Abstract
The bis-ortho-thioether 9,10-bis[(o-methylthio)phenyl]anthracene was synthesized as a syn-atropisomer, as revealed by X-ray diffraction. This alkylaryl thioether ligand (L) formed different macrocyclic complexes by coordination with silver(I) salts depending on the nature of the anion: M2L2 for AgOTf and AgOTFA, M6L4 for AgNO3. A discrete M2L complex was obtained in the presence of bulky PPh3AgOTf. These silver(I) complexes adopted similar structures in solution and in the solid state. As each sulfur atom in the ligand is prochiral, macrocycles L2M2 were obtained as mixtures of diastereoisomers, depending on the configurations of the sulfur atoms coordinated to silver cations. The X-ray structures of the two L2·(AgOTf)2 stereoisomers highlighted their different geometry. The catalytic activity of all silver(I) complexes was effective under homogeneous conditions in two tandem addition/cycloisomerization of alkynes using 0.5–1 mol % of catalytic loading.
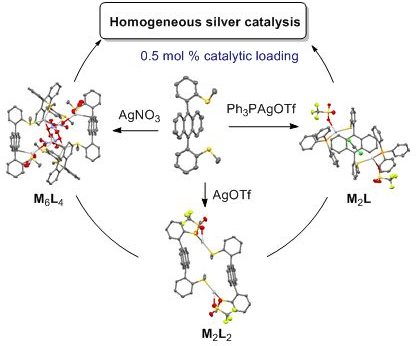
Graphical Abstract
Introduction
Since the early advances in the late eighties [1-10], silver(I) catalysis has been widely exploited based on the versatile redox and soft Lewis acid properties of this coinage metal cation. Silver catalysis has proved its effectiveness for numerous transformations involving unsaturated bond activation (allene, alkyne, alkene) [11-17], radical-based reactions [18-20] and several applications in asymmetric reactions [21,22].
This successful chemistry was usually conducted in the presence of commercially available inexpensive salts (AgOTf, AgNO3) and eventually a (chiral) ligand. Such silver(I) complexes were prepared by using (bi)pyridine [19,23-26], phosphine [22,27-30], ditopic N/P [22] ligands and a few S/P and S/N ones [31-34]. None of the silver(I) catalysts based on sulfur ligands were reported so far, although alkyl thioethers are soft σ-donor ligands such as crown thioethers that were largely developed as macrocyclic ligands for silver(I) [35-43]. Interestingly, depending on their design, these known silver(I) complexes can be discrete species [35-38], coordination driven supramolecules [35,42] or coordination polymers [39,40,42]. To the best of our knowledge, silver crown thioether complexes were never reported as catalysts.
Recently, we described a flexible bis-dialkyl thioether ligand for gold(III) chloride whose photoreduction to gold(I) was fast and controlled [44,45]. These gold complexes at different oxidation states showed efficient catalytic properties, that were highlighted in a one-pot cascade synthesis of 4H-benzoquinolizin-4-one. Now, we propose to investigate a rigid bis-alkylaryl thioether ligand to control the directionality and spatial orientation of its coordination to silver(I). Besides, 9,10-diphenylanthracenes (DPA) with ortho-substituted 9- and 10-aryl groups can exist as syn- or anti-atropisomers whose rotational barrier [46,47] ranges from 21 (ortho-H) to 25–29 kcal/mol (ortho-CH3). A few ortho-substituted DPA atropisomers have been described as potent molecular switches [48,49], building-blocks for self-assembled capsules [50,51] or optical materials [52-54]. However, due to their partially frozen structure, syn-isomers of DPA are ideal candidates for directional metal coordination. Herein, a new syn-atropisomer of 9,10-DPA ortho-substituted by two thioethers is exploited as a ligand for silver(I) salts. The impact of this bis-thioether ligand on silver(I) homogeneous catalysis is evaluated in two tandem addition/cycloisomerization reactions of alkynes.
Results and Discussion
Synthesis of silver(I) complexes
Ligand 1 was synthesized in one step, from commercially available 9,10-dibromoanthracene and 2-(methylthio)phenylboronic acid, using a Suzuki–Miyaura cross-coupling reaction. Notably, the yield was low (26%) [55], and the X-ray analysis of monocrystals revealed the syn-conformation of 1 (Scheme 1). A variable temperature 1H NMR (VT-NMR, Figures S10–12 in Supporting Information File 1) experiment conducted on syn-1 in the range of –30 to 110 °C in C2D2Cl4 showed the broadening of proton signals, due to the restricted rotation of the 9,10-aryl substituents in respect to the anthracene core, without any indication of a syn-to-anti isomerization.
![[1860-5397-15-239-i1]](/bjoc/content/inline/1860-5397-15-239-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of ligand 1, as its syn-atropisomer.
Scheme 1: Synthesis of ligand 1, as its syn-atropisomer.
Four silver(I) complexes 1a–d were prepared in excellent yield (77–92%) by mixing the ligand and the following silver salts in a 1:1 ratio in dichloromethane at room temperature: AgOTf for 1a, AgOTFA for 1b, AgNO3 for 1c and PPh3AgOTf for 1d.
Crystallographic structures of silver complexes
Single crystals of 1a, 1c and 1d suitable for X-ray diffraction analysis were grown from the slow diffusion of hexane into a solution of each complex in dichloromethane. Interestingly, each prochiral sulfur atom of the ligand becomes asymmetric by coordination to silver(I). In absence of any chiral source, the complexes were obtained either as nonchiral coordination products with a center or an axis of symmetry (1a and 1c, with (R,S)–1 ligands) or as a racemic mixture (1d, with (R,R)–1 or (S,S)–1 ligands).
The X-ray diffraction of monocrystals 1a revealed the formation of (R,S–1)2·(AgOTf)2 macrocycles driven by silver(I) coordination (Figure 1). The two ligands are facing through the coordination of one syn-thioether group to the same silver cation. Two different crystals were isolated and highlighted the two possible arrangements of the ligands that led to different diastereoisomeric macrocycles (Figure 1a,b). In Figure 1a, each silver cation was coordinated to two sulfur atoms with the same configuration (named head-to-head coordination mode for ligands) meanwhile in Figure 1b, each Ag(I) cation was coordinated to two sulfur atoms with (R)- and (S)-configuration, respectively (head-to-tail ligand coordination mode). These diastereoisomeric crystals presented a slightly different spatial arrangement.
![[1860-5397-15-239-1]](/bjoc/content/figures/1860-5397-15-239-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray structures of complex 1a, as two diastereoisomeric macrocycles (R,S-1)2·(AgOTf)2 with ligands assembled in: (a) a head-to-head fashion and, (b) a head-to-tail mode. Hydrogen atoms are omitted for clarity.
Figure 1: X-ray structures of complex 1a, as two diastereoisomeric macrocycles (R,S-1)2·(AgOTf)2 with ligands...
