Chem., Int. Ed. Engl. 1985, 24, 412–414. doi:10.1002/anie.198504121 |
| 15. | Nicewicz, D. A.; Nguyen, T. M. ACS Catal. 2014, 4, 355–360. doi:10.1021/cs400956a |
| 16. | Bellville, D. J.; Bauld, N. L.; Pabon, R.; Gardner, S. A. J. Am. Chem. Soc. 1983, 105, 3584–3588. doi:10.1021/ja00349a038 |
| 17. | Drew, S. L.; Lawrence, A. L.; Sherburn, M. S. Angew. Chem., Int. Ed. 2013, 52, 4221–4224. doi:10.1002/anie.201210084 |
| 9. | Woo, C. M.; Lu, L.; Gholap, S. L.; Smith, D. R.; Herzon, S. B. J. Am. Chem. Soc. 2010, 132, 2540–2541. doi:10.1021/ja910769j |
| 18. | Fukuzumi, S.; Kotani, H.; Ohkubo, K.; Ogo, S.; Tkachenko, N. V.; Lemmetyinen, H. J. Am. Chem. Soc. 2004, 126, 1600–1601. doi:10.1021/ja038656q |
| 9. | Woo, C. M.; Lu, L.; Gholap, S. L.; Smith, D. R.; Herzon, S. B. J. Am. Chem. Soc. 2010, 132, 2540–2541. doi:10.1021/ja910769j |
| 6. | Mehta, G.; Sunil Kumar, Y. C.; Khan, T. B. Tetrahedron Lett. 2010, 51, 5112–5115. doi:10.1016/j.tetlet.2010.07.110 |
| 7. | Arkoudis, E.; Lykakis, I. N.; Gryparis, C.; Stratakis, M. Org. Lett. 2009, 11, 2988–2991. doi:10.1021/ol901004e |
| 8. | Mehta, G.; Khan, T. B.; Sunil Kumar, Y. C. Tetrahedron Lett. 2010, 51, 5116–5119. doi:10.1016/j.tetlet.2010.07.109 |
| 10. | Gassman, P. G.; Singleton, D. A.; Wilwerding, J. J.; Chavan, S. P. J. Am. Chem. Soc. 1987, 109, 2182–2184. doi:10.1021/ja00241a047 |
| 11. | Gassman, P. G.; Lottes, A. C. Tetrahedron Lett. 1992, 33, 157–160. doi:10.1016/0040-4039(92)88038-7 |
| 12. | Sammakia, T.; Berliner, M. A. J. Org. Chem. 1994, 59, 6890–6891. doi:10.1021/jo00102a006 |
© 2019 Winter and Trauner; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)