The head-to head macrocycle 1a had a parallelepiped shape (Figure 1a): the interplanar distance between two anthracenes was ca. 6.31 Å and the dihedral angle between the anthracene core and its 9,10-aryl substituents was 89° and 104°, respectively. The head-to-tail macrocycle 1a adopted a V-shape (Figure 1b): the angle between the planes of the two anthracenes was 73.2° and the dihedral angles between the aryl substituents and the anthracene plane ranged between 77° and 87°, respectively.
In solution, the two diastereoisomeric macrocycles coexisted, possibly in different ratios as the chemical shift of protons on the 9,10-phenyl and anthracene moieties were found slightly different for three different batches of silver complexes prepared by the same procedure (see Supporting Information File 1, Figure S9).
The complex formed with AgNO3 had a different stoichiometry, due to the multidentate nitrate anions. The X-ray structure of 1c appeared as a large (R,S-1)4·(AgNO3)6 complex with three nitrate anions as coordinating bridges between the two symmetric M3L2 units (Figure 2). The resulting coordinated macrocycle M6L4 had a globular shape that displayed the polyaromatic ligands towards the exterior and fully encapsulated two nitrate anions and the silver(I) cations. This silver nitrate complex was thus soluble in chlorinated solvents.
![[1860-5397-15-239-2]](/bjoc/content/figures/1860-5397-15-239-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structure of complex 1c, as a (R,S-1)4·(AgNO3)6 cage with three nitrate anions as coordinating bridges. Hydrogen atoms are omitted for clarity.
Figure 2: X-ray structure of complex 1c, as a (R,S-1)4·(AgNO3)6 cage with three nitrate anions as coordinatin...
In the presence of the bulky triphenylphosphine silver triflate salt, a monocoordination occurred between Ag(I) and each sulfur atom of ligand 1 leading to a discrete complex (syn-1)·(Ph3PAgOTf)2, as revealed by 1H NMR and X-ray (Figure 3). The steric hindrance also induced the access to a racemic mixture of (R,R)- and (S,S)-complexes where triphenylphosphine groups were located at the opposite sides of the anthracene core.
![[1860-5397-15-239-3]](/bjoc/content/figures/1860-5397-15-239-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray structure of complex 1d, as a racemic mixture of (R,R)- and (S,S)-(syn-1)·(PPh3AgOTf)2.
Figure 3: X-ray structure of complex 1d, as a racemic mixture of (R,R)- and (S,S)-(syn-1)·(PPh3AgOTf)2.
The crystallographic structures of 1a, 1c and 1d showed that both ligand syn-1 and the nature of silver anion impacted on the stoichiometry of coordination complexes. Atropisomer 1 directed the self-assembly towards the same half-space (regarding the anthracene core). The triflate anion lead to a [2 + 2] macrocycle meanwhile the more coordinating nitrate anion induced the formation of a large globular macrocycle M6L4. A bulkier silver salt favored a mono-coordination on each binding site.
Silver(I) complexes in solution
Surprisingly, several batches of each complex 1a–d were compared by 1H NMR (CDCl3, 2 mM) (see Supporting Information File 1, Figure S9) and revealed slight variations in the chemical shifts that might originate from the presence of several species in solution, such as self-aggregates, mixture of conformers, mixture of diastereoisomers or complexes with different stoichiometry.
To investigate the possible self-aggregation of complexes 1a–c (7 mM), VT NMR experiments were conducted between –30 °C and 60 °C in CDCl3. For complex 1a, the aromatic protons were shifted for 0.1 to 0.2 ppm meanwhile the methyl of the thioether group was shifted for 0.12 ppm (Figure 4). The two most affected aromatic protons were located on the 9,10-substituents, the one adjacent to SMe group (H1) and the one closest to the anthracene core (H4). Similar changes on VT NMR were observed for complex 1b and 1c (see Supporting Information File 1, Figures S11 and 12), with chemical shift variations in the range of 0.1 to 0.3 ppm for 1c. The little differences observed on 1H NMR spectra at 60 °C accounted for a slight change in the geometry of the complexes probably due to the thermal motion, and not to the dissociation of self-aggregates. In the temperature range of –30 to 60 °C, none dissociated free ligand was observed, thus confirming that coordinated macrocycles 1a–c were thermally stable. At room temperature, the slight different 1H NMR spectra may account for the existence of several conformations for these constrained coordinated architectures, which still have a certain degree of freedom, that is required for further catalytic properties.
![[1860-5397-15-239-4]](/bjoc/content/figures/1860-5397-15-239-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Variable temperature 1H NMR of complex 1a in CDCl3 (7 mM) from −30 °C to 60 °C.
Figure 4: Variable temperature 1H NMR of complex 1a in CDCl3 (7 mM) from −30 °C to 60 °C.
Diffusion-ordered spectroscopy (DOSY) 1H NMR was also used on complexes 1a–d (5 mM) in CDCl3 (Supporting Information File 1, Table S7). Each complex appeared as a unique species whose diffusion coefficient (D) can be fitted. So the presence of species with different coordination modes can be excluded. Unexpectedly, the diffusion constants of both complexes 1a and 1b were disparate (0.6–1.9 × 10−9 m2/s) depending on the batches meanwhile the D values of complexes 1c and 1d at 5 mM were similar (0.80–0.93 × 10−9 m2/s) and not depending on the batch. The nonreproducibility of DOSY experiments for 1a and 1b might originate from the uncontrolled formation of different diastereoisomers by coordination which had different geometries. Thereafter, the silver(I) complexes are discussed with the proposed stoichiometry from X-ray and NMR data, without accounting for stereochemistry: M2L2 for AgOTf and AgOTFA complexes (1a,b), M6L4 for AgNO3 one (1c) and M2L complex with PPh3AgOTf (1d).
Finally, the photophysical properties of ligand syn-1 (20 μM) and complexes 1a–d (30 μM) were evaluated in dichloromethane (Supporting Information File 1, Figures S4–S8). The UV–visible and fluorescence emission spectra (λexc = 345 nm) of ligand and complexes were similar and correspond to those of 9,10-diphenylanthracene [56,57]. The silver(I) coordination on the ligand and the formation of supramolecular systems does not seem to affect the spectroscopic properties of the 9,10-diphenylanthracene system.
Silver catalysis
As silver(I) salts exhibit a high alkynophilicity [11-17], the new complexes 1a–d were evaluated as homogeneous catalysts in two tandem addition/cycloisomerization reactions using alkynes 2 and 3.
2-Alkynylbenzaldehyde 2 [58,59] was chosen as the first model substrate for a cyclization reaction in the presence of methanol as a second nucleophile. This tandem addition/cycloisomerization was previously described in high yields (>95%) using 5 mol % catalyst loadings starting from 2-(alkynyl)quinoline-3-carbaldehyde [60,61] with AgOTf catalyst and starting from 2-alkynylbenzaldehyde derivatives [62] in the presence of a macrocyclic pyridine-tetraaza complex of Ag(I) as a catalyst. In our control experiment, alkyne 2 was converted into product 4 in high yield (88 %) using AgOTf at 5 mol % (Table 1, entry 1). Interestingly, 1-methoxyisochromene 4 can also be isolated in 89% yield, using a AgOTf catalytic loading of 1 mol % (Table 1, entry 2). All silver complexes 1a–d (1 mol %) efficiently catalyzed the intramolecular cyclization with 65–92% yields (Table 1, entries 3–7). To our delight, the transformation also reached 92% yield by employing 1a at 0.5 mol % (Table 1, entry 4). Compared to literature [60-62], catalyst 1a is effective for the tandem cyclization of 2-alkynylbenzaldehyde 2 at lower catalyst loadings and under milder conditions (20 °C, full conversion after 12 h). As previously observed for inorganic Ag salts [60], the catalyst efficiency for this cyclization slightly depends on its anion nature (1a > 1c > 1b > 1d).
Table 1: Addition/cycloisomerization of alkyne 2.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-239-i2.svg?max-width=637&scale=1.18182)
|
|||
|---|---|---|---|
| Entry | [Ag] catalyst | Loading (mol %) | Yieldb (%) |
| 1 | AgOTf | 5 | 88 |
| 2 | AgOTf | 1 | 89 |
| 3 | 1a | 1 | 92 |
| 4 | 1a | 0.5 | 92 |
| 5 | 1b | 1 | 73 |
| 6 | 1c | 1 | 85 |
| 7 | 1d | 1 | 65 |
aUnless specified, all the reactions were carried out in dry CH2Cl2 at room temperature for 12–16 h with alkyne 2 (30 mg, 0.15 mmol), MeOH (0.45 mmol) and a silver(I) catalyst (0.5–5 mol %). bIsolated yield.
To further demonstrate the catalytic properties of silver(I) complexes 1a–d, we investigated their performance in the cyclization [63] of alkynone 3 [63-65] in the presence of benzylamine, as a nucleophile (Table 2) that lead to substituted pyrrole 5. This tandem condensation/cycloisomerization was previously reported in 78% yield using AgOTf at 5 mol % (reaction time 3.5 h, 50 °C) [63]. Noteworthy, at 50 °C the transformation occurs in 35% yield without any catalyst (Table 2, entry 1). In our hands, using AgOTf (2.5 mol %), the product was obtained in 73% yield meanwhile 67–76% yield were reached when silver complexes 1a–d at 1 mol % were employed (Table 2, entries 2–6). Interestingly, a lower catalytic loading of 0.5 mol % allowed the isolation of 73% of pyrrole 5 in the presence of catalysts 1a and 1b (Table 2, entries 7 and 8). Under the same conditions, the catalytic efficiency of 1c and 1d was slightly lower (64–67 % yield) but similar to AgOTf at 2.5 mol % (Table 2, entries 9 and 10). For this second tandem model cyclization, the effect of the anion on the catalysts’ efficiency was weak. Finally, this second model addition/cycloisomerization was successfully catalyzed by silver complexes 1a–b also at lower loadings (0.5 mol %) than AgOTf.
Table 2: Condensation/cycloisomerization of alkyne 3.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-239-i3.svg?max-width=637&scale=1.18182)
|
|||
|---|---|---|---|
| Entry | [Ag] catalyst | Loading (mol %) | Yieldb (%) |
| 1 | – | – | 35 |
| 2 | AgOTf | 2.5 | 73 |
| 3 | 1a | 1 | 76 |
| 4 | 1b | 1 | 67 |
| 5 | 1c | 1 | 72 |
| 6 | 1d | 1 | 76 |
| 7 | 1a | 0.5 | 73 |
| 8 | 1b | 0.5 | 73 |
| 9 | 1c | 0.5 | 67 |
| 10 | 1d | 0.5 | 64 |
aUnless specified, all the reactions were performed at 50 °C under argon atmosphere in dry 1,2-dichloroethane for 12–16 h, in the presence of alkyne 3 (27 mg, 0.2 mmol), benzylamine (0.3 mmol) and a silver(I) catalyst (0.5–2.5 mol %). bIsolated yield.
Conclusion
9,10-Diphenylanthracene with two ortho-substituted thioether functional groups is an attractive scaffold which allowed us to design and prepare a stable syn-atropisomer ligand 1 which can direct coordinate towards the same half-space. Three macrocyclic silver complexes were then synthesized and their coordination modes revealed by X-ray diffraction were depended on the nature of the anion, i.e., M2L2 for –OTf/–OTFA and M6L4 for NO3–. With prochiral thioether groups, M2L2 macrocycles were obtained as mixtures of diastereoisomers due to the two possible arrangements of the coordinated ligands (head-to-head or head-to-tail) resulting into different spatial arrangements. In solution, the architectures of silver(I) complexes with ligand syn-1 seemed to be similar to the solid-state structures. The silver(I) complexes were evaluated as homogeneous catalysts in two tandem addition/cycloisomerization reactions on model alkynes to give the expected cyclization products in excellent yields, also by using 0.5 mol % catalytic loading, with efficiencies similar to those reported in the literature [60-63] with inorganic silver salts and complexes employed at higher loadings (2.5–5 mol %). The use of original and effective silver complexes might be a way to lower the catalytic loading in silver-catalyzed transformations and opens perspectives to the design of new asymmetric ligands.
Experimental
General procedure for the synthesis of silver(I) complexes 1a–d: To a solution of ligand 1 (15 mg, 0.0355 mmol) in anhydrous CH2Cl2 (1 mL) was added the corresponding silver salt (0.0355 mmol, 1.0 equiv) under argon atmosphere at room temperature. The mixture was stirred for 4 h. The clear solution was concentrated to ca. 0.3 mL and diethyl ether (2.0 mL) was slowly added to afford a precipitation. After filtration, the isolated solid was washed with diethyl ether and dried under vacuum.
General procedure for the acetalization/cycloisomerization of alkyne 2: An oven-dried Schlenk tube was charged with the silver(I) catalyst (0.5–5 mol %), then degassed and backfilled with argon for three times. A solution of alkyne 2 (30 mg, 0.15 mmol) in anhydrous CH2Cl2 (1 mL) and dry MeOH (18 µL, 0.45 mmol) were successively added. The mixture was stirred at room temperature for 12 h. The solution was concentrated and the crude residue was purified by column chromatography on silica gel (eluent: petrol ether/ethyl acetate 50:1) to obtain 1-methoxyisochromene 4.
General procedure for condensation/cycloisomerization of alkyne 3: A dry Schlenk tube charged with silver catalyst (0.5–2.5 mol %) was degassed and backfilled with argon for three times. A solution of alkyne 3 (27 mg, 0.2 mmol) in anhydrous CH2Cl2 (1 mL) and benzylamine (33 µL, 0.3 mmol) were successively added. The mixture was stirred at 50 °C for 12 h. The solution was concentrated under reduced pressure and the crude product was purified by column chromatography on silica gel (eluent: petrol ether/ethyl acetate 70:1) to obtain pyrrole 5.
Supporting Information
| Supporting Information File 1: Synthesis and characterization data of ligand and silver complexes, details of X-ray structures. | ||
| Format: PDF | Size: 3.5 MB | Download |
| Supporting Information File 2: Crystallographic structures CCDC 1883532, 1883535, 1883536, 1883538 and 1883674 of ligand 1 and silver complexes 1a–d (.cif files). | ||
| Format: ZIP | Size: 1.8 MB | Download |
Acknowledgements
Financial support was provided by the China Scholarship Council (Z.C. Ph.D. fellowship), the University of Bordeaux and Centre National de la Recherche Scientifique. The authors acknowledge Damien Jardel and Clotilde Davies for their contribution to the synthesis of the syn-ligand, Stéphane Massip for the resolution of the corresponding crystallographic structure.
References
-
Castellino, S.; Sims, J. J. Tetrahedron Lett. 1984, 25, 4059–4062. doi:10.1016/s0040-4039(01)90181-9
Return to citation in text: [1] -
Lathbury, D.; Gallagher, T. Tetrahedron Lett. 1985, 26, 6249–6252. doi:10.1016/s0040-4039(00)95065-2
Return to citation in text: [1] -
Grimaldi, J.; Cormons, A. Tetrahedron Lett. 1986, 27, 5089–5090. doi:10.1016/s0040-4039(00)85140-0
Return to citation in text: [1] -
Pale, P.; Chuche, J. Tetrahedron Lett. 1987, 28, 6447–6448. doi:10.1016/s0040-4039(00)96884-9
Return to citation in text: [1] -
Sawamura, M.; Hamashima, H.; Ito, Y. J. Org. Chem. 1990, 55, 5935–5936. doi:10.1021/jo00311a007
Return to citation in text: [1] -
Hayashi, T.; Uozumi, Y.; Yamazaki, A.; Sawamura, M.; Hamashima, H.; Ito, Y. Tetrahedron Lett. 1991, 32, 2799–2802. doi:10.1016/0040-4039(91)85090-r
Return to citation in text: [1] -
Kimura, M.; Fugami, K.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 1991, 32, 6359–6362. doi:10.1016/0040-4039(91)80169-7
Return to citation in text: [1] -
Marshall, J. A.; Wang, X. J. J. Org. Chem. 1991, 56, 960–969. doi:10.1021/jo00003a013
Return to citation in text: [1] -
Cooper, D. M.; Grigg, R.; Hargreaves, S.; Kennewell, P.; Redpath, J. Tetrahedron 1995, 51, 7791–7808. doi:10.1016/0040-4020(95)00397-q
Return to citation in text: [1] -
Yanagisawa, A.; Nakashima, H.; Ishiba, A.; Yamamoto, H. J. Am. Chem. Soc. 1996, 118, 4723–4724. doi:10.1021/ja9603315
Return to citation in text: [1] -
Halbes-Letinois, U.; Weibel, J.-M.; Pale, P. Chem. Soc. Rev. 2007, 36, 759–769. doi:10.1039/b602151b
Return to citation in text: [1] [2] -
Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Rev. 2008, 108, 3174–3198. doi:10.1021/cr078361l
Return to citation in text: [1] [2] -
Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075–6089. doi:10.1002/ejoc.200900790
Return to citation in text: [1] [2] -
Muñoz, M. P. Chem. Soc. Rev. 2014, 43, 3164–3184. doi:10.1039/c3cs60408j
Return to citation in text: [1] [2] -
Fang, G.; Bi, X. Chem. Soc. Rev. 2015, 44, 8124–8173. doi:10.1039/c5cs00027k
Return to citation in text: [1] [2] -
Blanc, A.; Bénéteau, V.; Weibel, J.-M.; Pale, P. Org. Biomol. Chem. 2016, 14, 9184–9205. doi:10.1039/c6ob01468b
Return to citation in text: [1] [2] -
Sekine, K.; Yamada, T. Chem. Soc. Rev. 2016, 45, 4524–4532. doi:10.1039/c5cs00895f
Return to citation in text: [1] [2] -
Zheng, Q.-Z.; Jiao, N. Chem. Soc. Rev. 2016, 45, 4590–4627. doi:10.1039/c6cs00107f
Return to citation in text: [1] -
Alderson, J. M.; Corbin, J. R.; Schomaker, J. M. Acc. Chem. Res. 2017, 50, 2147–2158. doi:10.1021/acs.accounts.7b00178
Return to citation in text: [1] [2] -
Fang, G.; Cong, X.; Zanoni, G.; Liu, Q.; Bi, X. Adv. Synth. Catal. 2017, 359, 1422–1502. doi:10.1002/adsc.201601179
Return to citation in text: [1] -
Naodovic, M.; Yamamoto, H. Chem. Rev. 2008, 108, 3132–3148. doi:10.1021/cr068413r
Return to citation in text: [1] -
Pellissier, H. Chem. Rev. 2016, 116, 14868–14917. doi:10.1021/acs.chemrev.6b00639
Return to citation in text: [1] [2] [3] -
Türkmen, Y. E.; Montavon, T. J.; Kozmin, S. A.; Rawal, V. H. J. Am. Chem. Soc. 2012, 134, 9062–9065. doi:10.1021/ja302537j
Return to citation in text: [1] -
Zeng, Y.; Zhang, L.; Zhao, Y.; Ni, C.; Zhao, J.; Hu, J. J. Am. Chem. Soc. 2013, 135, 2955–2958. doi:10.1021/ja312711c
Return to citation in text: [1] -
Zeng, Y.; Hu, J. Chem. – Eur. J. 2014, 20, 6866–6870. doi:10.1002/chem.201402846
Return to citation in text: [1] -
Tseberlidis, G.; Dell'Acqua, M.; Valcarenghi, D.; Gallo, E.; Rossi, E.; Abbiati, G.; Caselli, A. RSC Adv. 2016, 6, 97404–97419. doi:10.1039/c6ra22231e
Return to citation in text: [1] -
Ji, J.-X.; Au-Yeung, T. T.-L.; Wu, J.; Yip, C. W.; Chan, A. S. C. Adv. Synth. Catal. 2004, 346, 42–44. doi:10.1002/adsc.200303148
Return to citation in text: [1] -
Yao, X.; Li, C.-J. Org. Lett. 2005, 7, 4395–4398. doi:10.1021/ol051575+
Return to citation in text: [1] -
Takechi, S.; Yasuda, S.; Kumagai, N.; Shibasaki, M. Angew. Chem., Int. Ed. 2012, 51, 4218–4222. doi:10.1002/anie.201200520
Return to citation in text: [1] -
Su, Y.; Lu, M.; Dong, B.; Chen, H.; Shi, X. Adv. Synth. Catal. 2014, 356, 692–696. doi:10.1002/adsc.201300785
Return to citation in text: [1] -
Chen, Q.-A.; Zeng, W.; Wang, D.-W.; Zhou, Y.-G. Synlett 2009, 2236–2241. doi:10.1055/s-0029-1217805
Return to citation in text: [1] -
Iglesias-Sigüenza, J.; Ros, A.; Díez, E.; Magriz, A.; Vázquez, A.; Álvarez, E.; Fernández, R.; Lassaletta, J. M. Dalton Trans. 2009, 8485–8488. doi:10.1039/b910846g
Return to citation in text: [1] -
Prakash, O.; Joshi, H.; Kumar, U.; Sharma, A. K.; Singh, A. K. Dalton Trans. 2015, 44, 1962–1968. doi:10.1039/c4dt02813a
Return to citation in text: [1] -
Rasheed, O. K.; Bawn, C.; Davies, D.; Raftery, J.; Vitorica-Yrzebal, I.; Pritchard, R.; Zhou, H.; Quayle, P. Eur. J. Org. Chem. 2017, 5252–5261. doi:10.1002/ejoc.201701033
Return to citation in text: [1] -
Küppers, H.-J.; Wieghardt, K.; Tsay, Y.-H.; Krüger, C.; Nuber, B.; Weiss, J. Angew. Chem., Int. Ed. Engl. 1987, 26, 575–576. doi:10.1002/anie.198705751
Return to citation in text: [1] [2] [3] -
De Groot, B.; Jenkins, H. A.; Loeb, S. J. Inorg. Chem. 1992, 31, 203–208. doi:10.1021/ic00028a015
Return to citation in text: [1] [2] -
Blake, A. J.; Gould, R. O.; Parsons, S.; Radek, C.; Schröder, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 2374–2376. doi:10.1002/anie.199523741
Return to citation in text: [1] [2] -
Alberto, R.; Nef, W.; Smith, A.; Kaden, T. A.; Neuburger, M.; Zehnder, M.; Frey, A.; Abram, U.; Schubiger, P. A. Inorg. Chem. 1996, 35, 3420–3427. doi:10.1021/ic951421y
Return to citation in text: [1] [2] -
Blake, A. J.; Li, W.-S.; Lippolis, V.; Schröder, M. Chem. Commun. 1997, 1943–1944.
Return to citation in text: [1] [2] -
Alberto, R.; Angst, D.; Schubiger, A. P.; Abram, U.; Ortner, K.; Kaden, T. A. Chem. Commun. 1999, 1513–1514. doi:10.1039/a903455b
Return to citation in text: [1] [2] -
Blake, A. J.; Champness, N. R.; Howdle, S. M.; Webb, P. B. Inorg. Chem. 2000, 39, 1035–1038. doi:10.1021/ic991145d
Return to citation in text: [1] -
Casula, A.; Nairi, V.; Fernández-Moreira, V.; Laguna, A.; Lippolis, V.; Garau, A.; Gimeno, M. C. Dalton Trans. 2015, 44, 18506–18517. doi:10.1039/c5dt02723c
Return to citation in text: [1] [2] [3] -
Chang, Y.-P.; Levason, W.; Reid, G. Dalton Trans. 2016, 45, 18393–18416. doi:10.1039/c6dt03409h
Return to citation in text: [1] -
Mongin, C.; Pianet, I.; Jonusauskas, G.; Bassani, D. M.; Bibal, B. ACS Catal. 2015, 5, 380–387. doi:10.1021/cs5016063
Return to citation in text: [1] -
Cao, Z.; Bassani, D. M.; Bibal, B. Chem. – Eur. J. 2018, 24, 18779–18787. doi:10.1002/chem.201804322
Return to citation in text: [1] -
Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Muldoon, J.; McGlinchey, M. J. Org. Lett. 2011, 13, 256–259. doi:10.1021/ol102665y
Return to citation in text: [1] -
Marriott, P. J.; Lai, Y.-H. J. Chromatogr. A 1998, 447, 29–41. doi:10.1016/0021-9673(88)90004-0
Return to citation in text: [1] -
Zehm, D.; Fudickar, W.; Linker, T. Angew. Chem., Int. Ed. 2007, 46, 7689–7692. doi:10.1002/anie.200700443
Return to citation in text: [1] -
Zehm, D.; Fudickar, W.; Hans, M.; Schilde, U.; Kelling, A.; Linker, T. Chem. – Eur. J. 2008, 14, 11429–11441. doi:10.1002/chem.200801355
Return to citation in text: [1] -
Suzuki, A.; Kondo, K.; Akita, M.; Yoshizawa, M. Angew. Chem., Int. Ed. 2013, 52, 8120–8123. doi:10.1002/anie.201302789
Return to citation in text: [1] -
Yazaki, K.; Akita, M.; Prusty, S.; Chand, D. K.; Kikushi, T.; Sato, H.; Yoshizawa, M. Nat. Commun. 2017, 8, 15914–15921. doi:10.1038/ncomms15914
Return to citation in text: [1] -
Iida, A.; Yamaguchi, S. Chem. Commun. 2009, 3002–3004. doi:10.1039/b901794a
Return to citation in text: [1] -
Moon, H.; Jun, Y. W.; Kim, D.; Ryu, H. G.; Wang, T.; Kim, K. H.; Huh, Y.; Jung, J.; Ahn, K. H. Chem. – Asian J. 2016, 11, 2518–2523. doi:10.1002/asia.201600986
Return to citation in text: [1] -
Belyaev, A.; Chen, Y.-T.; Su, S.-H.; Tseng, Y.-J.; Karttunen, A. J.; Tunik, S. P.; Chou, P.-T.; Koshevoy, I. O. Chem. Commun. 2017, 53, 10954–10957. doi:10.1039/c7cc06882d
Return to citation in text: [1] -
Du, C.; Ye, S.; Liu, Y.; Guo, Y.; Wu, T.; Liu, H.; Zheng, J.; Cheng, C.; Zhu, M.; Yu, G. Chem. Commun. 2010, 46, 8573–8575. doi:10.1039/c0cc04147e
Return to citation in text: [1] -
Meech, S. R.; Phillips, D. J. Photochem. 1983, 23, 193–217. doi:10.1016/0047-2670(83)80061-6
Return to citation in text: [1] -
Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. doi:10.1039/b912178a
Return to citation in text: [1] -
Sakamoto, T.; Kondo, Y.; Miura, N.; Hayashi, K.; Yamanaka, H. Heterocycles 1986, 24, 2311–2314. doi:10.3987/r-1986-08-2311
Return to citation in text: [1] -
Dyker, G.; Stirner, W.; Henkel, G. Eur. J. Org. Chem. 2000, 1433–1441. doi:10.1002/(sici)1099-0690(200004)2000:8<1433::aid-ejoc1433>3.0.co;2-7
Return to citation in text: [1] -
Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Chem. – Eur. J. 2007, 13, 5632–5641. doi:10.1002/chem.200700202
Return to citation in text: [1] [2] [3] [4] -
Parker, E.; Leconte, N.; Godet, T.; Belmont, P. Chem. Commun. 2011, 47, 343–345. doi:10.1039/c0cc02623a
Return to citation in text: [1] [2] [3] -
Dell’Acqua, M.; Castano, B.; Cecchini, C.; Pedrazzini, T.; Pirovano, V.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 3494–3505. doi:10.1021/jo5002559
Return to citation in text: [1] [2] [3] -
Harrison, T. J.; Kozak, J. A.; Corbella-Pané, M.; Dake, G. R. J. Org. Chem. 2006, 71, 4525–4529. doi:10.1021/jo060382c
Return to citation in text: [1] [2] [3] [4] -
Trost, B. M.; Rudd, M. T. J. Am. Chem. Soc. 2005, 127, 4763–4776. doi:10.1021/ja043097o
Return to citation in text: [1] -
Jazkewitsch, O.; Mondrzyk, A.; Staffel, R.; Ritter, H. Macromolecules 2011, 44, 1365–1371. doi:10.1021/ma1027627
Return to citation in text: [1]
| 63. | Harrison, T. J.; Kozak, J. A.; Corbella-Pané, M.; Dake, G. R. J. Org. Chem. 2006, 71, 4525–4529. doi:10.1021/jo060382c |
| 60. | Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Chem. – Eur. J. 2007, 13, 5632–5641. doi:10.1002/chem.200700202 |
| 61. | Parker, E.; Leconte, N.; Godet, T.; Belmont, P. Chem. Commun. 2011, 47, 343–345. doi:10.1039/c0cc02623a |
| 62. | Dell’Acqua, M.; Castano, B.; Cecchini, C.; Pedrazzini, T.; Pirovano, V.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 3494–3505. doi:10.1021/jo5002559 |
| 63. | Harrison, T. J.; Kozak, J. A.; Corbella-Pané, M.; Dake, G. R. J. Org. Chem. 2006, 71, 4525–4529. doi:10.1021/jo060382c |
| 1. | Castellino, S.; Sims, J. J. Tetrahedron Lett. 1984, 25, 4059–4062. doi:10.1016/s0040-4039(01)90181-9 |
| 2. | Lathbury, D.; Gallagher, T. Tetrahedron Lett. 1985, 26, 6249–6252. doi:10.1016/s0040-4039(00)95065-2 |
| 3. | Grimaldi, J.; Cormons, A. Tetrahedron Lett. 1986, 27, 5089–5090. doi:10.1016/s0040-4039(00)85140-0 |
| 4. | Pale, P.; Chuche, J. Tetrahedron Lett. 1987, 28, 6447–6448. doi:10.1016/s0040-4039(00)96884-9 |
| 5. | Sawamura, M.; Hamashima, H.; Ito, Y. J. Org. Chem. 1990, 55, 5935–5936. doi:10.1021/jo00311a007 |
| 6. | Hayashi, T.; Uozumi, Y.; Yamazaki, A.; Sawamura, M.; Hamashima, H.; Ito, Y. Tetrahedron Lett. 1991, 32, 2799–2802. doi:10.1016/0040-4039(91)85090-r |
| 7. | Kimura, M.; Fugami, K.; Tanaka, S.; Tamaru, Y. Tetrahedron Lett. 1991, 32, 6359–6362. doi:10.1016/0040-4039(91)80169-7 |
| 8. | Marshall, J. A.; Wang, X. J. J. Org. Chem. 1991, 56, 960–969. doi:10.1021/jo00003a013 |
| 9. | Cooper, D. M.; Grigg, R.; Hargreaves, S.; Kennewell, P.; Redpath, J. Tetrahedron 1995, 51, 7791–7808. doi:10.1016/0040-4020(95)00397-q |
| 10. | Yanagisawa, A.; Nakashima, H.; Ishiba, A.; Yamamoto, H. J. Am. Chem. Soc. 1996, 118, 4723–4724. doi:10.1021/ja9603315 |
| 19. | Alderson, J. M.; Corbin, J. R.; Schomaker, J. M. Acc. Chem. Res. 2017, 50, 2147–2158. doi:10.1021/acs.accounts.7b00178 |
| 23. | Türkmen, Y. E.; Montavon, T. J.; Kozmin, S. A.; Rawal, V. H. J. Am. Chem. Soc. 2012, 134, 9062–9065. doi:10.1021/ja302537j |
| 24. | Zeng, Y.; Zhang, L.; Zhao, Y.; Ni, C.; Zhao, J.; Hu, J. J. Am. Chem. Soc. 2013, 135, 2955–2958. doi:10.1021/ja312711c |
| 25. | Zeng, Y.; Hu, J. Chem. – Eur. J. 2014, 20, 6866–6870. doi:10.1002/chem.201402846 |
| 26. | Tseberlidis, G.; Dell'Acqua, M.; Valcarenghi, D.; Gallo, E.; Rossi, E.; Abbiati, G.; Caselli, A. RSC Adv. 2016, 6, 97404–97419. doi:10.1039/c6ra22231e |
| 48. | Zehm, D.; Fudickar, W.; Linker, T. Angew. Chem., Int. Ed. 2007, 46, 7689–7692. doi:10.1002/anie.200700443 |
| 49. | Zehm, D.; Fudickar, W.; Hans, M.; Schilde, U.; Kelling, A.; Linker, T. Chem. – Eur. J. 2008, 14, 11429–11441. doi:10.1002/chem.200801355 |
| 21. | Naodovic, M.; Yamamoto, H. Chem. Rev. 2008, 108, 3132–3148. doi:10.1021/cr068413r |
| 22. | Pellissier, H. Chem. Rev. 2016, 116, 14868–14917. doi:10.1021/acs.chemrev.6b00639 |
| 50. | Suzuki, A.; Kondo, K.; Akita, M.; Yoshizawa, M. Angew. Chem., Int. Ed. 2013, 52, 8120–8123. doi:10.1002/anie.201302789 |
| 51. | Yazaki, K.; Akita, M.; Prusty, S.; Chand, D. K.; Kikushi, T.; Sato, H.; Yoshizawa, M. Nat. Commun. 2017, 8, 15914–15921. doi:10.1038/ncomms15914 |
| 18. | Zheng, Q.-Z.; Jiao, N. Chem. Soc. Rev. 2016, 45, 4590–4627. doi:10.1039/c6cs00107f |
| 19. | Alderson, J. M.; Corbin, J. R.; Schomaker, J. M. Acc. Chem. Res. 2017, 50, 2147–2158. doi:10.1021/acs.accounts.7b00178 |
| 20. | Fang, G.; Cong, X.; Zanoni, G.; Liu, Q.; Bi, X. Adv. Synth. Catal. 2017, 359, 1422–1502. doi:10.1002/adsc.201601179 |
| 44. | Mongin, C.; Pianet, I.; Jonusauskas, G.; Bassani, D. M.; Bibal, B. ACS Catal. 2015, 5, 380–387. doi:10.1021/cs5016063 |
| 45. | Cao, Z.; Bassani, D. M.; Bibal, B. Chem. – Eur. J. 2018, 24, 18779–18787. doi:10.1002/chem.201804322 |
| 11. | Halbes-Letinois, U.; Weibel, J.-M.; Pale, P. Chem. Soc. Rev. 2007, 36, 759–769. doi:10.1039/b602151b |
| 12. | Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Rev. 2008, 108, 3174–3198. doi:10.1021/cr078361l |
| 13. | Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075–6089. doi:10.1002/ejoc.200900790 |
| 14. | Muñoz, M. P. Chem. Soc. Rev. 2014, 43, 3164–3184. doi:10.1039/c3cs60408j |
| 15. | Fang, G.; Bi, X. Chem. Soc. Rev. 2015, 44, 8124–8173. doi:10.1039/c5cs00027k |
| 16. | Blanc, A.; Bénéteau, V.; Weibel, J.-M.; Pale, P. Org. Biomol. Chem. 2016, 14, 9184–9205. doi:10.1039/c6ob01468b |
| 17. | Sekine, K.; Yamada, T. Chem. Soc. Rev. 2016, 45, 4524–4532. doi:10.1039/c5cs00895f |
| 46. | Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Muldoon, J.; McGlinchey, M. J. Org. Lett. 2011, 13, 256–259. doi:10.1021/ol102665y |
| 47. | Marriott, P. J.; Lai, Y.-H. J. Chromatogr. A 1998, 447, 29–41. doi:10.1016/0021-9673(88)90004-0 |
| 35. | Küppers, H.-J.; Wieghardt, K.; Tsay, Y.-H.; Krüger, C.; Nuber, B.; Weiss, J. Angew. Chem., Int. Ed. Engl. 1987, 26, 575–576. doi:10.1002/anie.198705751 |
| 36. | De Groot, B.; Jenkins, H. A.; Loeb, S. J. Inorg. Chem. 1992, 31, 203–208. doi:10.1021/ic00028a015 |
| 37. | Blake, A. J.; Gould, R. O.; Parsons, S.; Radek, C.; Schröder, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 2374–2376. doi:10.1002/anie.199523741 |
| 38. | Alberto, R.; Nef, W.; Smith, A.; Kaden, T. A.; Neuburger, M.; Zehnder, M.; Frey, A.; Abram, U.; Schubiger, P. A. Inorg. Chem. 1996, 35, 3420–3427. doi:10.1021/ic951421y |
| 39. | Blake, A. J.; Li, W.-S.; Lippolis, V.; Schröder, M. Chem. Commun. 1997, 1943–1944. |
| 40. | Alberto, R.; Angst, D.; Schubiger, A. P.; Abram, U.; Ortner, K.; Kaden, T. A. Chem. Commun. 1999, 1513–1514. doi:10.1039/a903455b |
| 41. | Blake, A. J.; Champness, N. R.; Howdle, S. M.; Webb, P. B. Inorg. Chem. 2000, 39, 1035–1038. doi:10.1021/ic991145d |
| 42. | Casula, A.; Nairi, V.; Fernández-Moreira, V.; Laguna, A.; Lippolis, V.; Garau, A.; Gimeno, M. C. Dalton Trans. 2015, 44, 18506–18517. doi:10.1039/c5dt02723c |
| 43. | Chang, Y.-P.; Levason, W.; Reid, G. Dalton Trans. 2016, 45, 18393–18416. doi:10.1039/c6dt03409h |
| 35. | Küppers, H.-J.; Wieghardt, K.; Tsay, Y.-H.; Krüger, C.; Nuber, B.; Weiss, J. Angew. Chem., Int. Ed. Engl. 1987, 26, 575–576. doi:10.1002/anie.198705751 |
| 42. | Casula, A.; Nairi, V.; Fernández-Moreira, V.; Laguna, A.; Lippolis, V.; Garau, A.; Gimeno, M. C. Dalton Trans. 2015, 44, 18506–18517. doi:10.1039/c5dt02723c |
| 31. | Chen, Q.-A.; Zeng, W.; Wang, D.-W.; Zhou, Y.-G. Synlett 2009, 2236–2241. doi:10.1055/s-0029-1217805 |
| 32. | Iglesias-Sigüenza, J.; Ros, A.; Díez, E.; Magriz, A.; Vázquez, A.; Álvarez, E.; Fernández, R.; Lassaletta, J. M. Dalton Trans. 2009, 8485–8488. doi:10.1039/b910846g |
| 33. | Prakash, O.; Joshi, H.; Kumar, U.; Sharma, A. K.; Singh, A. K. Dalton Trans. 2015, 44, 1962–1968. doi:10.1039/c4dt02813a |
| 34. | Rasheed, O. K.; Bawn, C.; Davies, D.; Raftery, J.; Vitorica-Yrzebal, I.; Pritchard, R.; Zhou, H.; Quayle, P. Eur. J. Org. Chem. 2017, 5252–5261. doi:10.1002/ejoc.201701033 |
| 39. | Blake, A. J.; Li, W.-S.; Lippolis, V.; Schröder, M. Chem. Commun. 1997, 1943–1944. |
| 40. | Alberto, R.; Angst, D.; Schubiger, A. P.; Abram, U.; Ortner, K.; Kaden, T. A. Chem. Commun. 1999, 1513–1514. doi:10.1039/a903455b |
| 42. | Casula, A.; Nairi, V.; Fernández-Moreira, V.; Laguna, A.; Lippolis, V.; Garau, A.; Gimeno, M. C. Dalton Trans. 2015, 44, 18506–18517. doi:10.1039/c5dt02723c |
| 22. | Pellissier, H. Chem. Rev. 2016, 116, 14868–14917. doi:10.1021/acs.chemrev.6b00639 |
| 22. | Pellissier, H. Chem. Rev. 2016, 116, 14868–14917. doi:10.1021/acs.chemrev.6b00639 |
| 27. | Ji, J.-X.; Au-Yeung, T. T.-L.; Wu, J.; Yip, C. W.; Chan, A. S. C. Adv. Synth. Catal. 2004, 346, 42–44. doi:10.1002/adsc.200303148 |
| 28. | Yao, X.; Li, C.-J. Org. Lett. 2005, 7, 4395–4398. doi:10.1021/ol051575+ |
| 29. | Takechi, S.; Yasuda, S.; Kumagai, N.; Shibasaki, M. Angew. Chem., Int. Ed. 2012, 51, 4218–4222. doi:10.1002/anie.201200520 |
| 30. | Su, Y.; Lu, M.; Dong, B.; Chen, H.; Shi, X. Adv. Synth. Catal. 2014, 356, 692–696. doi:10.1002/adsc.201300785 |
| 35. | Küppers, H.-J.; Wieghardt, K.; Tsay, Y.-H.; Krüger, C.; Nuber, B.; Weiss, J. Angew. Chem., Int. Ed. Engl. 1987, 26, 575–576. doi:10.1002/anie.198705751 |
| 36. | De Groot, B.; Jenkins, H. A.; Loeb, S. J. Inorg. Chem. 1992, 31, 203–208. doi:10.1021/ic00028a015 |
| 37. | Blake, A. J.; Gould, R. O.; Parsons, S.; Radek, C.; Schröder, M. Angew. Chem., Int. Ed. Engl. 1995, 34, 2374–2376. doi:10.1002/anie.199523741 |
| 38. | Alberto, R.; Nef, W.; Smith, A.; Kaden, T. A.; Neuburger, M.; Zehnder, M.; Frey, A.; Abram, U.; Schubiger, P. A. Inorg. Chem. 1996, 35, 3420–3427. doi:10.1021/ic951421y |
| 56. | Meech, S. R.; Phillips, D. J. Photochem. 1983, 23, 193–217. doi:10.1016/0047-2670(83)80061-6 |
| 57. | Suzuki, K.; Kobayashi, A.; Kaneko, S.; Takehira, K.; Yoshihara, T.; Ishida, H.; Shiina, Y.; Oishi, S.; Tobita, S. Phys. Chem. Chem. Phys. 2009, 11, 9850–9860. doi:10.1039/b912178a |
| 52. | Iida, A.; Yamaguchi, S. Chem. Commun. 2009, 3002–3004. doi:10.1039/b901794a |
| 53. | Moon, H.; Jun, Y. W.; Kim, D.; Ryu, H. G.; Wang, T.; Kim, K. H.; Huh, Y.; Jung, J.; Ahn, K. H. Chem. – Asian J. 2016, 11, 2518–2523. doi:10.1002/asia.201600986 |
| 54. | Belyaev, A.; Chen, Y.-T.; Su, S.-H.; Tseng, Y.-J.; Karttunen, A. J.; Tunik, S. P.; Chou, P.-T.; Koshevoy, I. O. Chem. Commun. 2017, 53, 10954–10957. doi:10.1039/c7cc06882d |
| 55. | Du, C.; Ye, S.; Liu, Y.; Guo, Y.; Wu, T.; Liu, H.; Zheng, J.; Cheng, C.; Zhu, M.; Yu, G. Chem. Commun. 2010, 46, 8573–8575. doi:10.1039/c0cc04147e |
| 63. | Harrison, T. J.; Kozak, J. A.; Corbella-Pané, M.; Dake, G. R. J. Org. Chem. 2006, 71, 4525–4529. doi:10.1021/jo060382c |
| 63. | Harrison, T. J.; Kozak, J. A.; Corbella-Pané, M.; Dake, G. R. J. Org. Chem. 2006, 71, 4525–4529. doi:10.1021/jo060382c |
| 64. | Trost, B. M.; Rudd, M. T. J. Am. Chem. Soc. 2005, 127, 4763–4776. doi:10.1021/ja043097o |
| 65. | Jazkewitsch, O.; Mondrzyk, A.; Staffel, R.; Ritter, H. Macromolecules 2011, 44, 1365–1371. doi:10.1021/ma1027627 |
| 60. | Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Chem. – Eur. J. 2007, 13, 5632–5641. doi:10.1002/chem.200700202 |
| 61. | Parker, E.; Leconte, N.; Godet, T.; Belmont, P. Chem. Commun. 2011, 47, 343–345. doi:10.1039/c0cc02623a |
| 62. | Dell’Acqua, M.; Castano, B.; Cecchini, C.; Pedrazzini, T.; Pirovano, V.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 3494–3505. doi:10.1021/jo5002559 |
| 60. | Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Chem. – Eur. J. 2007, 13, 5632–5641. doi:10.1002/chem.200700202 |
| 60. | Godet, T.; Vaxelaire, C.; Michel, C.; Milet, A.; Belmont, P. Chem. – Eur. J. 2007, 13, 5632–5641. doi:10.1002/chem.200700202 |
| 61. | Parker, E.; Leconte, N.; Godet, T.; Belmont, P. Chem. Commun. 2011, 47, 343–345. doi:10.1039/c0cc02623a |
| 62. | Dell’Acqua, M.; Castano, B.; Cecchini, C.; Pedrazzini, T.; Pirovano, V.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 3494–3505. doi:10.1021/jo5002559 |
| 11. | Halbes-Letinois, U.; Weibel, J.-M.; Pale, P. Chem. Soc. Rev. 2007, 36, 759–769. doi:10.1039/b602151b |
| 12. | Álvarez-Corral, M.; Muñoz-Dorado, M.; Rodríguez-García, I. Chem. Rev. 2008, 108, 3174–3198. doi:10.1021/cr078361l |
| 13. | Belmont, P.; Parker, E. Eur. J. Org. Chem. 2009, 6075–6089. doi:10.1002/ejoc.200900790 |
| 14. | Muñoz, M. P. Chem. Soc. Rev. 2014, 43, 3164–3184. doi:10.1039/c3cs60408j |
| 15. | Fang, G.; Bi, X. Chem. Soc. Rev. 2015, 44, 8124–8173. doi:10.1039/c5cs00027k |
| 16. | Blanc, A.; Bénéteau, V.; Weibel, J.-M.; Pale, P. Org. Biomol. Chem. 2016, 14, 9184–9205. doi:10.1039/c6ob01468b |
| 17. | Sekine, K.; Yamada, T. Chem. Soc. Rev. 2016, 45, 4524–4532. doi:10.1039/c5cs00895f |
| 58. | Sakamoto, T.; Kondo, Y.; Miura, N.; Hayashi, K.; Yamanaka, H. Heterocycles 1986, 24, 2311–2314. doi:10.3987/r-1986-08-2311 |
| 59. | Dyker, G.; Stirner, W.; Henkel, G. Eur. J. Org. Chem. 2000, 1433–1441. doi:10.1002/(sici)1099-0690(200004)2000:8<1433::aid-ejoc1433>3.0.co;2-7 |
© 2019 Cao et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)